一种非贵金属析氯阳极及其制备方法和应用
文献发布时间:2023-06-19 18:37:28
技术领域
本发明属于电化学技术领域,具体地说,是涉及一种析氯阳极及其制备方法和应用。
背景技术
氯碱工业是以盐为原料生产烧碱、氯气、氢气的基础原料工业。氯碱产品种类很多,关联度大,具有较高的经济延伸价值,产品广泛应用于农业、石油化工电力、冶金纺织等领域。氯碱工业通常使用金属钛为基底材料,由Ru、Ir混合其他金属氧化物(MMO)为高效催化组分来制作稳定的阳极材料。由于过电位低和析氯选择性高等优点,氧化铑和氧化铱依旧成为了商业析氯电极的首选材料,但由于Ru、Ir含量高价格贵,导致生产成本高昂。所以,如何制备高效又低成本的析氯阳极成为了氯碱工业的核心问题。
目前相关技术的研究重点集中在对涂层活性的改性上,如在涂层中参杂多种元素的氧化物形成三元或者多元混合氧化物的涂层,甚至在基底材料不同表面涂敷浓度不同的金属离子制备渐近变化的氧化物涂层,由此来提高电极的催化性能。
发明内容
本发明旨在降低氯碱工业中阳极材料对贵金属的依赖,提供了一种非贵金属析氯阳极及其制备方法和应用,通过Co
为了解决上述技术问题,本发明通过以下的技术方案予以实现:
根据本发明的一个方面,提供了一种非贵金属析氯阳极,包括Ti基底,所述Ti基底上生长有多层的Co
进一步地,多层的所述Co
根据本发明的另一个方面,提供了上述非贵金属析氯阳极的制备方法,包括如下步骤:
(1)将Ti基底在有机溶剂中进行超声处理,之后洗净烘干后在酸溶液中浸泡处理,之后清洗并真空干燥;
(2)将得到的Ti基底浸泡于钴盐溶液中,并进行电化学沉积;所述电化学沉积的沉积电压为-0.5V~-0.9V,沉积时间为0.5-1.5h;
(3)沉积完成后进行煅烧热分解处理,得到生长在Ti基底上的Co
(4)多次重复步骤(2)和步骤(3),直至多层的所述Co
进一步地,步骤(1)中,所述有机溶剂为乙醇和丙酮中的至少一种。
进一步地,步骤(1)中,所述超声处理的时间为1h-2.5h。
进一步地,步骤(1)中,所述酸溶液为盐酸溶液和草酸溶液中至少一种,所述酸溶液的浓度为0.5-3mol/L,温度为50~100℃。
进一步地,步骤(2)中,所述钴盐溶液为硝酸钴溶液、硫酸钴溶液、氯化钴溶液、乙酸钴溶液中的至少一种,所述钴盐溶液的浓度为0.1-0.3mol/L。
进一步地,步骤(2)中,沉积电压为0.7V,沉积时间为1h。
进一步地,步骤(3)中,所述煅烧热分解处理的温度为200℃-400℃,时间为1-3h。
根据本发明的另一个方面,提供了上述非贵金属析氯阳极在析氯电催化中的应用。
本发明的有益效果是:
本发明采用电化学技术与传统催化剂制备相结合的方法,在极简的步骤下便可获得性能优异的析氯电极;本发明所制备的Co
附图说明
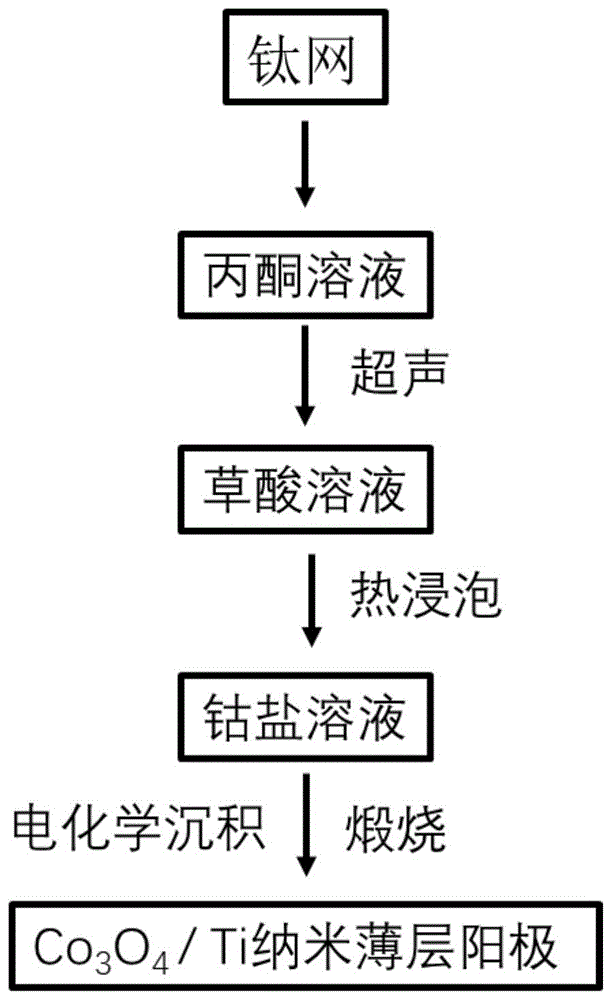
图1为本发明实施例1所提供的非贵金属析氯阳极的制备过程示意图;
图2为实施例1所制备的非贵金属析氯阳极中Co
图3为实施例1所制备的非贵金属析氯阳极中Co
具体实施方式
下面通过具体的实施例对本发明作进一步的详细描述,以下实施例可以使本专业技术人员更全面的理解本发明,但不以任何方式限制本发明。
实施例1
如图1所示,按照如下步骤制备非贵金属析氯阳极:
(1)将1.5cm*3.0cm的钛网浸泡于丙酮中,超声1.5h,并用去离子水冲洗干净;处理后的钛网浸入1.0mol/L的草酸溶液中并加热至80℃维持1.5h,之后清洗并真空干燥。
(2)配制好0.2mol/L的乙酸钴溶液,使用-0.7V的沉积电压沉积1h,使钴离子沉积在基底钛网上。
(3)将沉积完成的钛网烘干,放入马弗炉中;在空气氛围下以5℃/min的升温速率,达到300℃并保温3h,得到生长在Ti基底上的Co
(4)多次重复步骤(2)和步骤(3),直至多层的Co
样品XRD分析:
将负载钴金属的钛网进行干燥处理,对其进行XRD处理,采用布鲁克D2 PHASER型号的仪器进行检测,采用铜靶材料,具体扫描角度范围为10-80°,扫描速度5°/min,再采用JADE对其检测数据进行分析,结果如图1所示,该出峰为Co
材料的SEM图测试:
取块状材料1*1mm大小,清晰干净后干燥完毕,直接采用导电胶带把样品黏结在样品底座上,在扫描电镜下观察样品微观结构,结果如图2所示,从扫面电镜的结果可以看出,钛网表面长了一层颗粒状的固体,证明了电镀的方式让钛网表面附着了一层物质,进一步证明了电镀法的有效性。
实施例2
按照实施例1的方法制备非贵金属析氯阳极,区别在仅于步骤(2)中乙酸钴溶液的浓度为0.1mol/L。
实施例3
按照实施例1的方法制备非贵金属析氯阳极,区别在仅于步骤(2)中乙酸钴溶液的浓度为0.15mol/L。
实施例4
按照实施例1的方法制备非贵金属析氯阳极,区别在仅于步骤(2)中乙酸钴溶液的为0.25mol/L。
实施例5
按照实施例1的方法制备非贵金属析氯阳极,区别在仅于步骤(2)中乙酸钴溶液的为0.3mol/L
实施例6
按照实施例1的方法制备非贵金属析氯阳极,区别在仅于步骤(2)中的沉积电压为-0.5V。
实施例7
按照实施例1的方法制备非贵金属析氯阳极,区别在仅于步骤(2)中的沉积电压为-0.6V。
实施例8
按照实施例1的方法制备非贵金属析氯阳极,区别在仅于步骤(2)中的沉积电压为-0.8V。
实施例9
按照实施例1的方法制备非贵金属析氯阳极,区别在仅于步骤(2)中的沉积电压为-0.9V。
实施例10
按照实施例1的方法制备非贵金属析氯阳极,区别在仅于步骤(3)中的煅烧温度为200℃。
实施例11
按照实施例1的方法制备非贵金属析氯阳极,区别在仅于步骤(3)中的煅烧温度为250℃。
实施例12
按照实施例1的方法制备非贵金属析氯阳极,区别在仅于步骤(3)中的煅烧温度为350℃。
实施例13
按照实施例1的方法制备非贵金属析氯阳极,区别在仅于步骤(3)中的煅烧温度为400℃。
实施例14
按照实施例1的方法制备非贵金属析氯阳极,区别在仅于步骤(2)中的钴溶液替换成相同浓度的硝酸钴。
实施例15
按照实施例1的方法制备非贵金属析氯阳极,区别在仅于步骤(2)中的钴溶液替换成相同浓度的硫酸钴。
实施例16
按照实施例1的方法制备非贵金属析氯阳极,区别在仅于步骤(2)中的钴溶液替换成相同浓度的氯酸钴。
实施例17
按照实施例1的方法制备非贵金属析氯阳极,区别在仅于步骤(2)中沉积时间调整为0.5h。
实施例18
按照实施例1的方法制备非贵金属析氯阳极,区别在仅于步骤(2)中沉积时间调整为0.8h。
实施例19
按照实施例1的方法制备非贵金属析氯阳极,区别在仅于步骤(2)中沉积时间调整为1.2h。
实施例20
按照实施例1的方法制备非贵金属析氯阳极,区别在仅于步骤(2)中沉积时间调整为1.5h。
此外,本领域技术人员应当知晓:步骤(1)中用于超声处理的溶剂除丙酮之外,还可以是乙醇等有机溶剂;超声处理的时间通常为1h-2.5h。步骤(1)中的用于浸泡的除草酸溶液外还可以是盐酸溶液等;草酸溶液或盐酸溶液的浓度通常为0.5-3mol/L,浸泡温度优选为50~100℃。步骤(3)中煅烧热分解处理的时间一般为1-3h,升温速率也可以适当调整。
将以上所有实施例在氯碱-双氧水平台进行氯化钠电解效率测定,测定条件:阳极氯化钠起始浓度为310g/L(5L),阴极启动液为纯水(4L),电解电流I=75A,阴极液循环流速v=300ml/L,阴极进气量为4L/min,阳极液循环流速v=300ml/L,电解时间t=4h。
测试结果如下:
表1.对比实施例1-5不同钴盐浓度下氯化钠的电解效率
可见,随着钴盐浓度的增加,制备的电极材料在氯碱的电解生产中,其电解效率在不断的增加,当钴盐的浓度增加到0.3mol/L时,相较于0.2mol/L的钴盐,氯化钠的电解效率基本不变,因此在该电极制备工艺中钴盐浓度控制在0.1-0.2mol/L,0.2mol/L钴盐制备的电极电解效率最佳,归功于载体的比表面积有限,在0.2mol/L钴盐时电镀的钴金属已经达到其最大有效活性位点。
表2.对比实施例1与6-9不同沉积电压下氯化钠的电解效率
可见,在钴盐浓度为0.2mol/L,沉积电压为0.7V,制备的钴电极具有最佳的氯化钠电解效率,能够达到92.33%左右。
表3.对比实施例1与10-13不同煅烧温度下氯化钠的电解效率
可见,在盐浓度为0.2mol/L,沉积电压为0.7V,煅烧温度在300℃左右,制备的钴电极具有最佳的氯化钠电解效率,能够达到92.33%左右。
表4.对比实施例1与14-16不同钴盐下氯化钠的电解效率
可见,在盐浓度为0.2mol/L,沉积电压为0.7V,煅烧温度在300℃左右,采用不同的钴盐制备的钴电极的氯化钠电解效率均能达到92%左右,差别较小。
表5.对比实施例1与17-20不同沉积时间下氯化钠的电解效率
可见,在盐浓度为0.2mol/L,沉积电压为0.7V,煅烧温度在300℃左右,采用不同沉积时间制备不同的电极,结果显示随着沉积时间增加,氯化钠的电解效率在增加,沉积时间增加到1h,再继续沉积对氯化钠的电解效率增加的幅度较小,考虑到经济以及效率问题,该电极在1h的沉积效果最佳。
尽管上面结合附图对本发明的优选实施例进行了描述,但是本发明并不局限于上述的具体实施方式,上述的具体实施方式仅仅是示意性的,并不是限制性的,本领域的普通技术人员在本发明的启示下,在不脱离本发明宗旨和权利要求所保护的范围情况下,还可以作出很多形式的具体变换,这些均属于本发明的保护范围之内。