表面形貌可控的高粘附性改性纤维及其制备方法与应用
文献发布时间:2023-06-19 11:47:31
技术领域
本发明涉及一种改性纤维及其制备方法,特别涉及一种表面形貌可控、高粘附性及优异力学性能的改性纤维及其制备方法与应用,属于材料科学技术领域。
背景技术
在复合材料大家族中,纤维增强材料一直是人们关注的焦点,纤维增强复合材料具有比强度高、比模量大、可设计性、抗腐蚀性和耐久性能好等优点。这些特点使得纤维增强材料广泛地应用于各种民用、航空航天等领域中。但是传统的增强纤维,如玻璃纤维、超高分子量聚乙烯(UHMWPE)纤维及碳纤维等材料,表面光滑、极性基团含量较少,导致其与基体树脂之间界面粘结强度弱,受力时纤维易从基体中拔出,严重制约了其在复合材料领域中的应用。因此制备高表面形貌可控的纤维,提高其与树脂基体的粘结性能的同时保持纤维优异力学性能,一直是近年来的研究热点。
目前纤维的改性主要通过两类方法,一类是复合纺丝法,在纺丝时加入另一种含有极性基团的组分并纺丝;一类是表面改性法,主要通过氧化处理、等离子处理、辐照与涂覆等表面处理方法在纤维表面引入极性基团。公开号为CN112030563A的中国发明专利公开了一种单向导湿功能的异形截面纤维非织造布的制备方法,基于绿色的多巴胺化学在纤维表面形成粗糙结构,且多巴胺是一种天然的粘蛋白物质,比起传统的化学试剂更加具有环境友好性。公开号为CN112010572A的中国发明专利公开了一种导电玻璃纤维及其制备方法,该专利通过单宁酸的自身氧化在玻璃纤维表面沉积形成聚单宁酸功能涂层。公开号为CN108221389A的中国发明专利公开了一种提高涤纶通丝耐磨性能的涂层方法,它以水性聚氨酯浆料作为涂层浆料,将预处理后的涤纶浸渍于涂层浆料中进行浸轧,经烘干,热定型处理,得到涂层涤纶通丝,该发明选用水性聚氨酯有效改善其它涂层与涤纶间的粘附效果。公开号为CN103993479A的中国发明专利公开了一种硅烷交联改性超高分子量聚乙烯纤维的制备方法,将未经干燥处理的超高分子量聚乙烯UHMWPE冻胶纤维置于改性溶液中,超声处理,然后进行多级热拉伸,即得具有较高的表面粘结性能力学性能基本保持的UHMWPE纤维。尽管上述方法均能够制得粘附性提升的纤维,但是均依然存在一些不足:1、制备工艺繁琐,纺丝过程中改性需要对现有纺丝工艺进行改造,增加生产成本;2、氧化法或辐照改性会以降低纤维力学性能为代价,且需要额外的处理设备;3、具有多酚类仿生结构化合物(如多巴胺、单宁酸等)进行表面涂覆时,能够在保持纤维力学性能的前提下实现提升粘附性的目的,但是该方法得到的纤维表面较为平滑、形貌不可控,粘附性提升主要是依靠表面极性基团与基体树脂间的键接或非共价键作用得到的。
另外,目前传统的多酚类物质表面涂覆法依靠酚羟基与基体表面的氢键、范德华力等非共价键实现对基体表面的有效涂覆,且该涂覆条件温和,有效地保持了纤维的优异力学性能。但是,该方法依然存在一些问题:1、多酚类物质形成的涂层在纤维表面以薄膜形式存在,尽管相比于未改性的纤维表面变得粗糙,但是表面的形貌无法实现有效地控制;2、该涂层与基体树脂的作用力主要依靠的仍然是多酚类物质与基体树脂之间形成的共价键或非共价键,而粗糙表面与基体树脂之间的机械互锁作用无法调控。
因此有必要开发出一种新的粘附性提升的纤维及其制备方法。
发明内容
本发明的主要目的在于提供一种表面形貌可控、高粘附性及优异力学性能的改性纤维及其制备方法与应用,以克服现有技术中的不足。
为实现前述发明目的,本发明采用的技术方案包括:
本发明实施例提供了一种表面形貌可控的高粘附性改性纤维,包括纤维基体和覆盖纤维基体的涂层;所述涂层具有由纳米粒子构成的微纳结构,其中所述纳米粒子的尺寸为1-500nm,所述纳米粒子的形状包括球状、片状和柱状中的至少任意一种;以及,所述涂层中Si的含量占涂层总质量的2-15wt%,N的含量占涂层总质量的1-8wt%,并且在所述改性纤维的Si特征精细谱中,峰值为101.1eV处的特征峰面积与103.2eV处的特征峰面积的比值为5:1-50:1。
其中,所述纤维的总质量为纤维基体和涂层质量之和。
在一些实施方式中,所述改性纤维与水的接触角为50-95°。
在一些实施方式中,所述改性纤维的拉伸强度为3-6GPa,所述改性纤维的拉伸强度保持率为80%-99%,所述改性纤维的环氧树脂包埋测试单丝拔出力为10-50cN,所述改性纤维的环氧树脂/改性纤维复合材料界面剪切强度为5-30Mpa。
在一些实施方式中,所述纤维基体包括超高分子量聚乙烯纤维、碳纤维或芳纶纤维等。
本发明实施例还提供了一种表面形貌可控的高粘附性改性纤维的制备方法,包括如下步骤:
(1)对纤维基体进行清洗;
(2)将胺类化合物加入硅烷偶联剂反应溶液中,反应得到末端含有氨基的硅氧烷改性氨基化合物;
(3)将步骤(2)所获硅氧烷改性氨基化合物加入pH值为7-10的三羟甲基氨基甲烷缓冲溶液中反应,之后加入多酚类化合物和金属离子,其后加入经步骤(1)清洗后的纤维基体继续反应,从而获得所述表面形貌可控的高粘附性改性纤维。
在一些实施方式中,所述胺类化合物包括乙二胺、二亚乙基三胺、三亚乙基四胺、四亚乙基五胺和聚乙烯亚胺等中的任意一种或两种以上的组合。
在一些实施方式中,所述多酚类化合物包括多巴胺、邻苯二酚、邻苯三酚、5-羟基-1,4-萘醌、单宁酸等中的任意一种或两种以上的组合。
在一些实施方式中,所述金属离子包括Fe
本发明实施例还提供了由前述方法制备的表面形貌可控的高粘附性改性纤维。
本发明实施例还提供了所述表面形貌可控的高粘附性改性纤维于制备聚合物基复合材料中的用途。
与现有技术相比,本发明具有如下有益效果:
(1)本发明提供的表面形貌可控的高粘附性改性纤维表面产生大量极性基团,而且纤维表面形貌(球型、片状和柱状)可精确控制,有助于纤维与基体树脂之间发生共价键、非共价键及机械互锁提升粘附性,且该改性纤维的力学性能优异;
(2)本发明提供的改性纤维的制备工艺简便可行、成本可控、涂覆条件不苛刻,便于大规模生产。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明中记载的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
图1a-图1e是本发明一典型实施方案中表面改性纤维制备时采用的不同胺类化合物的结构及反应位点示意图;
图2是本发明一典型实施方案中一种表面形貌可控的高粘附性改性纤维的制备工艺原理图。
具体实施方式
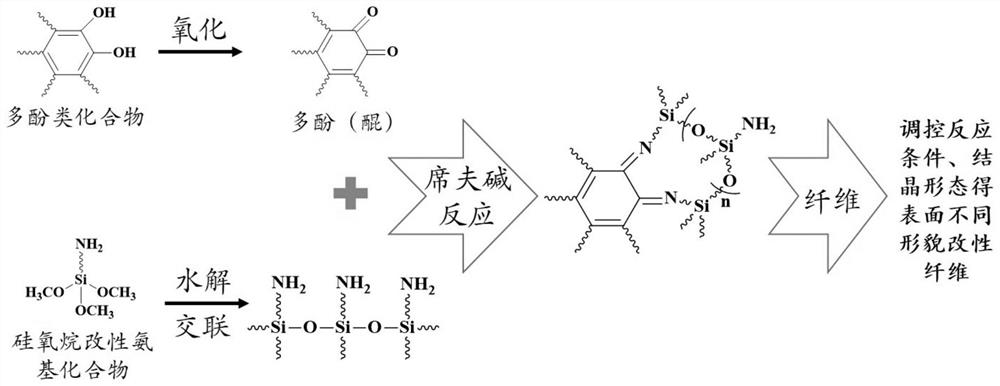
鉴于现有技术中的不足,本案发明人经长期研究和大量实践,得以提出本发明的技术方案,其主要是以多酚类化合物的氧化产物苯醌基与硅氧烷改性氨基化合物的两个端氨基发生席夫碱反应生成有机笼化合物,通过调控该化合物的结晶过程及形态实现对涂覆表面形貌的精确控制。如下将对该技术方案、其实施过程及原理等作进一步的解释说明。
本发明实施例的一个方面提供的是一种表面形貌可控的高粘附性及优异力学性能的改性纤维,该纤维表面形貌可控、力学性能优异,且该纤维与基体树脂间的粘附性强。
具体的,所述表面形貌可控、高粘附性及优异力学性能的改性纤维包括纤维基体和覆盖纤维基体的涂层;所述涂层具有由纳米粒子构成的微纳结构,其中所述纳米粒子的尺寸(直径或一端到另一端的最大值)为1-500nm,所述纳米粒子的形状包括球状、片状和柱状中的至少任意一种;以及,所述涂层中Si的含量占涂层总质量的2-15wt%,N的含量占涂层总质量的1-8wt%,并且使用XPS测试发现,在所述改性纤维的Si特征精细谱中,存在两个峰值在101.1eV和103.2eV附近,分别对应Si-O-C和Si-O-Si特征峰,峰值为101.1eV处的特征峰面积与103.2eV处的特征峰面积的比值为5:1-50:1。
进一步的,所述表面形貌可控、高粘附性及优异力学性能的改性纤维分为涂覆层和纤维基体,涂覆层是在纤维基体表面涂覆一层微纳形貌可控的涂层。
进一步的,所述涂层的质量为纤维基体质量的1-10wt%。
进一步的,所述涂层的厚度为2-500nm。
进一步的,所述改性纤维与水的接触角为50-95°。
进一步的,所述改性纤维的拉伸强度为3-6GPa。
进一步的,所述改性纤维的拉伸强度保持率(改性纤维与未改性纤维的比值)为80%-99%。
进一步的,所述改性纤维的环氧树脂包埋测试单丝拔出力为10-50cN。
进一步的,所述改性纤维的环氧树脂/改性纤维复合材料界面剪切强度为5-30Mpa。
进一步的,所述纤维基体包括但不限于超高分子量聚乙烯(UHMWPE)纤维、碳纤维或芳纶纤维中的任意一种。
本发明实施例的另一个方面提供了一种表面形貌可控、高粘附性及优异力学性能的改性纤维的制备方法,其包括:
(1)对纤维基体进行清洗;
(2)将胺类化合物加入硅烷偶联剂反应溶液中,反应得到末端含有氨基的硅氧烷改性氨基化合物;
(3)将步骤(2)所获硅氧烷改性氨基化合物加入pH值为7-10的三羟甲基氨基甲烷缓冲溶液中反应,之后加入多酚类化合物和金属离子,其后加入经步骤(1)清洗后的纤维基体继续反应,从而获得所述表面形貌可控的高粘附性改性纤维。
进一步的,步骤(1)包括:将纤维基体在溶剂中超声处理,之后取出干燥;所述溶剂包括但不限于乙醇、丙酮、丙二醇、丙三醇、正己烷、乙二醇等中的任意一种或多种的组合。
进一步的,所述纤维基体包括但不限于超高分子量聚乙烯(UHMWPE)纤维、碳纤维或芳纶纤维等中的任意一种。
在一些实施方式中,步骤(2)包括:在10-80℃(优选50-80℃)的温度下将胺类化合物加入到硅烷偶联剂反应溶液中,搅拌5-120min,之后冷冻干燥,得到所述硅氧烷改性氨基化合物。
进一步的,所述硅氧烷改性氨基化合物的重均分子量为100-2000。
进一步的,所述胺类化合物包括但不限于乙二胺、二亚乙基三胺、三亚乙基四胺、四亚乙基五胺和聚乙烯亚胺等中的任意一种或两种以上的组合;并且,乙二胺、二亚乙基三胺、三亚乙基四胺、四亚乙基五胺和聚乙烯亚胺等化合物中的反应位点示意图请参阅图1a-图1e所示,其中,所述聚乙烯亚胺的n值为5-200。
进一步的,所述硅烷偶联剂反应溶液包括水、醇中的任意一种或两种的组合与KH560的混合物,亦可称为KH560反应溶液。
进一步的,所述的KH560反应溶液为水、乙醇、乙二醇、丙三醇中的一种或几种的混合物。
进一步的,所述醇包括乙醇、乙二醇、丙三醇等中的任意一种或两种以上的组合(比例为任意比),但不限于此。
进一步的,所述混合物中KH560的质量占总质量的1%-20%,水的质量占总质量的0.1%-10%,醇的质量占总质量的98.9%-70%。
进一步的,所述胺类化合物中氨基与KH560中环氧基团的摩尔比为0.8:1-2:1。
进一步的,所述硅氧烷改性氨基化合物是硅烷偶联剂的环氧基团与氨基化合物的氨基(或亚氨基)发生开环反应,得到末端含有氨基的化合物,以乙二胺和二亚乙基三胺的①号反应位点反应产物为例(见如下结构),不同胺类化合物的不同反应位点均能够参与反应得到末端含有氨基的硅氧烷改性氨基化合物。
进一步的,所述硅氧烷改性氨基化合物的结构可表示为:
进一步的,在所述硅氧烷改性氨基化合物的Si特征精细谱中,存在两个峰值在101.1eV和103.2eV附近,分别对应Si-O-C和Si-O-Si特征峰的面积比大于500:1,亦即,峰值为101.1eV处的特征峰面积与103.2eV处的特征峰面积的比值大于500:1。
在一些实施方式中,步骤(3)包括:将步骤(2)所获硅氧烷改性氨基化合物加入pH值为7-10的三羟甲基氨基甲烷缓冲溶液(亦可称为Tris缓冲溶液)中形成第一反应体系,并进行水解交联反应0.1-1小时,之后加入多酚类化合物和金属离子形成第二反应体系进行席夫碱反应生成有机笼化合物,其后迅速加入经步骤(1)清洗后的纤维基体继续进行反应1-48小时,将所获纤维洗涤干燥,从而获得所述表面形貌可控、高粘附性及优异力学性能的改性纤维。
在一些实施方式中,所述多酚类化合物包括但不限于多巴胺、邻苯二酚、邻苯三酚、5-羟基-1,4-萘醌、单宁酸等中的任意一种或两种以上的组合。
在一些实施方式中,所述金属离子包括但不限于Fe
进一步的,所述第二反应体系中金属离子的浓度为0.1-10mg/ml。
进一步的,所述第二反应体系中多酚类化合物的浓度为0.5-5mg/ml。
进一步的,所述第一反应体系中硅氧烷改性氨基化合物的浓度为0.1-4mg/ml。
进一步的,所述水解交联反应的温度为20-60℃,优选的,所述水解交联反应的温度为20-50℃,更优选的,所述水解交联反应的温度为30-50℃。
进一步的,所述席夫碱反应的温度为20-60℃,优选为30-50℃。
进一步的,步骤(3)中,所述三羟甲基氨基甲烷缓冲溶液(即Tris缓冲溶液)包含浓度为0.05-0.2mol/L的三羟甲基氨基甲烷、水和醇类化合物,亦即,所述的Tris缓冲溶液中Tris的物质的量浓度为0.05-0.2mol/L,缓冲溶液为水和醇类物质的混合物。
进一步的,所述醇类化合物包括但不限于乙醇、乙二醇、丙三醇等中的任意一种或两种以上的组合。
进一步的,所述三羟甲基氨基甲烷缓冲溶液中水的质量占水与醇类物质二者总质量的5%-30%,醇类化合物的质量占水与醇类物质二者总质量的70%-95%。
本发明实施例的另一个方面提供了由前述方法制备的表面形貌可控的高粘附性改性纤维。
本发明实施例的另一个方面提供了所述表面形貌可控的高粘附性改性纤维于制备聚合物基复合材料中的用途。
进一步的,所述聚合物基复合材料为纤维增强树脂复合材料。
请参阅图1,本发明实施例利用多酚类物质能够在空气作用下氧化为醌类化合物,醌类化合物的碳氧双键(C=O)能够与硅氧烷改性氨基化合物的氨基发生席夫碱反应,生成有机笼化合物(请参阅图2)。通过控制反应条件、有机笼化合物结晶过程实现对有机笼化合物不同形貌的精确调控。
其中,在一些更为具体的实施案例之中,本发明的表面形貌可控、高粘附性及优异力学性能的改性纤维的制备方法的具体步骤可以是:
步骤1、纤维的清洗:
纤维表面清洗过程为纤维基体在溶剂中超声处理,之后取出干燥;所述溶剂包括但不限于乙醇、丙酮等中的任意一种或多种的组合。
所述纤维是指UHMWPE纤维、碳纤维或芳纶纤维中的任一种。
步骤2、硅氧烷改性氨基化合物的配制:在10-80℃(优选50-80℃)的温度下将胺类化合物加入到KH560反应溶液中,搅拌5~120min,冷冻干燥,得到硅氧烷改性氨基化合物。
步骤3、表面形貌可控改性纤维的制备:首先将步骤2制得的硅氧烷改性氨基化合物加入到pH为7-10的Tris缓冲溶液中,反应0.1-1小时后加入多酚类化合物和金属离子,然后将步骤1得到的清洗后纤维浸入到上述混合液中,继续反应1-48小时后将纤维洗涤干燥,得表面形貌可控、高粘附性及优异力学性能的改性纤维。
其中,本发明中硅氧烷改性氨基化合物的结构受到制备条件的影响,KH560反应溶液中水的含量过少时,难以发生环氧基团与氨基的开环反应,水的含量过多时,硅氧烷基团会发生水解交联反应,无法得到目标产物。此外,硅氧烷改性氨基化合物的分子量在一定范围内(重均分子量为100-2000)才能起到控制纤维表面形貌的目的,这是由于分子量过高时,含有氨基的分子链会对硅氧烷基团形成包裹,阻碍水解交联反应的发生。
在本发明的以上实施例中,硅氧烷改性氨基化合物水解交联产物的结构影响产物的形貌与结构,一方面,该水解交联产物为两端氨基的化合物,方便后续发生席夫碱反应得到成环的有机笼化合物;另一方面水解交联产物的结构能够通过改变水解交联条件进行调控,得到具有不同分子链结构(网状或直线状)、分子量大小的水解交联产物,从而达到控制有机笼化合物结晶形貌及晶体大小的目的。
在本发明的以上实施例中,硅氧烷改性氨基化合物在Tris缓冲液中的预反应时间对于有机笼化合物的生成产生影响,该预反应主要目的是硅氧烷的水解交联,水解交联时间过短无法得到分子链两端具有氨基的化合物,时间过长硅氧烷改性氨基化合物会发生大量的自聚合,无法进行下一步的席夫碱反应。Tris缓冲溶液中醇类化合物和水溶液的比值也会对有机笼化合物的生成产生影响,醇类物质过高,硅氧烷无法进行水解交联,醇类物质过低,硅氧烷的水解交联过程很快完成,无法对水解交联产物的结构进行调控,进而难以形成结构形貌可控的有机笼化合物。多酚类化合物和硅氧烷改性氨基化合物的浓度也会影响产物的形貌,浓度过高,生成结晶过快,对形貌的精确调控难以实现;浓度过低,发生反应的概率较低,表面纳米粒子分布过少,纤维的粘附性提升较弱。此外,加入金属离子可以起到诱导结晶及控制结晶形貌的作用,这也是我们经过大量实验得到的,同样的,金属离子的种类和浓度也会影响改性纤维表面形貌及其粘附性。
综述之,本发明提供的表面形貌可控的高粘附性改性纤维表面产生大量极性基团,而且纤维表面形貌(球型、片状和柱状)可精确控制,有助于纤维与基体树脂之间发生共价键、非共价键及机械互锁提升粘附性,且该改性纤维的力学性能优异;同时,该改性纤维的制备工艺简便可行、成本可控、涂覆条件不苛刻,便于大规模生产。
如下将结合若干具体实施例对本发明的技术方案作更为详尽的说明。若非特别说明,如下各对比例及各实施例所采用的纤维及其它原料均从市场途径获取,且其中采用的各类反应设备、测试设备、测试方法等也均是本领域人员已知的。
下面实施例中所述的试验方法,如无特殊说明,均为常规方法;所述试剂和材料,如无特殊说明,均可从商业途径获得。
实施例1
(1)纤维的清洗以及硅氧烷改性氨基化合物的配制:将UHMWPE纤维浸入丙酮中超声处理1h,洗涤后室温下干燥;10℃下将乙二胺加入到KH560反应溶液(乙醇和水混合溶液)中,其中水质量占KH560、乙醇、水三者总质量的0.1%,KH560的质量占总质量的1%,乙醇质量占总质量的98.9%,乙二胺中氨基与KH560中环氧基团的摩尔比为0.8:1,搅拌5min后冷冻干燥,得到重均分子量为100的硅氧烷改性氨基化合物,该化合物的Si特征精细谱中,Si-O-C和Si-O-Si特征峰的面积比为501:1。
(2)表面形貌可控改性纤维的制备:首先将步骤2制得的硅氧烷改性氨基化合物加入到pH值为7、浓度为0.05mol/L的Tris缓冲溶液(水和乙醇的混合物,水的质量占乙醇、水二者总质量的5%,乙醇质量占二者总质量的95%)中,得到硅氧烷改性氨基化合物浓度为0.1mg/ml的混合溶液,20℃反应0.1小时后加入多巴胺(浓度为0.5mg/ml)和金属离子Fe
改性后UHMWPE纤维表面的纳米粒子形状为球型,尺寸为1nm,涂层的质量为纤维基体质量的1wt%,厚度为2nm,表面Si含量占涂层总质量的2wt%,N含量占涂层总质量的1wt%,使用XPS测试发现,在Si的特征精细谱中,存在两个峰值在101.1eV和103.2eV附近,分别对应Si-O-C和Si-O-Si特征峰的面积比为50:1。改性后纤维与水的接触角为95°,改性纤维的拉伸强度为3GPa,改性后拉伸强度保持率(改性纤维与未改性纤维比值)为80%,基体树脂包埋测试单丝拔出力为10cN,基体树脂/改性纤维复合材料界面剪切强度为5Mpa。
实施例2
(1)纤维的清洗以及硅氧烷改性氨基化合物的配制:将碳纤维浸入乙醇中超声处理1h,洗涤后室温下干燥;80℃下将二亚乙基三胺加入到KH560反应溶液中(乙醇和水混合溶液),其中水质量占KH560、乙醇、水三者总质量的10%,KH560的质量占总质量的20%,乙醇质量占总质量的70%,乙二胺中氨基与KH560中环氧基团的摩尔比为2:1,搅拌120min后冷冻干燥,得到重均分子量为2000的硅氧烷改性氨基化合物,该化合物的Si特征精细谱中,Si-O-C和Si-O-Si特征峰的面积比为600:1。
(2)表面形貌可控改性纤维的制备:首先将步骤2制得的硅氧烷改性氨基化合物加入到pH值为10、浓度为0.2mol/L的Tris缓冲溶液(水和乙二醇的混合物,水的质量占乙二醇、水二者总质量的30%,乙醇质量占缓冲溶液总质量的70%)中,得到硅氧烷改性氨基化合物浓度为4mg/ml的混合溶液,60℃反应0.5小时后加入邻苯二酚(浓度为5mg/ml)和金属离子Al
改性后碳纤维表面的纳米粒子形状为片状,尺寸为500nm,涂层的质量为纤维基体质量的10wt%,厚度为500nm,表面Si含量占涂层总质量的15wt%,N含量占涂层总质量的8wt%,使用XPS测试发现,在Si的特征精细谱中,存在两个峰值在101.1eV和103.2eV附近,分别对应Si-O-C和Si-O-Si特征峰的面积比为5:1。改性后纤维与水的接触角为50°,改性纤维的拉伸强度为6GPa,改性后拉伸强度保持率(改性纤维与未改性纤维比值)为99%,基体树脂包埋测试单丝拔出力为50cN,基体树脂/改性纤维复合材料界面剪切强度为30Mpa。
实施例3
(1)纤维的清洗以及硅氧烷改性氨基化合物的配制:将碳纤维浸入丙二醇中超声处理1h,洗涤后室温下干燥;45℃下将三亚乙基四胺加入到KH560反应溶液(乙二醇和水混合溶液)中,其中水质量占KH560、乙二醇、水三者总质量的5%,KH560的质量占总质量的10%,乙二醇质量占总质量的85%,三亚乙基四胺中氨基与KH560中环氧基团的摩尔比为1.4:1,搅拌60min后冷冻干燥,得到重均分子量为1000的硅氧烷改性氨基化合物,该化合物的Si特征精细谱中,Si-O-C和Si-O-Si特征峰的面积比为510:1。
(2)表面形貌可控改性纤维的制备:首先将步骤2制得的硅氧烷改性氨基化合物加入到pH值为8、浓度为0.12mol/L的Tris缓冲溶液(水和丙三醇的混合物,水的质量占丙三醇、水二者总质量的18%,丙三醇质量占缓冲溶液总质量的82%)中,得到硅氧烷改性氨基化合物浓度为2mg/ml的混合溶液,50℃反应1小时后加入邻苯二酚(浓度为5mg/ml)和金属离子Ag
改性后碳纤维表面的纳米粒子形状为柱状,尺寸为250nm,涂层的质量为纤维基体质量的5wt%,厚度为280nm,表面Si含量占涂层总质量的8wt%,N含量占涂层总质量的4wt%,使用XPS测试发现,在Si的特征精细谱中,存在两个峰值在101.1eV和103.2eV附近,分别对应Si-O-C和Si-O-Si特征峰的面积比为28:1。改性后纤维与水的接触角为72°,改性纤维的拉伸强度为4.5GPa,改性后拉伸强度保持率(改性纤维与未改性纤维比值)为90%,基体树脂包埋测试单丝拔出力为30cN,基体树脂/改性纤维复合材料界面剪切强度为18Mpa。
实施例4
(1)纤维的清洗以及硅氧烷改性氨基化合物的配制:将芳纶纤维浸入丙三醇中超声处理1h,洗涤后室温下干燥;50℃下将四亚乙基五胺加入到KH560反应溶液(丙三醇和水混合溶液)中,其中水质量占KH560、丙三醇、水三者总质量的8%,KH560的质量占总质量的15%,丙三醇质量占总质量的77%,四亚乙基五胺中氨基与KH560中环氧基团的摩尔比为1.5:1,搅拌100min后冷冻干燥,得到重均分子量为1200的硅氧烷改性氨基化合物,该化合物的Si特征精细谱中,Si-O-C和Si-O-Si特征峰的面积比为700:1。
(2)表面形貌可控改性纤维的制备:首先将步骤2制得的硅氧烷改性氨基化合物加入到pH值为9、浓度为0.12mol/L的Tris缓冲溶液(水和乙二醇、丙三醇的混合物,水的质量占乙二醇、丙三醇、水三者总质量的25%,乙二醇、丙三醇质量占缓冲溶液总质量的75%)中,得到硅氧烷改性氨基化合物浓度为2mg/ml的混合溶液,30℃反应1小时后加入邻苯三酚(浓度为4mg/ml)和金属离子为Cu
改性后碳纤维表面的纳米粒子形状为片状,尺寸为400nm,涂层的质量为纤维基体质量的8wt%,厚度为300nm,表面Si含量占涂层总质量的10wt%,N含量占涂层总质量的4wt%,使用XPS测试发现,在Si的特征精细谱中,存在两个峰值在101.1eV和103.2eV附近,分别对应Si-O-C和Si-O-Si特征峰的面积比为40:1。改性后纤维与水的接触角为80°,改性纤维的拉伸强度为4.6GPa,改性后拉伸强度保持率(改性纤维与未改性纤维比值)为90%,基体树脂包埋测试单丝拔出力为38cN,基体树脂/改性纤维复合材料界面剪切强度为25Mpa。
实施例5
(1)纤维的清洗以及硅氧烷改性氨基化合物的配制:将UHMWPE纤维浸入正己烷中超声处理1h,洗涤后室温下干燥;65℃下将聚乙烯亚胺(n值为5)加入到KH560反应溶液(乙醇、乙二醇、丙三醇和水混合溶液)中,其中水质量占KH560、乙醇、乙二醇、丙三醇、水五者总质量的8%,KH560的质量占总质量的8%,醇类(乙醇、乙二醇、丙三醇任意比混溶)质量占总质量的84%,聚乙烯亚胺中氨基与KH560中环氧基团的摩尔比为1.2:1,搅拌80min后冷冻干燥,得到重均分子量为1900的硅氧烷改性氨基化合物,该化合物的Si特征精细谱中,Si-O-C和Si-O-Si特征峰的面积比为650:1。
(2)表面形貌可控改性纤维的制备:首先将步骤2制得的硅氧烷改性氨基化合物加入到pH值为9、浓度为0.16mol/L的Tris缓冲溶液(水和乙醇、乙二醇的混合物,水的质量占乙醇、乙二醇、水三者总质量的15%,乙醇、乙二醇质量占缓冲溶液总质量的85%)中,得到硅氧烷改性氨基化合物浓度为2.4mg/ml的混合溶液,40℃反应0.5小时后加入5-羟基-1,4-萘醌(浓度为3mg/ml)和金属离子为Zn
改性后UHMWPE纤维表面的纳米粒子形状为球型,尺寸为200nm,涂层的质量为纤维基体质量的3wt%,厚度为210nm,表面Si含量占涂层总质量的10wt%,N含量占涂层总质量的6wt%,使用XPS测试发现,在Si的特征精细谱中,存在两个峰值在101.1eV和103.2eV附近,分别对应Si-O-C和Si-O-Si特征峰的面积比为33:1。改性后纤维与水的接触角为80°,改性纤维的拉伸强度为5GPa,改性后拉伸强度保持率(改性纤维与未改性纤维比值)为89%,基体树脂包埋测试单丝拔出力为32cN,基体树脂/改性纤维复合材料界面剪切强度为20Mpa。
实施例6
(1)纤维的清洗以及硅氧烷改性氨基化合物的配制:将芳纶纤维浸入乙二醇中超声处理1h,洗涤后室温下干燥;40℃下将聚乙烯亚胺(n值为200)加入到KH560反应溶液(丙三醇和水混合溶液)中,其中水质量占KH560、丙三醇、水三者总质量的18%,KH560的质量占总质量的6%,丙三醇质量占总质量的76%,乙二胺中氨基与KH560中环氧基团的摩尔比为1.8:1,搅拌90min后冷冻干燥,得到重均分子量为800的硅氧烷改性氨基化合物,该化合物的Si特征精细谱中,Si-O-C和Si-O-Si特征峰的面积比为800:1。
(2)表面形貌可控改性纤维的制备:首先将步骤2制得的硅氧烷改性氨基化合物加入到pH值为9、浓度为0.16mol/L的Tris缓冲溶液(水和乙二醇的混合物,水的质量占乙二醇、水二者总质量的30%,乙醇质量占缓冲溶液总质量的70%)中,得到硅氧烷改性氨基化合物浓度为4mg/ml的混合溶液,60℃反应1小时后加入单宁酸(浓度为4mg/ml)和金属离子Fe
改性后芳纶纤维表面的纳米粒子形状为柱状,尺寸为200nm,涂层的质量为纤维基体质量的6wt%,厚度为300nm,表面Si含量占涂层总质量的11wt%,N含量占涂层总质量的6wt%,使用XPS测试发现,在Si的特征精细谱中,存在两个峰值在101.1eV和103.2eV附近,分别对应Si-O-C和Si-O-Si特征峰的面积比为35:1。改性后纤维与水的接触角为58°,改性纤维的拉伸强度为5.6GPa,改性后拉伸强度保持率(改性纤维与未改性纤维比值)为87%,基体树脂包埋测试单丝拔出力为39cN,基体树脂/改性纤维复合材料界面剪切强度为25Mpa。
对比例1(未改性的UHMWPE纤维)
未改性纤维的清洗:将UHMWPE纤维浸入丙酮中超声处理1h,洗涤后室温下干燥。
该UHMWPE纤维未经处理,表面无涂层存在,其表面孔径为900nm,BET比表面积为20m
该UHMWPE纤维与水的接触角为120°,拉伸强度为3.5GPa,拉伸应力为118cN,环氧树脂包埋测试单丝拔出力为4cN,环氧树脂/改性纤维复合材料界面剪切强度为3Mpa。
对比例2
本对比例与实施例1相比,不同之处在于:步骤(1)中未加入乙二胺。
改性后UHMWPE纤维表面没有观察到形状规则的纳米颗粒,涂层质量为纤维基体质量的0.8wt%,厚度为3nm,表面Si含量占涂层总质量的为16wt%,N含量占涂层总质量的为10wt%,使用XPS测试发现,在Si的特征精细谱中,存在两个峰值在101.1eV和103.2eV附近,分别对应Si-O-C和Si-O-Si特征峰的面积比为60:1。
所获改性UHMWPE纤维与水的接触角为100°,拉伸强度为5GPa,改性后拉伸强度保持率(改性纤维与未改性纤维比值)为79%,基体树脂包埋测试单丝拔出力为8cN,基体树脂/改性纤维复合材料界面剪切强度为4Mpa。
对比例3
本对比例与实施例1相比,不同之处在于:步骤(2)中未加入金属离子Fe
改性后UHMWPE纤维表面没有观察到形状规则的纳米颗粒,涂层质量为纤维基体质量的12wt%,厚度为600nm,表面Si含量占涂层总质量的为19wt%,N含量占涂层总质量的为12wt%,使用XPS测试发现,在Si的特征精细谱中,存在两个峰值在101.1eV和103.2eV附近,分别对应Si-O-C和Si-O-Si特征峰的面积比为4:1。
所获改性UHMWPE纤维与水的接触角为101°,拉伸强度为3.5GPa,改性后拉伸强度保持率(改性纤维与未改性纤维比值)为76%,基体树脂包埋测试单丝拔出力为4cN,基体树脂/改性纤维复合材料界面剪切强度为2Mpa。
对比例4
本对比例与实施例1相比,不同之处在于:步骤(2)中未加入多巴胺。
改性后UHMWPE纤维表面没有观察到形状规则的纳米颗粒,涂层质量为纤维基体质量的0.3wt%,厚度为1nm,表面Si含量占涂层总质量的为1wt%,N含量占涂层总质量的为0.8wt%,使用XPS测试发现,在Si的特征精细谱中,无法观察到两个峰值在101.1eV和103.2eV附近的Si-O-C和Si-O-Si特征峰。
所获改性UHMWPE纤维与水的接触角为110°,拉伸强度为5.4GPa,改性后拉伸强度保持率(改性纤维与未改性纤维比值)为60%,基体树脂包埋测试单丝拔出力为4cN,基体树脂/改性纤维复合材料界面剪切强度为2Mpa。
尽管已参考说明性实施例描述了本发明,但所属领域的技术人员将理解,在不背离本发明的精神及范围的情况下可做出各种其它改变、省略及/或添加且可用实质等效物替代所述实施例的元件。另外,可在不背离本发明的范围的情况下做出许多修改以使特定情形或材料适应本发明的教示。因此,本文并不打算将本发明限制于用于执行本发明的所揭示特定实施例,而是打算使本发明将包含归属于所附权利要求书的范围内的所有实施例。
- 表面形貌可控的高粘附性改性纤维及其制备方法与应用
- 一种具有高粘附性复合功能水凝胶的制备方法及其应用