一种sgRNA文库构建方法
文献发布时间:2023-06-19 11:29:13
技术领域
本发明属于基因工程领域,具体的,涉及一种sgRNA文库构建方法。
背景技术
基于CRISPR-Cas系统的靶向基因组编辑技术是研究生物学和基因治疗的强 大工具,能够完成RNA导向的靶基因识别及编辑(敲除、插入、突变)。CRISPR-Cas 系统最初发现于细菌体内,CRISPR(clustered regularly interspaced short palindromic repeats)与Cas蛋白一起实现针对外源基因的免疫防御。CRISPR 是细菌内一段小段规律成簇间隔短回文序列,CRISPR-Cas9基因编辑系统主要由 Cas9和sgRNA组成,Cas9是与CRISPR序列相关的核酸内切酶,sgRNA为一段序 列特异性向导RNA分子(sequence-specific guide RNA),可引导Cas9到达靶 基因处,从而完成基因组的编辑。
除了多克隆筛选方法外,CRISPR-Cas可用于快速高效地生成大量的敲除细 胞,针对全基因组sgRNA的基因文库做大规模的功能基因筛选,如利用人源细 胞的慢病毒敲除sgRNA文库,筛选出疽热毒素(chimaeric anthrax,PA/LFnDTA) 和白喉毒素(diphtheriatoxin,DT)引起致病性的相关基因,这种基因筛选技 术不仅能用于探索致病机制,还能应用于基因治疗等相关领域。
发明内容
本发明的目的在于提供一种一种sgRNA文库构建方法。
本发明需要解决的技术问题为:
现有技术中,sgRNA为一段序列特异性向导RNA分子,可引导Cas9到达靶基 因处,从而完成基因组的编辑,针对全基因组sgRNA的基因文库做大规模的功能 基因筛选,筛选出疽热毒素和白喉毒素引起致病性的相关基因,这种基因筛选 技术不仅能用于探索致病机制,还能应用于基因治疗等相关领域。
本发明的目的可以通过以下技术方案实现:
一种sgRNA文库构建方法,包括以下步骤:
第一步、sgRNA设计:针对需编辑的基因设计sgRNA,每个基因通常设计3条 sgRNA,每条sgRNA 20bp,20个碱基作为靶基因的特异性识别序列;
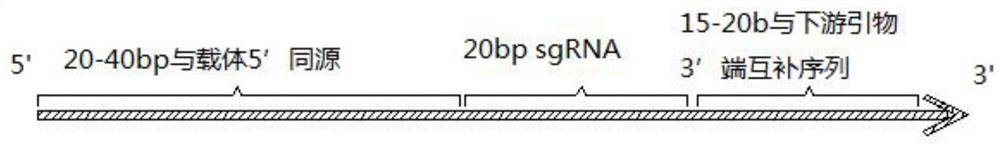
第二步、上下游引物合成:上游引物50-80bp,从5’端至3’端依次为与载 体的5’端20-40bp同源序列,20bp sgRNA,15-20b与下游引物3’端互补序列; 下游引物为通用引物,35-60bp,从5’端至3’端依次为与载体的5’端20-40bp 同源序列,15-20b与上游引物3’端互补序列;
第三步、构建sgRNA文库:(1)将第二步中N条上游引物等量混合均匀,再 与下游引物一起混合均匀,最终浓度为10μm;将其作为PCR扩增的引物和模板, 添加dNTP、PCR反应Buffer、高保真DNA聚合酶等,反应体系为10-100uL,进行 PCR扩增;PCR产物的大小在60-140bp之间,采用乙醇/异丙醇沉淀的方式回收pcr 产物;(2)以BsmBI酶切商品化质粒lentiCRISPR v2,并与纯化后的pcr产物通 过Gibson重组的方式进行连接;转化至感受态细胞后37℃下过夜培养,对慢病 毒载体lentiCRISPR v2优选重组酶recA13突变型菌株Stbl3,为提高转化效率, 宜选择电转感受态;(3)挑选单克隆,摇菌抽质粒,测序验证;(4)对测序 结果进行分析统计。
本发明的有益效果:
与现有技术相比,本发明构建sgRNA文库过程中发明采用乙醇/异丙醇沉淀 的方式回收pcr产物,较常规的pcr产物回收试剂盒对短片断的回收率较低,针 对全基因组sgRNA的基因文库做大规模的功能基因筛选,可以筛选出引起致病性 的相关基因,这种基因筛选技术不仅能用于探索致病机制,还能应用于基因治 疗等相关领域,与传统的退火连接方法相比,通过PCR的方法得到混合sgRNA库, 大大节省引物合成成本,提高合成产量。
附图说明
为了更清楚地说明本发明实施例的技术方案,下面将对实施例描述所需要 使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一 些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还 可以根据这些附图获得其他的附图。
图1为本发明上游特异性引物F的示意图;
图2为下游通用引物R的示意图;
图3为实施案例中sgRNA文库的Lenti-Crispr-v2载体图谱;
图4为实施案例中sgRNA文库的PCR产物琼脂糖凝胶电泳检测图;
图5为实施案例中随机挑选的14个克隆的测序验证结果。
图中:M为Marker5000;1和2为混合PCR产物。
具体实施方式
下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描 述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中 的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其 它实施例,都属于本发明保护的范围。
请参阅图1-5所示,本发明为一种sgRNA文库构建方法,本案例中选取了 张峰实验室提供的Mouse_GeCKOv2_Library_A中前20条sgRNA做实施。
1、引物合成。
合成上下游引物,引物序列如表1所示,其中MGLibA_F1至MGLibA_F20为 上游引物(含sgRNA序列),MGLibA-R为该文库的通用下游引物。
表1.sgRNA上游文库引物F
2、sgRNA文库构建
1)将上述20条上游引物等量混合均匀,终浓度为10um。
PCR反应体系如下:
添加dNTP、PCR反应Buffer、GPV8高保真DNA聚合酶(通用生物产品), 反应体系为50ul,进行PCR扩增,PCR体系如下:
PCR反应条件为:96℃3min;(96℃15s,58℃20s,72℃10s)X 25Cycles; 72℃1min;4℃hold。
PCR产物的理论大小为81bp,检测图如图4所示。
2)以BsmBI酶切商品化质粒lentiCRISPRv2,并与纯化后的PCR产物通过 Gibson重组的方式进行连接。
重组体系如下:
50℃反应30min。
3)将上述重组产物异丙醇沉淀法纯化
4)将纯化后的重组产物电转至感受态细胞Stbl3(购自唯地生物),37℃ 过夜培养。
5)次日挑选16个单克隆,摇菌抽质粒,测序验证。送测的16个克隆中, 2个为空载体,14个为阳性克隆。下表为16个克隆的测序结果,其中2个克隆 为空载克隆,其余14个均为成功克隆,从初步的实施结果来看,验证了通过 PCR的方式获取sgRNA方法的可行性。为构建更高通量的sgRNA文库,用芯片合 成引物池、二代测序法验证sgRNA文库通量提供了一种方法。
表2. 16个克隆的测序结果统计
以上内容仅仅是对本发明的构思所作的举例和说明,所属本技术领域的技 术人员对所描述的具体实施例做各种各样的修改或补充或采用类似的方式替 代,只要不偏离发明的构思或者超越本权利要求书所定义的范围,均应属于本 发明的保护范围。