一种基于转录组中相邻微卫星序列的植物鉴别方法
文献发布时间:2023-06-19 11:13:06
技术领域
本发明涉及生物技术领域中区分植物样本,更具体的说是涉及一种基于转录组中相邻微卫星序列的植物鉴别方法。
背景技术
植物样本的精细鉴别在生产实践中有着重要的意义,尤其在经济作物和中药材领域,许多优良的品系遗传差异很小,需要高分辨力的鉴别方法。目前植物的鉴别一般采用形态鉴别法和基于DNA序列的分子鉴别方法。但是这两种方法在进行遗传差异非常小的植物品系,或同种植物由于不同化学处理、环境因素、生长周期等因素的不同,表达不同特定基因的样本时,经常会遇到分辨力不足的情况。
形态学鉴别方法应用范围广,但有些遗传差异并不会表现为形态学上的差异,这就无法运用形态学方式进行鉴别。而基于DNA序列的鉴别方法,如基于线粒体或叶绿体基因保守序列的分析方法,一般可以对属以上的分类单元进行分类。在对于种以下的植物分类单元进行鉴别时,处于成本考虑,一般采用分子标记的方法,如RAPD、SSR、ISSR、RFLP和AFLP等。这样的分子标记方法对于亲缘关系相对较近的植物具有较好的分辨率。但对于亲缘关系非常接近的品系,也会遇到分辨力不足的情况,因为许多存在于调控序列中的基因并不一定与分子标记相关联,从而无法用分子标记的方法鉴别。
然而,在实际的生产实践中,需要对植物组织样本进行更为细致的区分,如亲缘关系十分相近的植物,或无明显性状差异的优良品系;但上述方式就显得无能为力了。
植物中RNA的表达具有多态性,可以提供相较于DNA更多的遗传差异信息。由于目前对于RNA测序的转录组测序服务成本快速下降,该技术也将有更广泛的应用前景。微卫星序列是存在于基因组中的短串重复序列,这些序列可以转录,从而存在于植物的转录组数据中,由于RNA的转录受内部调控序列影响和外部环境影响,所以具有较高的多态性,微卫星序列按照重复单元中碱基的数目和类型一般可以分为单碱基重复、二碱基重复、三碱基重复、四碱基重复、五碱基重复、六碱基重复和相邻微卫星序列。其中相邻微卫星序列是指在两个微卫星序列相邻,之间一般有一段短的碱基序列,将两个微卫星序列隔开,因为中间短的微卫星序列有种属特异性,所以相较于其它微卫星序列类型具有较高的区分度。针对植物转录组中相邻微卫星序列相似度分析,可以揭示植物的亲缘关系,从而作为辅助植物鉴别的一种方法。
发明内容
本发明的目的是提供一种基于转录组中相邻微卫星序列的植物鉴别方法,该鉴别方法,相较于其它微卫星序列类型具有较高的区分度。针对植物转录组中相邻微卫星序列相似度分析,可以作为辅助植物鉴别的一种方法。
为解决上述技术问题,本发明通过一种基于转录组中相邻微卫星序列的植物鉴别方法进行实现,该方法包括以下步骤:
提取待鉴定植物组织样本的RNA,使用高通量测序的方法对转录组进行测序,将测得的原始序列Rawdata经过去掉接头序列,过滤掉不合格的序列后得到有效数据Cleandata序列,由有效数据Clean data序列根据已知基因组进行拼接或根据重复序列拼接Unigene,使用MISA软件对拼接除重后得到的序列进行微卫星序列进行分析,得到微卫星序列统计数据,包括微卫星序列的类型和微卫星序列的具体序列。在不同样本间进行相似度比对时,在微卫星序列的类型中选择有相邻的微卫星序列(c型),统计序列相同的微卫星序列,以相同微卫星序列个数除以除重后总的微卫星序列数得出相似度,利用相似度构建聚类图。从而实现不同植物样本的区分。
本发明设计了基于转录组中相邻微卫星序列的植物鉴别方法。本发明所使用的方法基于植物基因的转录本,具有多态性高、分辨力强和用量少的优点。对于亲缘关系十分接近的植物或不同外界环境因素处理下的同种植物样本的区分鉴别有着重要的意义。
本发明的其他特征和优点将在随后的说明书中阐述,或者,部分特征和优点可以从说明书推知或毫无疑义地确定,或者通过实施本发明的上述技术即可得知。
为使本发明的上述目的、特征和优点能更明显易懂,下文特举较佳实施方式,并配合所附附图,作详细说明如下。
附图说明
为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
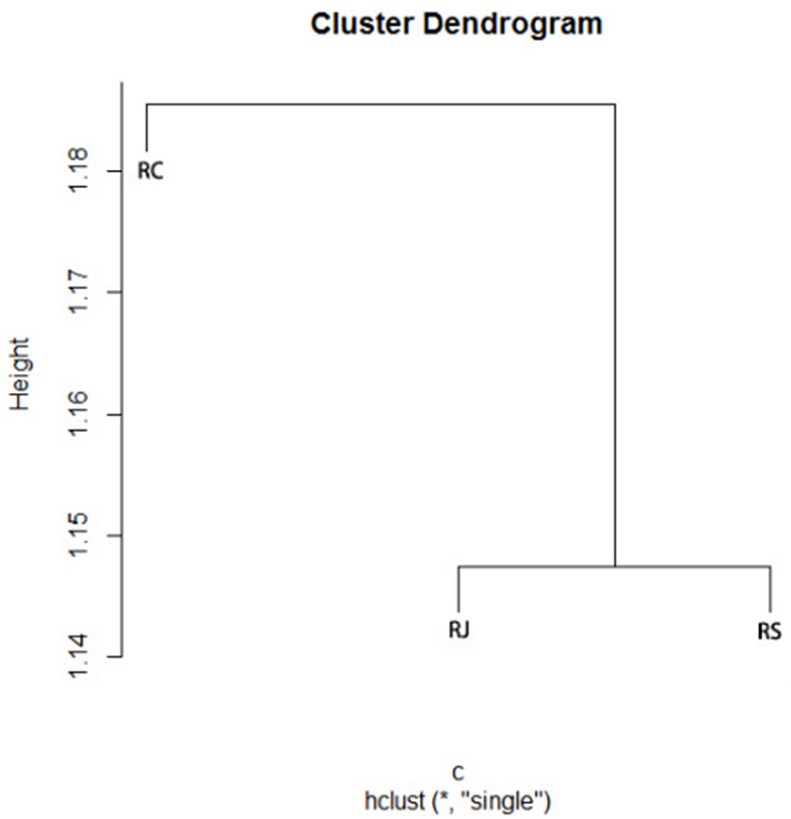
图1为本发明的最短距离法构建的RC、RJ和RS样本聚类图。
图2为本发明的最短距离法构建的RC、RJ、CKg、HH、CKh和LP样本聚类图。
具体实施方式
为使本发明实施例的目的、技术方案和优点更加清楚,下面将结合附图对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
下述实施例中所使用的实验方法如无特殊说明,均为常规方法。
下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
目前对于植物组织样本进行更为细致的区分,如亲缘关系十分相近的植物,或无明显性状差异的优良品系进行区分鉴别时,是十分困难的,基于这一原因,本发明提供了一种基于转录组中相邻微卫星序列的植物鉴别方法;该方法主要是通过如下步骤加以实施:提取待鉴定植物组织样本的RNA,使用高通量测序的方法对转录组进行测序,将测得的原始序列Rawdata经过去掉接头序列,过滤掉不合格的序列后得到有效数据Clean data序列,由有效数据Clean data序列根据已知基因组进行拼接或根据重复序列拼接Unigene,使用MISA软件对拼接除重后得到的序列进行微卫星序列进行分析,得到微卫星序列统计数据,包括微卫星序列的类型和微卫星序列的具体序列。在不同样本间进行相似度比对时,在微卫星序列的类型中选择有相邻的微卫星序列(c型),统计序列相同的微卫星序列,以相同微卫星序列个数除以除重后总的微卫星序列数得出相似度,利用相似度构建聚类图。从而实现不同植物样本的区分。
为了更加方便本领域技术人员的理解,下面申请人以下面两个实施例加以说明。
实施例1:鉴别正常生长、水杨酸处理和茉莉酸甲酯处理的微型月季。
一、植物材料的准备
选取处于开花期的盆栽微型月季(Rosa hybrid minima),水杨酸处理组(RS)使用100 mL 1 mmol/L的水杨酸进行叶面喷施,茉莉酸甲酯处理组(RJ)使用100 mL 1 mmol/L的茉莉酸甲酯进行叶面喷施,对照组(RC)使用100 mL的蒸馏水,于24 h后取花瓣,取样后液氮速冻,用于总RNA的提取。
二、RNA提取
采用Invitrogen Trizol试剂提取微型月季花瓣的总RNA,提取方法按照试剂说明书操作。RNA的质量和完整性使用Bioanalyzer 2100和RNA 1000 Nano LabChip kit检测。总RNA使用连接有poly-T的磁珠进行两轮纯化。
三、RNA转录组文库的构建和测序
将RNA片段进一步分割后按Illumina的mRNA Seq sample preparation kit说明书反转录来创建cDNA文库。双链产物使用AMPure XP beads进行纯化,将双链产物的粘性末端修复为平末端并加接头,使用AMPureXP beads进行片段选择,将含有U的cDNA第二链酶解后进行PCR扩增以获得最终测序文库。测序文库在质检合格后进行测序,采用IlluminaHiseq平台。
四、序列的组装
测序产生的原始数据(raw data)使用cutadapt去除reads中的接头序列。使用fqtrim过滤不合格的序列,去除poly-A/T和截短后长度小于100 bp或N的含量在5%以上的序列,得到有效数据clean data。使用clean data根据重叠区域单独组装Unigene。最后进行归一化得到Unigene。
五、微卫星序列的统计
使用微卫星序列分析软件MISA分别对不同样本的Unigene,得到不同样本的微卫星序列统计数据。采用两两对比的方式,在微卫星序列的类型中选择有相邻微卫星序列类型(c型),统计其中序列相同的微卫星序列,除去重复序列后,各样本相邻微卫星序列数分别为RC510条,RJ394条,RS419条;RC与RJ相同序列为126条,RC与RS相同序列为127条,RJ与RS相同序列为129条。
以相同微卫星序列个数除以除去重复后总的微卫星序列数得出相似度,
公式为相似度=相同微卫星序列个数/(样本1微卫星序列个数+样本2微卫星序列个数-相同微卫星个数)
样本间相似度如表1所示:
表1 RC、RJ和RS样本间两两相似度
使用R语言的ggplot2包进行绘图,聚类计算函数采用hclust,计算方法采用最短距离法(single),得到聚类图如图1所示。
实施例2
一、植物材料的准备
盆栽蒙古黄芪(
两年生甘草幼苗,CKg处理叶片喷施蒸馏水,HH处理叶面喷施10mg/L高分子量几丁质,24小时后采集叶片,洗净后液氮速冻,用于总RNA的提取。
二、RNA提取
采用Invitrogen Trizol试剂提取植物组织的总RNA,提取方法按照试剂说明书操作。RNA的质量和完整性使用Bioanalyzer 2100和RNA 1000 Nano LabChip kit检测。总RNA使用连接有poly-T的磁珠进行两轮纯化。
三、RNA转录组文库的构建和测序
将RNA片段进一步分割后按Illumina的mRNA Seq sample preparation kit说明书反转录来创建cDNA文库。双链产物使用AMPure XP beads进行纯化,将双链产物的粘性末端修复为平末端并加接头,使用AMPureXP beads进行片段选择,将含有U的cDNA第二链酶解后进行PCR扩增以获得最终测序文库。测序文库在质检合格后进行测序,采用IlluminaHiseq平台。
四、序列的组装
测序产生的原始数据(raw data)使用cutadapt去除reads中的接头序列。使用fqtrim过滤不合格的序列,去除poly-A/T和截短后长度小于100 bp或N的含量在5%以上的序列,得到有效数据clean data。使用clean data根据重叠区域单独组装,最后进行归一化得到Unigene。
五、微卫星序列的统计
使用微卫星序列分析软件MISA分别对不同样本的Unigene,得到不同样本的微卫星序列统计数据。在分析中同时也使用实施例1中的数据RC和RJ。
采用两两对比的方式,在微卫星序列的类型中选择有相邻微卫星序列类型(c型),统计其中序列相同的微卫星序列,除去重复序列后,各样本相邻微卫星序列数分别为RC510条,RJ394条,CKg,H1345条,HH1328条,CKh167条,LP152条;
CKg与CKh相同序列为1条,CKg与RC相同序列为3条,CKh与RC相同序列为3条,CKg与HH相同序列为181条,CKh与LP相同序列为17条,RC与RJ相同序列为126条,CKg与LP相同序列为1条,CKg与RJ相同序列为0条,CKh与HH相同序列为0条,CKh与RJ相同序列为1条,RC与HH相同序列为2条,RC与LP相同序列为0条,HH与LP相同序列为1条,HH与RJ相同序列为3条,LP与RJ相同序列为0条。
以相同微卫星序列个数除以除去重复后总的微卫星序列数得出相似度,
公式为相同微卫星序列个数/(样本1微卫星序列个数+样本2微卫星序列个数-相同微卫星个数)
样本间相似度如表2所示:
表2 CKg、CKh、RC、HH、LP和RJ样本间两两相似度
使用R语言的ggplot2包进行绘图,聚类计算函数采用hclust,计算方法采用最短距离法(single),得到聚类图如图2所示。
最后应说明的是:以上所述实施例,仅为本发明的具体实施方式,用以说明本发明的技术方案,而非对其限制,本发明的保护范围并不局限于此,尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,其依然可以对前述实施例所记载的技术方案进行修改或可轻易想到变化,或者对其中部分技术特征进行等同替换;而这些修改、变化或者替换,并不使相应技术方案的本质脱离本发明实施例技术方案的精神和范围,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应所述以权利要求的保护范围为准。
- 一种基于转录组中相邻微卫星序列的植物鉴别方法
- 一种基于转录组测序的微卫星不稳定性检测方法