FFPE样本建库的方法及其应用
文献发布时间:2023-06-19 11:34:14
技术领域
本发明涉及高通量测序文库构建领域,具体而言,涉及一种FFPE样本建库的方法及其应用。
背景技术
福尔马林固定石蜡包埋(Formalin Fixed andParaffin Embedde,FFPE)是病理学、法医病理学等进行样本保存的最主要手段。对组织样本进行FFPE的保存,有助于研究人员更加全面地了解经多年保存后的病理标本中所包含的遗传机制和病理成因。目前,全球约有2-4亿FFPE组织样本,这些组织样本是疾病诊断和科学研究的最为宝贵的的生物学资源之一。
基因组学检测技术的快速发展,尤其是主流的二代测序技术(NGS)的飞速发展,在细胞分子水平上提供了前所未有的检测能力。研究人员可以使用这一技术手段,重新开启FFPE样本宝藏的大门,寻找疾病与基因关系,进而去解开疾病的未解之谜。然而,FFPE样本十分珍贵且不可替代,在FFPE组织标本的制备和贮存过程中,组织经福尔马林固定、石蜡包埋后很容易引起DNA的交联和降解,研究人员很难获得足量的高质量的DNA样本,这对二代测序中的文库制备技术提出新的要求。
在实际的FFPE样本的构建文库时,会有1/3左右的样本是很难建库成功的,建库不成功的主要因素是样本降解严重。尽管现有技术中针对降解严重的FFPE样本也提出了一些改进的建库方案,但改善有限,仍存在部分FFPE样本难以建库或建库测序后所提供的信息有限等问题。
发明内容
本发明的目的是提供一种FFPE的建库方法及其应用,以解决现有技术中对低质量的FFPE样本难以建库的问题。
本发明提供了一种FFPE样本建库的方法,该方法包括:采用内切酶将FFPE样本DNA片段化,其中,内切酶具有如下功能:a)在DNA缺口处切割;b)保留双链末端突出碱基部分;对片段化DNA依次进行末端修复加A及接头连接;对连上接头的DNA片段进行PCR扩增,得到测序文库。
进一步地,内切酶选自T7内切酶。
进一步地,按50ng DNA计,内切酶的用量为1~5U。
根据本发明的第二个方面,提供了一种FFPE测序文库,该FFPE测序文库通过任一种方法构建而成。
根据本发明的第三个方面,提供了一种高通量测序方法,该方法包括:采用任一种方法进行建库,获得测序文库;将测序文库进行上机测序。
应用本发明的技术方案,根据降解严重FFPE的特点,有针对性的优化打断流程,使打断在FFPE损伤处断裂,通过利用FFPE降解样本自身有伤的部位断裂,充分利用可利用的片段,使相同的DNA投入量用本发明的方法更好的提升建库成功率和测序数据表现,做到最大限度的实现样本的可被检测性和降低获取有用检测信息的成本,这样就解决了FFPE自身损伤和打断的矛盾问题,为低质量FFPE建库的方法及应用提供了解决方案。
附图说明
构成本申请的一部分的说明书附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。在附图中:
图1示出了不同等级的FFPE样本DNA琼脂糖凝胶电泳图质控;
图2示出了超声或普通酶切的降解DNA片段利用率;
图3示出了T7内切酶I和S1在处理FFPE样本的区别;
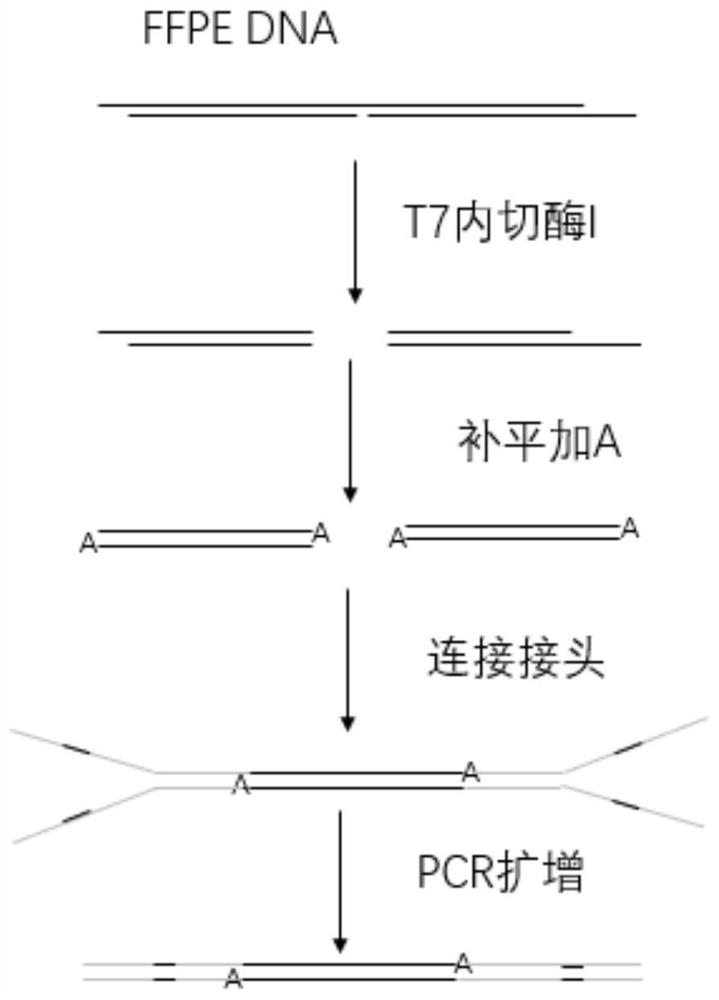
图4示出了本发明的建库流程示意图;
图5示出了不同方案的建库产出效果图。
具体实施方式
需要说明的是,在不冲突的情况下,本申请中的实施例及实施例中的特征可以相互组合。下面将结合实施例来详细说明本申请。
如背景技术所提到的,现有技术中仍存在部分FFPE样本建库不成功的情况,而经分析认为,这与目前主流通过超声打断或酶切打断方式进行建库存在不兼容有很大关系。由于本身样本就降解严重,再加超声打断或酶切打断进一步破碎,导致有很多片段进一步断裂成更小的片段而不能被建库利用,因而建库不成功或即使可以建成文库,文库的丰富度也不够,导致测序信息收集不全,进而导致测序成本较高。
为解决这一现状,发明人对不同降解等级的样本进行了分析,并通过实验比较提出了一种新的改进方案,具体如下:
根据FFPE样本DNA的降解情况,将FFPE样本DNA等级划分为B+级、B级和C级,如图1所示的FFPE的不同质量等级的样本进行琼脂糖凝胶电泳的结果,B+级的样本是主带清晰,几乎没有多少降级;B级样本是主带不清晰,大部分是弥散条带,大片段部分多于小片段部分;C级样本是大部分弥散,弥散部分主要集中在1kb附近。
目前一般质量相对较好的FFPE样本,如图1中所示的B+和B级级别的样本,DNA用传统的超声打断和酶切打断建库就能够成功建库,且所建文库质量高,因为这部分样本的DNA完整度比较好,但是这类样本只占所有FFPE的样本的一半左右,还有一半左右的样本是C级,甚至比C级降解更严重的样本,由于这类样本本身DNA降解比较严重,如果仍是用超声和普通酶切打断,会存在如图2所示的三种可能:第一种,在损伤附近断裂,会有两个片段可被建库利用;第二种在损伤的左侧或右侧断裂,会有一个片段被建库利用;第三种,在损伤两边都断裂,由于剩余片段过小而没有片段被利用,所以传统的超声和酶切打断并没有充分考虑到样本的降解情况,所以建库效率不高导致建库失败或建库质量不高,导致有大概1/3的样本无法检测。
为了解决降解严重的FFPE的DNA建库问题,本发明方案充分考虑FFPE的降解特点,在降解的样本中主要有单链部分和缺刻两种损伤情况。有文献报道S1(S1 nuclease)进行酶切建库,能够使传统的超声打断建库成功率和质量都有很大的提升。在本发明中,发明人也对该改进方案进行了测试,发现如图3所示,利用S1酶切建库,虽然可以解决部分的打断与损伤难以兼容的问题,但是对样本的利用率仍相抵较低。进一步分析,发现S1酶在建库时是去除单链部分的,而事实上有很多单链部分是可以通过后期的延伸加以利用,比如,单链部分时可以通过修复酶补平加以利用。因而在此基础上,发明人提供了一种新的改进方案,即既充分利用在缺口处进行打断以解决打断与损伤难以兼容的问题,又对部分单链通过修复延伸后加以利用。比如,通过采用T7内切酶I进行酶切处理的方式进行建库,这样打断时既考虑了FFPE的本身断裂方式的利用,也充分考虑保留单链部分能被利用的信息,在有单链5’突出部分单链的片段时,T7内切酶I比S1酶更有优势。
如图4所示,整个完成的建库过程就是通过T7内切酶I打断再加传统的建库补平加A,然后再连接头,连完接头后进行扩增建库。由于在降解的样本中能被保留的信息都是很可贵的信息,所以通过T7内切酶I处理降级比较严重的FFPE样本,使得5’凸出单链部分后被后续的补平加A环节给修复,这样既充分考虑了在断裂的缺口处打断,也能充分利用5’突出的单链部分片段信息,从而提高建库成功率的同时,也提高了所建文库信息的丰富程度。
进一步地,通过选择图1所示的C级样本,取相同量50ng的样本投入建库,分别用T7内切酶I处理建库、S1处理建库、超声打断建库和普通酶切打断建库,都扩增了7个循环,最终的文库产出,如图5所示。超声打断建库效果最差,只有T7内切酶I的1/5,说明超声打断对降解严重的样本建库效率最低;普通酶切打断的效果要好于超声打断建库方式,但也只有T7内切酶I的1/2不到;S1酶处理的和T7内切酶I的效果都较好,但显然T7内切酶I处理的产出要高于S1处理的建库方式,说明T7内切酶I打断不仅很好的解决了降解严重的样本的打断问题,而且也能比S1更好利用5’突出单链部分片段进行建库。
在上述研究结果基础上,申请人提出了本发明的技术方案---一种FFPE样本建库的方法,该方法包括:采用内切酶将FFPE样本DNA片段化,其中,内切酶具有如下功能:a)在DNA缺口处切割;b)保留双链末端突出碱基部分;对片段化DNA依次进行末端修复加A及接头连接;对连上接头的DNA片段进行PCR扩增,得到测序文库。
该建库方法,通过将FFPE样本存在缺口及末端碱基脱落等严重降解的不利情况,转化为可利用的优点,采用既能充分利用缺口进行酶切片段化DNA,又不降解末端存在部分脱落碱基的单链的内切酶进行酶切片段化,能够最大化利用降解的FFPE DNA,使得不仅能够成功建库,而且所构建的文库包含的信息更丰富。
本发明中,尽管实施例部分仅以T7内切酶为例对具有此类功能的内切酶进行了验证,但从验证结果可以得知,具有与T7内切酶功能相同的其他内切酶同样适用于本发明。根据具体操作中所使用的内切酶的种类及其活性高低的不同,其用量也有所差异。在本发明优选实施例中,按50ng DNA计,内切酶的用量为1~5U。
根据本发明的第二个方面,提供了一种FFPE测序文库,该FFPE测序文库通过任一种方法构建而成。该FFPE测序文库不仅库容较高,而且所包含的DNA的信息更丰富。
根据本发明的第三个方面,提供了一种高通量测序方法,该方法包括:采用任一种方法进行建库,获得测序文库;将测序文库进行上机测序。采用上述方法构建的FFPE测序文库不仅库容较高,而且所包含的DNA的信息更丰富,因而更适合临床FFPE样本的高通量测序检测应用。
下面将结合具体的实施例进一步说明本申请的有益效果。需要说明的是,以下实施例仅是示例性说明,并不限定本申请的方法仅能采用如下方法。
实施例:
方案一:FFPE-C级样本DNA经T7内切酶I打断建库
采用FFPE-C级DNA样本ND18131,除酶切步骤外,其余使用纳昂达通用建库试剂盒(for Illumina,货号为#1002131)进行建库。
实验步骤如下:
步骤一:DNA经T7核酸内切酶I修复及产物纯化
1、取出10X NEBufferⅡ常温融解,混合均匀,瞬时离心置于冰上备用。
2、取出T7核酸内切酶I置于冰上,混合均匀,瞬时离心备用。
3、按照下表,在已标记的0.2mL PCR管中,依次加入样本和下列试剂。
4、混合均匀,瞬时离心使全部反应液置于PCR管底部。
5、在PCR仪上启动如下反应程序,待温度稳定至37℃时将反应管放进PCR仪:
注意:热盖温度设置为50℃。
6、提前将
7、从PCR仪中取出反应产物,加入100μL
8、将PCR管瞬时离心后放置于磁力架上5-10min至液体完全澄清,使用移液器吸取移弃上清。
注意:必须完全澄清,不同品牌磁力架须调整放置时间。
9、沿PCR管侧壁缓慢加入150μL 80%乙醇,注意勿扰动磁珠,静置30sec,使用移液器吸取移弃上清。
10、重复步骤9一次。
11、将PCR管瞬时离心后放置于磁力架上,使用10μL吸头移去少量残留乙醇,注意勿吸到磁珠。
12、打开PCR管盖,并于室温静置约2-3min,至乙醇挥发完全。
13、移出PCR管,向PCR管中加入40μL Nuclease Free Water,涡旋振荡混匀,室温孵育5min。
14、将PCR管瞬时离心后置于磁力架上2min至液体完全澄清,使用移液器小心将上清转移至新的0.2mLPCR管中进行保存,注意勿吸到磁珠。
步骤二:末端修复&加A
1、取出End Repair&A-Tailing Buffer常温融解,混合均匀,瞬时离心置于冰上备用。
2、取出End Repair&A-Tailing Enzyme置于冰上自然融解,混合均匀,瞬时离心备用。
3、按照下表,在置于冰上的0.2ml PCR管中进行反应体系配制:
4、混合均匀,瞬时离心使全部反应液置于PCR管底部。
5、在PCR仪上启动如下反应程序,等温度稳定至20℃时将反应管放进PCR仪:
步骤三:接头连接
1、取出Ligation Buffer常温融解,混合均匀,瞬时离心置于冰上备用。
注意:Ligation Buffer非常粘稠,移液器吸取与释放应缓慢平稳以确保体积准确。
2、取出DNA Ligase置于冰上自然融解,混合均匀,瞬时离心备用。
3、从PCR仪上取出步骤二PCR反应管,置于冰上,按照下表进行反应体系配制:
注意1:为避免接头自连,
注意2:多样本操作时,为避免因操作缓慢导致接头自连,应按比例预配置Ligation Buffer和DNA Ligase混合液。
4、混合均匀,瞬时离心使全部反应液置于PCR管底部。
5、在PCR仪上启动如下反应程序,等温度稳定至20℃时将反应管放进PCR仪:
注意:此程序无需热盖。
步骤四:连接产物纯化
1、提前将
2、向步骤三连接反应产物加入40μL
3、将PCR管瞬时离心后放置于磁力架上5min至液体完全澄清,使用移液器吸取移弃上清。
注意:必须完全澄清,不同品牌磁力架须调整放置时间。
4、沿PCR管侧壁缓慢加入150μl 80%乙醇,注意勿扰动磁珠,静置30s,使用移液器吸取移弃上清。
5、重复步骤4一次。
6、将PCR管瞬时离心后放置于磁力架上,使用10μl吸头移去少量残留乙醇,注意勿吸到磁珠。
7、打开PCR管管盖,并于室温静置约5min,至乙醇挥发完全。
注意:切勿过分干燥,否则会降低得率。
8、从磁力架上移出PCR管,向PCR管中加入20μL Nuclease Free Water,带磁珠进入下一步PCR扩增。
步骤五:PCR扩增
1、取出2X HiFi PCR Master Mix和Amplification Primer Mix置于冰上自然融解,混合均匀,瞬时离心备用。
2、按照下表在置于冰上的0.2mL PCR管中进行反应体系配制:
3、将PCR管放置在PCR仪中启动如下程序:
步骤六:扩增文库纯化
1、向步骤五扩增反应产物PCR管加入50μl的
2、将PCR管瞬时离心后放置于磁力架上5min至液体完全澄清,使用移液器吸取移弃上清。
注意:必须完全澄清,不同品牌磁力架须调整放置时间。
3、沿PCR管侧壁缓慢加入150μl 80%乙醇,注意勿扰动磁珠,静置30s,使用移液器吸取移弃上清。
4、重复步骤3一次。
5、PCR管瞬时离心后放置于磁力架上,使用10μl吸头移去少量残留乙醇,注意勿吸到磁珠。
6、打开PCR管管盖,并于室温静置约5min,至乙醇挥发完全。
注意:切勿过分干燥,否则会降低得率。
7、移出PCR管,向PCR管加入30μl Nuclease Free Water,使用移液器将磁珠悬浮均匀,25℃孵育2min。
8、将PCR管瞬时离心后放置于磁力架上2min至液体完全澄清,使用移液器小心将上清转移至一个新的0.2ml PCR管中进行保存,注意勿吸到磁珠。
步骤七:文库定量和质检
使用Qubit方法对文库进行产量质检。
使用Qsep100对文库进行片段化质检。
方案二:FFPE-C级样本DNA经S1处理打断建库
采用FFPE-C级DNA样本ND18131,除酶切步骤外,其余使用纳昂达通用建库试剂盒(for Illumina,货号为#1002131)进行建库。
实验步骤如下:
步骤一:DNA经S1修复及产物纯化
1、取出5X Reaction Buffer常温融解,混合均匀,瞬时离心置于冰上备用。
2、取出S1核酸酶置于冰上,混合均匀,瞬时离心备用。
3、按照下表,在已标记的0.2mL PCR管中,依次加入样本和下列试剂。
4、混合均匀,瞬时离心使全部反应液置于PCR管底部。
5、在PCR仪上启动如下反应程序,待温度稳定至37℃时将反应管放进PCR仪:
注意:热盖温度设置为50℃。
6、从PCR仪中取出反应产物,加入2μL0.5M EDTA,混合均匀,瞬时离心使全部反应液置于PCR管底部。
7、在PCR仪上启动反应程序,待温度稳定至70℃时将反应管放进PCR仪:
注意:热盖温度设置为105℃。
8、提前将
9、从PCR仪中取出反应产物,加入64μL
10、将PCR管瞬时离心后放置于磁力架上5-10min至液体完全澄清,使用移液器吸取移弃上清。
注意:必须完全澄清,不同品牌磁力架须调整放置时间。
11、沿PCR管侧壁缓慢加入150μL 80%乙醇,注意勿扰动磁珠,静置30sec,使用移液器吸取移弃上清。
12、重复步骤11一次。
13、将PCR管瞬时离心后放置于磁力架上,使用10μL吸头移去少量残留乙醇,注意勿吸到磁珠。
14、打开PCR管盖,并于室温静置约2-3min,至乙醇挥发完全。
15、移出PCR管,向PCR管中加入40μL Nuclease Free Water,涡旋振荡混匀,室温孵育5min。
16、将PCR管瞬时离心后置于磁力架上2min至液体完全澄清,使用移液器小心将上清转移至新的0.2mLPCR管中进行保存,注意勿吸到磁珠。
步骤二:末端修复&加A
1、取出End Repair&A-Tailing Buffer常温融解,混合均匀,瞬时离心置于冰上备用。
2、取出End Repair&A-Tailing Enzyme置于冰上自然融解,混合均匀,瞬时离心备用。
3、按照下表,在置于冰上的0.2ml PCR管中进行反应体系配制:
4、混合均匀,瞬时离心使全部反应液置于PCR管底部。
5、在PCR仪上启动如下反应程序,等温度稳定至20℃时将反应管放进PCR仪:
步骤三:接头连接
1、取出Ligation Buffer常温融解,混合均匀,瞬时离心置于冰上备用。
注意:Ligation Buffer非常粘稠,移液器吸取与释放应缓慢平稳以确保体积准确。
2、取出DNA Ligase置于冰上自然融解,混合均匀,瞬时离心备用。
3、从PCR仪上取出步骤二PCR反应管,置于冰上,按照下表进行反应体系配制:
注意1:为避免接头自连,
注意2:多样本操作时,为避免因操作缓慢导致接头自连,应按比例预配置Ligation Buffer和DNA Ligase混合液。
4、混合均匀,瞬时离心使全部反应液置于PCR管底部。
5、在PCR仪上启动如下反应程序,等温度稳定至20℃时将反应管放进PCR仪:
注意:此程序无需热盖。
步骤四:连接产物纯化
1、提前将
2、向步骤三连接反应产物加入40μL
3、将PCR管瞬时离心后放置于磁力架上5min至液体完全澄清,使用移液器吸取移弃上清。
注意:必须完全澄清,不同品牌磁力架须调整放置时间。
4、沿PCR管侧壁缓慢加入150μl 80%乙醇,注意勿扰动磁珠,静置30s,使用移液器吸取移弃上清。
5、重复步骤4一次。
6、将PCR管瞬时离心后放置于磁力架上,使用10μl吸头移去少量残留乙醇,注意勿吸到磁珠。
7、打开PCR管管盖,并于室温静置约5min,至乙醇挥发完全。
注意:切勿过分干燥,否则会降低得率。
8、从磁力架上移出PCR管,向PCR管中加入20μL Nuclease Free Water,带磁珠进入下一步PCR扩增。
步骤五:PCR扩增
1、取出2X HiFi PCR Master Mix和Amplification Primer Mix置于冰上自然融解,混合均匀,瞬时离心备用。
2、按照下表在置于冰上的0.2mL PCR管中进行反应体系配制:
3、将PCR管放置在PCR仪中启动如下程序:
步骤六:扩增文库纯化
1、向步骤五扩增反应产物PCR管加入50μl的
2、将PCR管瞬时离心后放置于磁力架上5min至液体完全澄清,使用移液器吸取移弃上清。
注意:必须完全澄清,不同品牌磁力架须调整放置时间。
3、沿PCR管侧壁缓慢加入150μl 80%乙醇,注意勿扰动磁珠,静置30s,使用移液器吸取移弃上清。
4、重复步骤3一次。
5、PCR管瞬时离心后放置于磁力架上,使用10μl吸头移去少量残留乙醇,注意勿吸到磁珠。
6、打开PCR管管盖,并于室温静置约5min,至乙醇挥发完全。
注意:切勿过分干燥,否则会降低得率。
7、移出PCR管,向PCR管加入30μl Nuclease Free Water,使用移液器将磁珠悬浮均匀,25℃孵育2min。
8、将PCR管瞬时离心后放置于磁力架上2min至液体完全澄清,使用移液器小心将上清转移至一个新的0.2ml PCR管中进行保存,注意勿吸到磁珠。
步骤六:文库定量和质检
使用Qubit方法对文库进行产量质检。
使用Qsep100对文库进行片段化质检。
方案三和四:普通酶切建库和超声打断建库纳昂达产品说明书
采用FFPE-C级DNA样本ND18131,使用纳昂达快速酶切建库试剂盒(for Illumina,货号为#1002131)进行建库。
采用FFPE-C级DNA样本ND18131,使用纳昂达超声打断建库试剂盒(for Illumina,货号为#1002101)进行建库。
如图5所示,对上述四种建库方案所产出的文库进行比较可知,T7内切酶I处理的产出最高,是超声打断建库库容的近5倍,是常规酶切建库库容的两倍多,比S1酶切建库的库容还高18%。
从以上实施例中的比较结果可以看出,本发明提供的一种解决低质量FFPE的建库方式,通过T7内切酶I处理的方式打断降解的DNA样本,此方法在充分利用缺口位置断裂DNA同时也保留了5’突出的单链部分信息,使降解严重的样本相同投入量和相同扩增循环数的产出更高,能够保留更多的样本信息,可以解决以前建库效果不好的样本有效建库,解决低质量FFPE不能建库的问题,使更多的临床样本可以实现有效建库检测。
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。