二维金属-全硫醇化的六苯并苯框架催化剂上二氧化碳电化学还原的计算方法
文献发布时间:2023-06-19 18:37:28
技术领域
本发明涉及二氧化碳氢化为甲酸和甲烷的计算化学。更具体地,涉及到二维金属-全硫醇化的六苯并苯框架催化剂(TM-PTC)在二氧化碳电化学还原机理的计算与研究。
背景技术
化石燃料燃烧产生的二氧化碳排放到大气中,造成了能源和环境问题,如温室效应和海洋酸化。寻找处理甚至利用CO
二维(2D)导电金属-有机骨架(MOF)由于其超高的表面积,灵活的结构和化学可调性,近年来引起了人们的极大兴趣。对于2D-MOFs,许多研究致力于研究它们作为CO
除了实验研究外,还有许多关于二维MOFs上CO
上述文献虽然给出了模拟计算催化反应机理的方法,但是精确度度=还有待提高,对于实际化学反应过程,催化机理、最优反应路径、决速步骤等机理研究和作用机制是至关重要的,为进一步提高催化机理模拟计算的精度,开发一种更适合的模拟计算方法,对于二维MOF型CO
发明内容
本发明在实验合成的导电性良好的Ni-PTC和Co-PTC的基础上,基于密度泛函理论(DFT)研究了一系列用于CO
为达到上述目的,本发明提供一种二维金属-全硫醇化的六苯并苯框架上CO
(1)构建催化剂活性中心的计算模型:包括模型选定、建立单层超晶胞、设置真空层以及模型优化几个步骤。
所述模型为Material Studio材料数据库中的石墨烯(graphite)模型;所述单层超晶胞为5×5×1的超细胞;所述真空层为
本发明的一个实施方式中,利用Material Studio材料数据库中的石墨烯(graphite)模型,由于实验中Co-PTC和Ni-PTC的晶格参数为
所述模型优化是指,在经过固定z轴优化x轴和y轴进行的晶格参数的优化和晶胞内原子在x,y,z三个方向作用力的优化,得到TM-PTC催化剂的晶胞参数,使其与实验结果相符。
在本发明的一个实施方式中,经过模型优化后,Co-PTC和Ni-PTC的晶胞参数为
(2)计算方法与参数设定:包括泛函密度理论、平面波基的截止能量、K点、收敛标准以及反应物与底物的范德华作用、分析原子间成键强度、建立隐式溶剂化模型、确定电子间库伦排斥参数U、计算吸附能与极限电位,并利用从头算分子动力学(AIMD)模拟,使用Nose–Hoover方法来控制温度。
所述泛函密度理论是在VASP软件包中基于Perdeu-Burke-Ernzerhof泛函的广义梯度近似中使用自旋极化密度泛函理论(DFT)。
所述平面波基的截止能量被设定为400eV。
几何优化和电子结构的计算过程中,所述K点大小分别设置为5×5×1和7×7×1。
能量和力优化的收敛标准分别设定为1×10
所述范德华相互作用采用Becke-Jonson阻尼法(DFT-D3)。
所述原子间的成键强度通过LOBSTER代码中晶体轨道哈密顿布居(COHP)来分析,COHP的正值和负值分别对应反键态和成键态。进一步地,可以采用积分COHP(ICOHP)用来估计结合强度,即ICOHP的负值越大,结合强度越大。
所述隐式溶剂化模型为VASPsol模型。
所述电子间库伦排斥参数U值是根据轨道占有率相对于变化U的变化计算出来的,按照公式(i)-(iii)计算得到:其基本思路是对原子相关(d或f轨道)能级施加一个电位,并观察这个施加的电位如何改变d轨道的占有率。轨道电子占有率可以简单地从轨道的电荷密度和原子分解中计算出来。相对于变化的电位,轨道占有率的变化取决于两个响应函数的值:一个自洽响应和一个非自洽响应。前者是在允许电荷密度放松的情况下计算的,而后者是在保持电荷密度固定的情况下计算的。
非自洽响应函数可写为:
其中
同样,自洽反应函数的具体形式如下:
所计算的有效U值即为:
根据上述计算公式,本发明涉及催化剂中计算得出的每个金属原子的有限U值见表1。
表1
利用公式(I)、(II)计算形成能(E
E
U
其中E
不同金属催化剂Uodiss(metal,balk)和n的数值见表2。
表2
吸附能根据公式(III)计算得到:
△E
其中E
所述每个基元反应的自由能是基于
所述极限电位(U
U
其中ΔG
对于从头算分子动力学(AIMD)模拟,使用Nose–Hoover方法来控制温度,所述温度为500K。
具体来说,本发明所有理论计算都是在VASP软件包中基于Perdeu-Burke-Ernzerhof泛函的广义梯度近似中使用自旋极化密度泛函理论(DFT)进行的。在计算过程中,平面波基的截止能量被设定为400eV。对于几何优化和电子结构的计算的K点大小分别设置为5×5×1和7×7×1。将能量和力优化的收敛标准分别设定为1×10
为了考虑电子间的库仑排斥,我们选用了DFT+U的方法。该方法的基本思想是将势能应用于原子位置的相关(d或f轨道)能级,并观察这个变化的U是如何改变d轨道占有率的。有效U值是根据轨道占有率相对于变化U的变化计算出来的。相对于变化的电位,轨道占有率的变化取决于两个响应函数的值:一个自洽响应和一个非自洽响应。前者是在允许电荷密度放松的情况下计算的,而后者是在保持电荷密度固定的情况下计算的。非自洽响应可写为:
其中
所计算的有效U值即为:
根据下列公式计算了形成能(E
E
U
其中E
吸附能根据公式(III)计算得到:
△E
其中E
所述每个基元反应的自由能是基于
所述极限电位(U
U
其中ΔG
对于从头算分子动力学(AIMD)模拟,使用Nose–Hoover方法来控制温度。
(3)判断材料的结构和稳定性
通过计算电子局域化(ELF)和差分电荷密度研究TM-PTC的成键情况,表明材料的结构。已知ELF介于0和1之间,ELF>0.5表示共价键特征,而ELF<0.5代表离子键,ELF=0.5对应于金属键。
本发明的一个实施方式中,以Mo-PTC为例(如图2a所示),C-C和C-S键具有共价键特征,而Mo-S键是离子键的性质。这确保了框架的稳定性。另一方面,Mo-PTC的差分电荷密度清楚地描述了从Mo原子到S原子的电荷转移过程,也证实了Mo-S键为离子键(如图2b所示)。
稳定性是催化剂的关键属性之一。所述稳定性包括热力学稳定性、电化学稳定性以及动态稳定性。
进一步地,利用步骤(2)计算得到催化剂的形成能和溶解电位来评价催化剂的热力学稳定性和电化学稳定性。
催化剂的形成能是正值,则热力学上是不稳定的;反之,形成能≤0,则热力学上是稳定的。催化剂的溶解电位是负值,则表明电化学不稳定;反之,溶解电位≥0,则电化学是稳定的。
进一步地,通过AIMD模拟验证催化剂的动态稳定性。所述AIMD模拟在温度500K下进行,时间步长为1.0fs,总共10ps。
本发明的一个实施方式中,Ag-PTC的生成能是正值(如图3a所示),表明它在热力学上是不稳定的。对于电化学稳定性,结果表明TM-PTCs(TM=Sc,Ti,V,Mn,Fe,Y,Zr,Nb,Mo)具有负的解离电位,表明它们是不稳定的(图3a)。进一步地,以Mo-PTC作为一个例子进行了AIMD模拟检验了其的动态稳定性。计算结果表明,在模拟过程中经过10000步后没有明显的结构变形(图3b),表明Mo-PTC是动态稳定的,因此Mo-PTC在环境条件下具有良好的热稳定性。
(4)CO
作为CO
所述参考标准为0.61eV,并进一步计算*HCOO途径后续反应的自由能大小。
在本发明的一个实施方式中,如图5所示。计算结果表明,(TM=Sc-Cr,Fe-Co,Zn,Y-Mo)低于HER,而其余催化剂更倾向于HER。将CO
另一方面,从以前的研究中也知道,铜是CO
经过上述筛选过程,只剩下TM-PTC(TM=Sc,Ti,V,Cr,Mn,Fe,Zn,Zr,Nb,Mo)催化剂作进一步的机理研究。对于这些催化剂,中间体*HCOO的自由能远低于*COOH,这意味着质子电子对会优选攻击CO
(5)催化反应机制分析:将步骤(4)筛选得到催化剂进行反应机制分析,主要包括中间体以及产物、反应途径分析。
所述中间体分析包括中间体建模、确定中间体结构,并计算中间体的自由能,分析气相和溶液中自由能变化;根据自由能变化确定气相和溶液相的反应途径、关键中间体和决速步骤。
本发明的一个实施方式中,对所有CO
计算表明经步骤(4)筛选出的催化剂经过CO
所述产物分析,是根据催化反应产物甲醛HCOOH和甲烷CH
对于产物甲烷来说,根据自由能变化确定气相中的反应途径、主要中间体等。
本发明的一个实施方式中,以甲烷为产物的五种催化剂:TM=Sc,Ti,Y,Zr,Nb。最优的途径是相同的,即CO
对于产物甲醛,还需要考虑甲醛解吸,计算两种反应的自由能,确定气相中催化反应路径和决速步骤。
具体来说,甲醛(H
进一步地,由于电化学反应都在电解液中进行,因此,需要考虑溶剂效应。本发明采用隐式溶剂化模型计算反应在溶液环境中的反应路径。
本发明的一个实施方式中,计算结果表明,五种TM-PTC催化剂在溶液中的最优路径与在气相中的不同,但是产物保持不变。
(6)轴向氧修饰TM-PTC:TM-O-PTC
为了避免在CH
本发明的一个实施方式种,五种以甲烷作为CO
对于其余三种催化剂TM-O-PTC(TM=Ti,Nb,Mo),计算结果显示它们与TM-PTC(TM=Ti,Nb,Mo)相比具有不同的反应途径(图7vs图8)。首先,轴向O原子的修饰使得CO
进一步地,本发明通过以HCOOH和CH
本发明的一个实施方式中,以CH
(7)电荷转移分析
本发明进一步分析中间体和催化剂之间的电荷转移,进行Bader电荷分析,确定CO
本发明的一个实施方式中,为了深入了解反应中间体和催化剂之间的电荷转移,我们以Mo-PTC为例进行了Bader电荷分析。如图10a所示,研究对象大致分为三个部分。部分1是吸附在催化剂表面上的反应中间体(C
本发明取得的有益效果:
本发明利用密度泛函理论对CO
本发明通过分析二维TM-PTC催化剂的可能的反应路径催化剂稳定性等筛选适宜的催化剂,并分析产物、最优反应路径、决速步骤和中间体结构等,经多因素充分考量,精确模拟分析CO
附图说明
图1为TM-PTC催化剂优化的几何结构;
图2为TM-PTC催化剂成键情况,其中(a)为TM-PTC的电子局域函数图,(b)为TM-PTC的差分电荷图;
图3为TM-PTC催化剂稳定性分析图,其中(a)为TM-PTC和TM-O-PTC的电化学稳定性和热力学稳定性,(b)为Mo-PTC的分子动力学(AIMD)图;
图4为TM-PTC催化剂上的CO
图5为气相中TM-PTC催化剂上的CO
图6为TM-PTC催化剂上CO
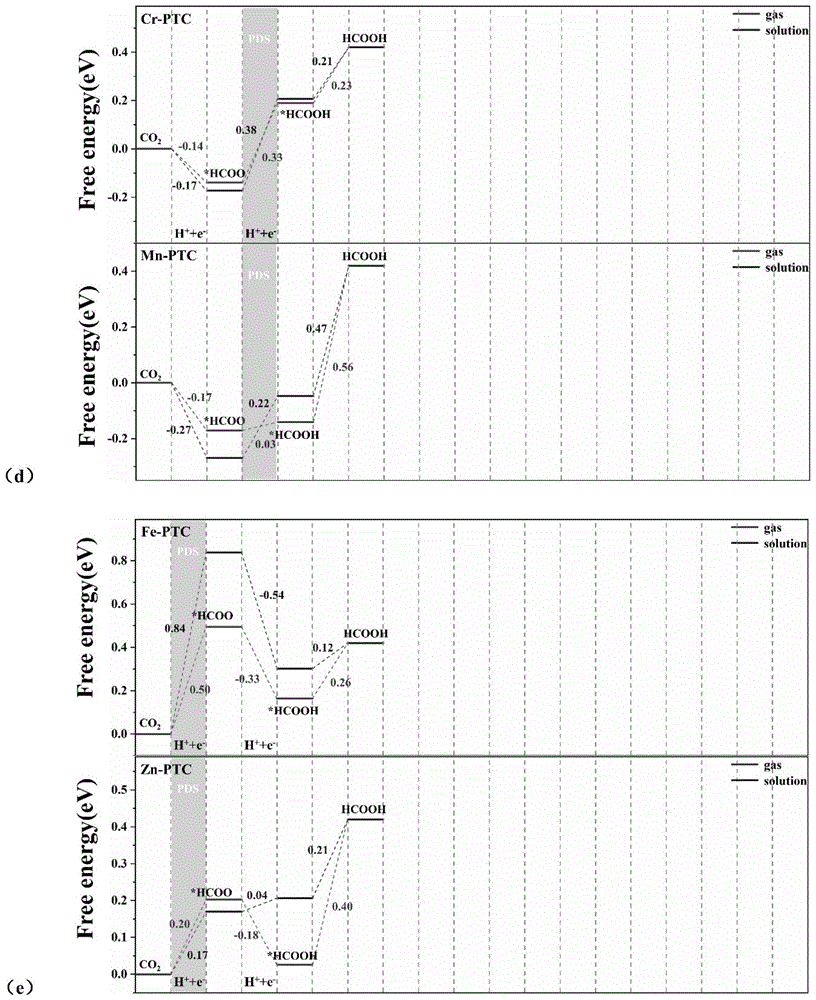
图7为TM-PTC催化剂在溶液中和气相中的自由能对比图,其中(a)为Sc-PTC和Ti-PTC催化剂在溶液和气相中反应自由能对比图,(b)为Zr-PTC和Nb-PTC催化剂在溶液和气相中反应自由能对比图,(c)为Mo-PTC催化剂在溶液和气相中反应自由能对比图,(d)为Cr-PTC和Mn-PTC催化剂在溶液和气相中反应自由能对比图,(e)为Fe-PTC和Zn-PTC催化剂在溶液和气相中反应自由能对比图;
图8为TM-O-PTC在气相中的自由能图和在溶液和气相中自由能对比图,其中;(a)为Ti-O-PTC、Nb-O-PTC和Mo-O-PTC催化剂气相中反应自由能图,(b)为Ti-O-PTC、Nb-O-PTC和Mo-O-PTC催化剂在溶液和气相中反应自由能对比图;
图9为催化剂的过电势统计图,其中(a)以CH
图10为Mo-PTC催化剂电荷转移情况,其中(a)Mo-PTC的三部分,(b)整个CO
具体实施方式
下面结合具体实施例对本发明作进一步说明,但本发明并不限于以下实施例。
下述实施例中所述实验方法,如无特殊说明,均为常规方法;所述试剂和材料,如无特殊说明,均可从商业途径获得。
本发明的二维TM-PTC催化剂(TM=Sc-Zn,Y-Mo,Ru-Ag,PTC=1,2,3,4,5,6,7,8,9,10,11,12-pertholated coronene)的计算、筛选、分析过程如下。
实施例1
一种二维金属-全硫醇化的六苯并苯框架上CO
(1)构建催化剂活性中心的计算模型:包括模型选定、建立单层超晶胞、设置真空层以及模型优化。
利用Material Studio材料数据库中的石墨烯(graphite)模型,由于实验中Co-PTC和Ni-PTC的晶格参数为
所述模型优化是指,在经过固定z轴优化x轴和y轴进行的晶格参数的优化和晶胞内原子在x,y,z三个方向作用力的优化,得到TM-PTC催化剂的晶胞参数,使其与实验结果相符。
经过模型优化后,Co-PTC和Ni-PTC的晶胞参数为
(2)计算方法与参数设定:具体包括泛函密度理论、平面波基的截止能量、K点、收敛标准以及反应物与底物的范德华作用、分析原子间成键强度、建立隐式溶剂化模型、确定电子间库伦排斥参数U、计算吸附能与极限电位,并利用从头算分子动力学(AIMD)模拟,使用Nose–Hoover方法来控制温度。
本发明所有理论计算都是在VASP软件包中基于Perdeu-Burke-Ernzerhof泛函的广义梯度近似中使用自旋极化密度泛函理论(DFT)进行的。在计算过程中,平面波基的截止能量被设定为400eV。对于几何优化和电子结构的计算的K点大小分别设置为5×5×1和7×7×1。将能量和力优化的收敛标准分别设定为1×10
为了考虑电子间的库仑排斥,我们选用了DFT+U的方法。有效U值是根据轨道占有率相对于变化U的变化计算出来的,具体见表1。
根据下列公式计算了形成能(E
E
U
其中E
吸附能定义:
△E
其中E
基于
极限电位(U
对于从头算分子动力学(AIMD)模拟,使用Nose–Hoover方法来控制温度,并且在500K的温度下进行模拟。
(3)判断材料的结构和稳定性
通过计算电子局域化(ELF)和差分电荷密度研究TM-PTC的成键情况,表明材料的结构。已知ELF介于0和1之间,ELF>0.5表示共价键特征,而ELF<0.5代表离子键,ELF=0.5对应于金属键。
本发明的一个实施方式中,以Mo-PTC为例(如图2a所示),C-C和C-S键具有共价键特征,而Mo-S键是离子键的性质。这确保了框架的稳定性。另一方面,Mo-PTC的差分电荷密度清楚地描述了从Mo原子到S原子的电荷转移过程,也证实了Mo-S键为离子键(如图2b所示)。
利用步骤(2)计算得到催化剂的形成能和溶解电位来评价催化剂的热力学稳定性和电化学稳定性。
Ag-PTC的生成能是正值(如图3a所示),表明它在热力学上是不稳定的。对于电化学稳定性,结果表明TM-PTCs(TM=Sc,Ti,V,Mn,Fe,Y,Zr,Nb,Mo)具有负的解离电位,表明它们是不稳定的(图3a)。进一步地,以Mo-PTC作为一个例子进行了AIMD模拟检验了其的动态稳定性。计算结果表明,在模拟过程中经过10000步后没有明显的结构变形(图3b),表明Mo-PTC是动态稳定的,因此Mo-PTC在环境条件下具有良好的热稳定性。
(4)CO
通过比较CO
另一方面,铜是CO
经过上述筛选过程,只剩下TM-PTC(TM=Sc,Ti,V,Cr,Mn,Fe,Zn,Zr,Nb,Mo)催化剂作进一步的机理研究。对于这些催化剂,中间体*HCOO的自由能远低于*COOH,这意味着质子电子对会优选攻击CO
(5)催化反应机制分析:主要包括中间体以及产物、反应途径分析。
所述中间体分析包括中间体建模、确定中间体结构,并计算中间体的自由能,分析气相和溶液中自由能变化;根据自由能变化确定气相和溶液相的反应途径、关键中间体和决速步骤。
对所有CO
电化学反应都在电解液中进行,因此,需要考虑溶剂效应。本发明采用隐式溶剂化模型计算反应在溶液环境中的反应路径。
计算表明经步骤(4)筛选出的催化剂经过CO
所述产物分析,是根据催化反应产物甲醛HCOOH和甲烷CH
对于筛选过后的8个TM-PTC(TM=Sc、Ti、Cr、Fe、Zn、Zr、Nb、Mo),我们的计算表明有两种最终产品,即HCOOH和CH
HCOOH:CO
CH
H
到目前为止,我们发现对于TM=Sc、Ti、Zr、Nb、Mo,反应路径都包含*OH中间体,其中四个(Sc、Zr、Nb、Mo)的速率决定步骤是*OH氢化成H2O分子。同时,以往的研究发现,对于具有*OH中间体的二维CO2RR催化剂,通常需要高能量来再生催化剂表面以便于催化剂的循环利用。此外,经过六次氢化后,中间体*H3CO释放出一个CH4分子并留下一个O原子,在催化剂表面形成轴向配位的O。
计算结果表明,五种催化剂在溶液中的最优路径与在气相中的相同,但是决速步骤有所不同。其中Ti-PTC和Nb-PTC在溶液中的限速步骤与气相中的相同,对应的限制电压分别为-0.94V和-0.54V。对于Sc-PTC和Zr-PTC,溶剂效应使它们在溶液中的决速步骤与气相不同,为*HCOO→*H
(6)轴向氧修饰TM-PTC:TM-O-PTC
为了避免在CH
五种以甲烷作为CO
对于其余三种催化剂TM-O-PTC(TM=Ti,Nb,Mo),计算结果显示它们与TM-PTC(TM=Ti,Nb,Mo)相比具有不同的反应途径(图7vs图8)。首先,轴向O原子的修饰使得CO
通过以HCOOH和CH
以CH
(7)电荷转移分析
为了深入了解反应中间体和催化剂之间的电荷转移,我们以Mo-PTC为例进行了Bader电荷分析。如图10(a)所示,研究对象大致分为三个部分。部分1是吸附在催化剂表面上的反应中间体(C
综上所述,本发明通过理论模拟研究了一系列二维TM-PTC(TM=Sc-Zn、Y-Mo、Ru-Ag)作为CO
本领域的技术人员应当理解,上述实施方式仅仅是为了清楚地说明本公开,而并非是对本公开的范围进行限定。对于所属领域的技术人员而言,在上述公开的基础上还可以做出其它变化或变型,并且这些变化或变型仍处于本公开的范围内。