一种冷冻睾丸组织精原干细胞分离方法
文献发布时间:2023-06-19 18:46:07
技术领域
本发明属于细胞生物学领域,具体涉及一种冷冻睾丸组织精原干细胞分离方法。
背景技术
精子发生包括精原细胞有丝分裂、精母细胞减数分裂和精子形成三个阶段。有丝分裂过程包括未分化精原细胞(单个型(A
然而,精原干细胞在睾丸中的占比非常少,在牛中占比仅为0.2%~0.3%。因此,需要对分离的精原干细胞进行富集纯化以满足培养和移植需求。精原干细胞分离主要采用两步酶消化法,富集和纯化的主要方法包括:免疫磁珠分选、流式细胞分选、差异贴壁法、密度梯度离心法等。与其他富集方法相比,差异贴壁法对精原干细胞富集产生了最佳结果,但它特异性较低。免疫磁珠和流式细胞分选可以富集高纯度的精原干细胞。例如Li等利用流式细胞分选或免疫磁珠分离以获得Thy1
发明内容
有鉴于此,本发明的目的在于提供一种冷冻睾丸组织精原干细胞分离方法,可最大限度保持精原干细胞内环境,维持细胞活性,减少细胞损伤。
为了实现上述发明目的,本发明提供了以下技术方案:
本发明提供了一种冷冻睾丸组织精原干细胞分离方法,包括以下步骤:
冷冻睾丸组织复苏,复苏后加入胶原酶Ⅳ和DNA酶Ⅰ进行消化,至曲细精管暴露、管壁毛糙终止;加入DPBS缓冲液洗涤,离心,培养至细胞迁出;收集迁出细胞,进行精原干细胞富集纯化。
优选的,所述复苏方法包括:将冻存管放在36~38℃水浴锅中进行复苏至完全解冻。
优选的,所述冷冻睾丸组织包括冷冻牛睾丸组织。
优选的,所述睾丸组织依次加入等体积的DPBS缓冲液、胶原酶Ⅳ溶液和DNA酶Ⅰ溶液进行消化。
优选的,所述胶原酶Ⅳ溶液浓度为0.5~2mg/mL,所述DNA酶Ⅰ溶液浓度为0.5~2mg/mL。
优选的,所述消化条件为:36~38℃水浴孵育。
优选的,所述培养条件为:36~38℃,5%CO
优选的,所述精原干细胞富集纯化方法为:采用Percoll梯度离心,纯化,得到精原干细胞。
优选的,离心管中依次加入40%Percoll溶液、20%Percoll溶液和迁出细胞悬液,离心后取40%Percoll溶液和20%Percoll溶液中间相,得到精原干细胞。
优选的,所述40%Percoll溶液、20%Percoll溶液和迁出细胞悬液的体积比为1:1:2。
与现有技术相比,本发明具有如下有益效果:
本发明通过对曲细精管短暂培养可以维持精原干细胞内环境稳定,同时使细胞自然迁出,可减少两步酶消化法中胰酶对细胞的损伤,保持细胞活性,更适用于冷冻睾丸组织中精原干细胞的分离,不仅解决了用于分离细胞的组织长时间保存问题,还为珍稀濒危物种的遗传信息保存及个体复原更新提供了重要支持。
本发明所述实验方法简单便捷,易于操作,利于推广实施。
附图说明
图1为酶消化后暴露的曲细精管;
图2为曲细精管细胞迁出贴壁;
图3为精原干细胞团簇状生长,黑色箭头指示精原干细胞;
图4为精原干细胞荧光染色鉴定结果。
具体实施方式
本发明提供了一种冷冻睾丸组织精原干细胞分离方法,包括以下步骤:
冷冻睾丸组织复苏,复苏后加入胶原酶Ⅳ和DNA酶Ⅰ进行消化,至曲细精管暴露、管壁毛糙终止;加入DPBS缓冲液洗涤,离心,培养至细胞迁出;收集迁出细胞,进行精原干细胞富集纯化。
本发明所述冷冻睾丸组织优选为冷冻牛睾丸组织。
本发明所述冷冻睾丸组织,需要先经复苏,然后再用于精原干细胞分离,所述复苏方法可选为:将冻存管从液氮中取出,立即放在36~38℃水浴锅中进行复苏,等完全解冻后进行后续细胞分离实验。所述水浴锅温度更优选为37℃。
本发明先对复苏后的睾丸组织中的曲细精管进行分离,在分离前优选为先将解冻后睾丸组织加入DPBS缓冲液洗涤。作为一种可选的实施方式,本发明将解冻后睾丸组织放入离心管中,加入含10%青霉素、链霉素溶液(P/S)的DPBS缓冲液,冰上静置4~6min,去上清,重复2~3遍。
本发明向睾丸组织中依次加入DPBS缓冲液、胶原酶Ⅳ溶液和DNA酶Ⅰ溶液进行消化,所述DPBS缓冲液、胶原酶Ⅳ溶液和DNA酶Ⅰ溶液的体积比优选为1:1:1。所述胶原酶Ⅳ溶液浓度为0.5~2mg/mL,优选为1mg/mL;所述DNA酶Ⅰ溶液浓度为0.5~2mg/mL,优选为1mg/mL。本发明所述消化条件为:36~38℃水浴孵育;作为一种可选的实施方式,本发明将待消化体系放在37℃水浴锅中孵育,期间多次摇晃,并在显微镜下观察,直至曲细精管暴露,管壁毛糙为止,终止消化。
本发明向分离得到的曲细精管中加入DPBS缓冲液终止消化,冰上静置8~15min,弃上清,重复操作2~3遍,以去除间质细胞。然后再次加入DPBS缓冲液重悬沉淀,离心,用DFS培养基重悬细胞,进行细胞培养,培养至细胞迁出。本发明所述培养条件为36~38℃,优选为37℃,所述培养时间优选为22~24h。作为可选的实施方式,本发明采用DFS培养基重悬后接种到6孔板中,于5%CO
本发明所述DFS培养基的配制优选为:88.5%~90%DMEM培养基+8.5%~10.5%FBS+1%~1.5%青霉素、链霉素溶液(P/S)。
本发明取出接种有曲细精管的培养板,在显微镜下观察细胞迁出,观察细胞迁出后,用吸管吹打,收集细胞悬液,静置8~12min,取上清过40μm细胞筛,收集过筛后的细胞。
本发明将收集后的细胞洗涤,制备DPBS细胞悬液。作为可选的实施方式,本发明将过筛后的细胞离心,去上清,再次加入DPBS缓冲液洗涤,离心,去上清,加入DPBS缓冲液重悬得到DPBS细胞悬液;所述离心条件优选为600g,5min。
本发明所述精原干细胞富集纯化方法为:采用Percoll梯度离心,纯化,得到精原干细胞。
本发明对DPBS细胞悬液采用Percoll梯度离心。所述Percoll溶液为20%Percoll溶液和40%Percoll溶液。本发明在离心管中依次加入40%Percoll溶液、20%Percoll溶液和迁出细胞悬液,所述40%Percoll溶液、20%Percoll溶液和迁出细胞悬液的体积比为1:1:2;离心后取40%Percoll溶液和20%Percoll溶液中间相(离心后由于密度不同会呈现分层现象,精原干细胞位于40%Percoll溶液和20%Percoll溶液中间相),得精原干细胞;所述离心条件可选为600g,7min。
本发明中,40%Percoll和20%Percoll制备方法为:
40%Percoll:10%10×PBS缓冲液,50%超纯水,40%Percoll溶液,加入20μL酚红溶液,用浓盐酸调整pH到7.0。
20%Percoll:10%10×PBS缓冲液,70%超纯水,20%Percoll溶液,加入20μL酚红溶液,用浓盐酸调整pH到7.0。
本发明优选为将40%Percoll溶液和20%Percoll溶液中间相加入DPBS缓冲液洗涤2~3遍,用精原干细胞培养基重悬得到细胞悬液。作为可选的实施方式,本发明加入DPBS缓冲液后吹打均匀,离心,弃上清,重复洗涤;所述离心条件可选为600g,5min。
本发明将所得精原干细胞培养基重悬液接种到铺有滋养层细胞的培养板中进行培养。优选为将细胞放培养箱中培养,2~3天换一次培养液,5~7天传代培养。
作为可选的实施方式,本发明所述滋养层细胞制备方法为:使用DFS培养基培养小鼠胚胎成纤维(MEF)细胞,待细胞融合度达到70%~80%时,使用丝裂霉素C(10ng/mL)处理MEF细胞2h,加入0.25%胰酶消化3min后,加入DFS培养基终止消化,600g,5min离心收集细胞,用DFS培养基重悬MEF细胞并进行细胞计数,以5×10
本发明分离得到的精原干细胞可进行传代培养,所述传代培养方法可选为:等培养板细胞融合度达到70%~80%时,吸弃培养基,加入0.25%胰酶进行消化,约3min后,加入DFS培养基终止消化,吹打细胞到离心管中,600g,5min离心,吸弃上清,加入精原干细胞培养基重悬,接种到提前铺有滋养层细胞的培养板中进行培养。
下面结合实施例对本发明提供的技术方案进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
本发明具体实施例中,
DFS培养基:89%DMEM培养基+10%FBS+1%青霉素、链霉素溶液(P/S);
40%Percoll:10%10×PBS缓冲液,50%超纯水,40%Percoll溶液,加入20μL酚红溶液,用浓盐酸调整pH到7.0;
20%Percoll:10%10×PBS缓冲液,70%超纯水,20%Percoll溶液,加入20μL酚红溶液,用浓盐酸调整pH到7.0;
滋养层细胞制备:使用DFS培养基培养小鼠胚胎成纤维(MEF)细胞,待细胞融合度达到70%~80%时,使用丝裂霉素C(10ng/mL)处理MEF细胞2h,加入0.25%胰酶2mL消化3min后,加入2mLDFS培养基终止消化,600g,5min离心收集细胞,用1mLDFS培养基重悬MEF细胞并进行细胞计数,以5×10
精原干细胞培养基:MEM-Alpha基础培养基(Gibco,12561-049)、牛血清白蛋白(sigma,A6003)5mg/mL、全转铁蛋白(sigma,T0665)100μg/mL、胰岛素(sigma,12643)25μg/mL、L-谷氨酰胺溶液100×(普诺赛,PB180419)1×、腐胺(sigma,P5780)20μg/mL、亚硒酸钠(sigma,214485)0.3nM、2-硫基乙醇(Gibco,21985)55μM、MEM非必需氨基酸100×溶液(Gibco,11140)1×、MEM维生素100×溶液(Gibco,11120052)1×、StemProTM添加剂50×溶液(Gibco,A1050801)1×、GDNF(Peprotech,AF-450-10)20ng/mL、FGF(Peprotech,AF-100-18B)5ng/mL。
下述实施例中,如无特殊说明,均为常规方法。
下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
实施例1
本实施例提供了冷冻保存的牛睾丸组织精原干细胞分离培养方法:
(1)睾丸组织冷冻保存
冷冻保存液配制:
将DMEM、FBS和DMSO按照7:2:1的比例配制成冷冻保存液,并用0.22μm滤器过滤备用。
睾丸组织采集及冻存:
将采集的3~5月龄睾丸组织去掉白膜,在睾丸中间位置横切,用刀片沿横切面缓慢刮取睾丸组织薄片(黄豆大小)放入冻存管中,加入约1.5mL冷冻保存液并混匀,然后采用梯度降温法,将冻存管横向放置在4℃1~2h,-20℃1~2h,-80℃12~16h,最后在液氮中长期保存。
(2)冷冻组织复苏
将液氮中保存的睾丸组织冻存管取出,立即放37℃水浴锅中解冻,待完全融化后,将冻存管中组织转移至50mL离心管中。
(3)精原干细胞分离培养
曲细精管分离培养:
用DPBS缓冲液进行洗涤,每次加入10mLDPBS缓冲液(含10%P/S)缓冲液,洗3遍,期间多次吹打,使里面组织尽量散开。然后分别加入5mL DPBS缓冲液、5mL胶原酶Ⅳ溶液(1mg/mL)、5mLDNA酶Ⅰ溶液(1mg/mL),放37℃水浴锅中消化15min,期间多次吹打混匀,使组织完全分散开,直至曲细精管暴露,管壁毛糙为止(见图1)。然后加入30mL的DPBS缓冲液终止消化,将离心管放在冰上重力沉降10min,去掉上清液,加入10mLDPBS缓冲液洗涤,如此重复3遍,尽量去掉间质细胞,保留曲细精管。400g,离心5min,弃上清。加入3mLDFS培养基重悬曲细精管,接种到6孔板中,放37℃,5%CO
精原干细胞富集纯化:
24h后取出培养板,此时曲细精管贴壁,管内细胞大量迁出(见图2)。用吸管吹打,收集细胞悬液,静置10min,取上清过40μm细胞筛。将过筛后的细胞收集在一起,600g,5min离心,去上清,加入DPBS缓冲液洗涤,600g,5min离心,然后用2mLDPBS缓冲液重悬。取15mL离心管,按先后顺序分别加入1mL40%Percoll溶液、1mL20%Percoll溶液、2mL细胞悬液。600g离心7min,取40%Percoll溶液和20%Percoll溶液的中间相,加入8mL DPBS缓冲液吹打混匀,600g,离心5min。去上清液,重复两遍,用3mL精原干细胞培养基重悬,然后接种到铺有滋养层细胞的培养板中,放培养箱中培养,2~3天换一次培养液,5~7天传代。
(4)精原干细胞传代
等细胞融合度达到70%~80%时,吸弃培养基,加入0.25%胰酶2mL进行消化,等细胞消化变圆时,加入2mLDFS培养基终止消化,吹打细胞到15mL离心管中,600g,5min离心,吸弃上清,加入精原干细胞培养基重悬,接种到提前铺有滋养层细胞的培养板中培养,在P1代时,精原干细胞开始呈现团簇状生长(见图3)。
(5)精原干细胞鉴定
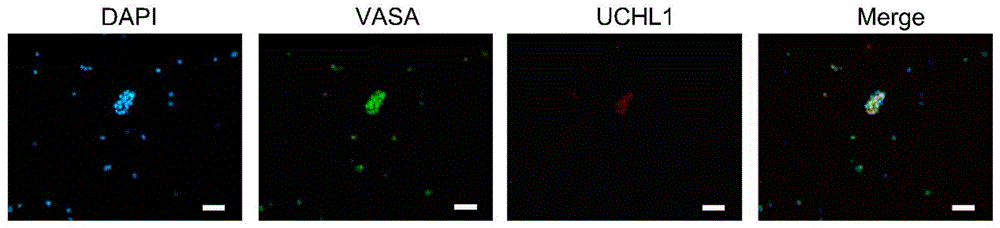
取出培养板,小心缓慢吹打使部分细胞脱落,吸取培养基,转移到15mL离心管中,向培养板中加入新的精原干细胞培养基。将吹打脱落的细胞600g,5min离心,吸弃上清,加入DPBS缓冲液重悬,600g,5min离心,弃上清,向沉淀中加入4%多聚甲醛固定液重悬,转移到黏附性载玻片上,冰上静置20min,待细胞沉降到载玻片表面时,吸弃固定液,加入DPBS缓冲液,放摇床上缓慢摇动洗涤,重复3遍,每遍5min。加入0.1%TritonX-100溶液,通透20min后吸弃,加入DPBS缓冲液洗3次,每次5min。吸弃DPBS缓冲液,加入10%驴血清封闭1h,然后加入稀释好的一抗,即鼠源UCHL1和兔源VASA,4℃孵育16h,加入DPBS缓冲液洗3次,每次10min,加入二抗,即驴抗兔488,驴抗鼠594,室温孵育2h,然后用DPBS缓冲液洗3次,每次10min。加入DAPI染液,染色1min后吸弃,50%甘油封片,荧光显微镜下观察(见图4)。
由图4可知,通过免疫荧光染色,生殖细胞标记基因VASA(绿色)和精原干细胞标记基因UCHL1(红色)对所分离培养的细胞均能明显着色,因此分离培养的细胞为精原干细胞。
目前对于牛睾丸组织分离培养均采用新鲜睾丸组织,暂未发现利用冷冻的牛睾丸组织进行精原干细胞分离培养的方法。本发明创新提出可适用于冷冻保存牛睾丸组织,经过复苏后培养精原干细胞的方法。此外,本发明还创新提出牛曲细精管片段培养法,使细胞自然迁出后富集纯化精原干细胞。经过本发明所述方法分离培养的牛精原干细胞,在P1代观察到了团簇状增殖的精原干细胞。
实施例2
本实施例与实施例1的区别在于:步骤(3)中,所述胶原酶Ⅳ溶液浓度为0.5mg/mL,所述DNA酶Ⅰ溶液浓度为0.5mg/mL。
实施例3
本实施例与实施例1的区别在于:步骤(3)中,所述胶原酶Ⅳ溶液浓度为2mg/mL,所述DNA酶Ⅰ溶液浓度为2mg/mL。
实施例4
本实施例与实施例1的区别在于:放38℃水浴锅中消化13min。
实施例5
本实施例与实施例1的区别在于:放36℃水浴锅中消化16min。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。