化合物以及包含其的聚合物、混合物、组合物和有机电子器件
文献发布时间:2024-04-18 20:01:55
技术领域
本申请涉及光电领域,具体涉及一种化合物以及包含其的聚合物、混合物、组合物和有机电子器件。
背景技术
现有用于蓝光有机发光二极管(organic light-emitting diode,OLED)器件的发光材料多为芘,蒽,苝类等传统荧光分子,该类分子的发光光谱半峰宽(full width athalf maximum,FWHM)较宽,显示效果难以实现深蓝光,不满足高显色指数显示技术的要求,且该类材料器件效率较低,不利于实现低功耗与长使用寿命的显示器件。目前,对于蓝光OLED的研究热点为硼-氮环化合物,该类在分子设计上采用多重共振效应,即将分子结构保持在一个特殊的平面刚性共轭结构内并利用硼原子上特有的可以参与电子云共轭的空轨道与氮原子上的可以参与电子云共轭的孤对电子所带来的不同的电负性通过共轭效应相互增强,从而形成分子内短程的电荷转移态,从而实现高效发光。而且相比于其他发光材料,这种平面结构的化合物可使FWHM变窄,但是基于目前的报道,已有化合物在提高了分子刚性的同时,难以平衡兼顾光色、发光效率与发光光谱FWHM等方面的问题,难以同时满足高效、高显色指数的蓝光OLED显示技术的需求。
发明内容
本申请提供了一种化合物以及包含其的聚合物、混合物、组合物和有机电子器件,以提高获得一种高发光效率、高显示指数的蓝光OLED器件。
第一方面,本申请提供一种化合物,所述化合物具有如式(I)所示结构,
其中,L
所述式(I-1)和所述式(I-2)中,X选自B、N、P、P=O或P=S;
Y
R
Y
A1-A8选自C5~C30碳原子的取代或未取代的芳香基团或C5~C30碳原子的取代或未取代的杂芳香基团;Y
A4可与A2和/或A3成环,A5可与A6和/或A8成环;
A1-A4的任一个通过L与A5-A8中的任一个相连,L为二价桥联基团。
本申请通过部分引入氧或硫原子,将硼-氮体系材料修饰为特定结构的硼-氮-氧或硼-氮-硫体系材料,可适当减少氮原子带来的苯基,提高分子刚性。与此同时,本申请也引入二价键桥,将硼-氮-氧或硼-氮-硫分子链接形成二聚体,进一步提高分子刚性。其中,氧或硫原子的引入所带来的共轭长度减小现象恰好可以与键桥引入所带来的的共轭长度增加现象相互弥补,从而减小额外的分子设计对材料发光光色的影响。
在一种可能的实现方式中,所述化合物选自式(II-1)至式(II-3)中的一种:
在一种可能的实现方式中,所述A1至所述A8各自独立地选自苯环、萘环、呋喃环或噻吩环中的至少一种。
在一种可能的实现方式中,所述化合物具有如式(III)所示结构:
其中,R
m、n各自独立地选自0-3的整数。
其中,R
以上基团中,任一可与式(III)键合的碳键、N键均可为连接位点。
进一步地,R
以上基团中,任一可与式(III)键合的碳键、N键均可为连接位点。
在一种可选实现方式中,R
其中,以上各所述桥接基团中的虚线表示与相邻的结构单元键合。
在一种可能的实现方式中,所述L选单键或以下结构中的一种:
其中,------代表L在式(I)、式(II-1)至式(II-3)、以及式(III)中的连接键;
Z选自O或S;
Q选自O、S、Se、BR
R
O选自1-6的整数。
在一种可能的实现方式中,所述L
其中,以上基团中的氢可被C1~C20碳原子的直链烷基取代;
以上基团中,任一可与式(I)键合的碳键均可为连接位点。
在一种可能的实现方式中,所述化合物选自以下结构中的一种:
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
进一步地,化合物具有如下结构式中的一种:
/>
第二方面,本申请还提供一种聚合物,该聚合物包括重复单元,所述重复单元包括由本申请第一方面所述的化合物形成的结构单元。
第三方面,本申请还提供一种混合物,该混合物包括第一有机物和第二有机物,所述第一有机物选自本申请第一方面的化合物或如本申请第二方面的聚合物,所述第二有机物为有机功能材料,所述第二有机物选自空穴注入材料、电子注入材料、空穴传输材料、电子传输材料、空穴阻挡材料、电子阻挡材料、发光材料、主体材料或有机染料中的至少一种。
第四方面,本申请提供一种组合物,该组合物包含至少一种有机溶剂,所述有机溶剂中含有至少一种上述化合物、上述聚合物或上述混合物中的至少一种。
第五方面,本申请还提供一种有机电子器件,该所述有机电子器件包含一功能层,所述功能层中包含一种上述有机化合物、上述高聚物或上述混合物。
其中,有机电子器件包括但不限于有机发光二极管、有机光伏电池、有机发光电池、有机场效应管、有机发光场效应管、有机激光器、有机自旋电子器件、有机传感器及有机等离激元发射二极管等。
上述第二方面至第五方面可以达到的技术效果,可以参照上述第一方面中的相应效果描述,这里不再重复赘述。
附图说明
图1为化合物1的甲苯稀溶液(1*10
图2为化合物2的甲苯稀溶液(1*10
图3为化合物3的甲苯稀溶液(1*10
图4为化合物4的甲苯稀溶液(1*10
图5为化合物5的甲苯稀溶液(1*10
图6为化合物6的甲苯稀溶液(1*10
具体实施方式
为了使本申请的目的、技术方案和优点更加清楚,下面将结合附图对本申请作进一步地详细描述。
以下实施例中所使用的术语只是为了描述特定实施例的目的,而并非旨在作为对本申请的限制。如在本申请的说明书和所附权利要求书中所使用的那样,单数表达形式“一个”、“一种”、“所述”、“上述”、“该”和“这一”旨在也包括例如“一个或多个”这种表达形式,除非其上下文中明确地有相反指示。
在本说明书中描述的参考“一个实施例”或“一些实施例”等意味着在本申请的一个或多个实施例中包括结合该实施例描述的特定特征、结构或特点。由此,在本说明书中的不同之处出现的语句“在一个实施例中”、“在一些实施例中”、“在其他一些实施例中”、“在另外一些实施例中”等不是必然都参考相同的实施例,而是意味着“一个或多个但不是所有的实施例”,除非是以其他方式另外特别强调。术语“包括”、“包含”、“具有”及它们的变形都意味着“包括但不限于”,除非是以其他方式另外特别强调。
以下将结合具体实施例对本申请的化合物做具体说明。
实施例1-化合物632
化合物632的制备过程包括如下步骤:
S1)制备化合物1-3
在25mL的干燥史莱克管中加入化合物1-1(193.0mg,1.0mmol)、化合物1-2(279.4mg,1.0mmol)、碳酸铯(488.7mg,1.5mmol),抽真空氮气置换三次后加入3mL的N,N’-二甲基甲酰胺,加热至150℃回流反应。通过硅胶薄层色谱监测反应结束,用饱和食盐水和乙酸乙酯萃取,收集有机相用无水硫酸镁干燥,减压蒸馏除去有机溶剂,使用色谱柱纯化得到化合物1-3。
利用大气固体分析探针质谱(atmospheric solids analysis probe
核磁数据
S2)制备化合物1-5
在25mL的干燥史莱克管中依次加入化合物1-3(452.4mg,1.0mmol)、化合物1-4(175.2mg,0.5mmol)、碳酸铯(488.7mg,1.5mmol),抽真空氮气置换三次后加入3mL的N,N’-二甲基甲酰胺,加热至100℃反应。通过硅胶薄层色谱监测反应结束,用饱和食盐水和乙酸乙酯萃取,收集有机相用无水硫酸镁干燥,减压蒸馏除去有机溶剂,使用色谱柱纯化得到化合物1-5。MS(ASAP):1212.4。
S3)制备化合物632
在25mL的干燥史莱克管中加入化合物1-5(364.6mg,0.3mmol),抽真空氮气置换三次后加入3mL叔丁基苯,冷却至-30℃,将正丁基锂的正己烷溶液(2.5M,252μL)逐滴加入到反应管内,在-30℃下保温30min后,恢复室温继续反应。通过硅胶薄层色谱监测反应完全后冷却至-30℃,逐滴加入三溴化硼(225.5mg,0.9mmol),自然恢复至室温继续反应1h。将混合物冷却至-30℃,逐滴加入N,N’-二异丙基乙胺(206.8mg,1.6mmol),在-30℃下保温30min后,加热到170℃回流反应8h后结束反应。用饱和食盐水和乙酸乙酯萃取,收集有机相用无水硫酸镁干燥,减压蒸馏除去有机溶剂,使用色谱柱纯化得到化合物632,产率3%。MS(ASAP):1072.5。
化合物632的核磁数据如下:
1
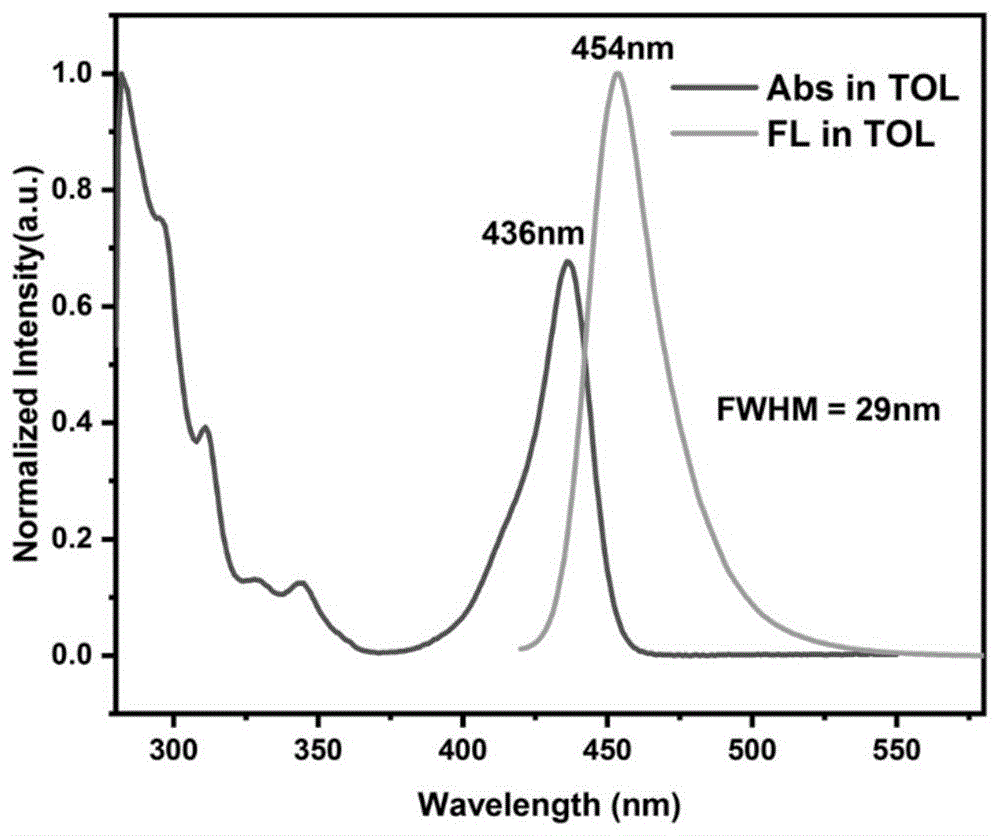
发光光谱测试:图1为化合物632的甲苯稀溶液(1*10-5M)在光致发光光谱图,如图1所示,化合物632的甲苯稀溶液(1*10-5M)在光致发光测试中展现出了纯蓝色发光,化合物632的发光峰值为454nm,且发光光谱FWHM仅29nm,具有高的发光效率与窄的发光光谱FWHM,具有实现高效蓝光显示的潜力。
对比于已有的硼-氮-氧体系材料或硼-氮二聚体体系材料,在发光峰值没有明显变化的情况下FWHM明显变窄,更具备实现高效高显色指数蓝光显示的潜力。
实施例2-化合物623
化合物623的制备过程包括以下步骤:
S1)制备化合物2-3
在25mL的干燥史莱克管中加入化合物2-1(193.0mg,1.0mmol)、化合物2-2(167.2mg,1.0mmol)、碳酸铯(488.7mg,1.5mmol),抽真空氮气置换三次后加入3mL的N,N’-二甲基甲酰胺,加热至150℃回流反应。通过硅胶薄层色谱监测反应结束,用饱和食盐水和乙酸乙酯萃取,收集有机相用无水硫酸镁干燥,减压蒸馏除去有机溶剂,使用色谱柱纯化得到化合物2-3。MS(ASAP):339.0。
S2)制备化合物2-5
在25mL的干燥史莱克管中依次加入化合物2-3(340.2mg,1.0mmol)、化合物2-4(175.2mg,0.5mmol)、碳酸铯(488.7mg,1.5mmol),抽真空氮气置换三次后加入3mL的N,N’-二甲基甲酰胺,加热至100℃反应。通过硅胶薄层色谱监测反应结束,用饱和食盐水和乙酸乙酯萃取,收集有机相用无水硫酸镁干燥,减压蒸馏除去有机溶剂,使用色谱柱纯化得到2-5。MS(ASAP):988.1。
S3)制备化合物623
在25mL的干燥史莱克管中加入化合物2-5(330.2mg,0.3mmol),抽真空氮气置换三次后加入3mL叔丁基苯,冷却至-30℃,将正丁基锂的正己烷溶液(2.5M,252μL)逐滴加入到反应管内,在-30℃下保温30min后,恢复室温继续反应。通过硅胶薄层色谱监测反应完全后冷却至-30℃,逐滴加入三溴化硼(225.5mg,0.9mmol),自然恢复至室温继续反应1h。将混合物冷却至-30℃,逐滴加入N,N’-二异丙基乙胺(206.8mg,1.6mmol),在-30℃下保温30min后,加热到170℃回流反应8h后结束反应。用饱和食盐水和乙酸乙酯萃取,收集有机相用无水硫酸镁干燥,减压蒸馏除去有机溶剂,使用色谱柱纯化得到化合物623,产率4%。MS(ASAP):848.3。
化合物623的核磁数据如下:
1
发光光谱测试:图2为化合物623的甲苯稀溶液(1*10-5M)在光致发光光谱图,如图2所示,化合物623的甲苯稀溶液(1*10-5M)在光致发光测试中展现出了纯蓝色发光,化合物623的发光峰值为446nm,且发光光谱FWHM仅30nm,具有高的发光效率与窄的发光光谱FWHM,具有实现高效蓝光显示的潜力。
对比于已有的硼-氮-氧体系材料或硼-氮二聚体体系材料,在发光峰值没有明显变化的情况下FWHM明显变窄,更具备实现高效高显色指数蓝光显示的潜力。
实施例3
化合物1054的制备过程包括以下步骤:
S1)制备化合物1-3
与实施例1中的化合物1-3的制备过程相同,在此不再重复描述。
S2)制备化合物3-5
在25mL的干燥史莱克管中依次加入化合物1-3(452.4mg,1.0mmol)、化合物3-4(185.2mg,0.5mmol)、碳酸铯(488.7mg,1.5mmol),抽真空氮气置换三次后加入3mL的N,N’-二甲基甲酰胺,加热至100℃反应。通过硅胶薄层色谱监测反应结束,用饱和食盐水和乙酸乙酯萃取,收集有机相用无水硫酸镁干燥,减压蒸馏除去有机溶剂,使用色谱柱纯化得到3-5。MS(ASAP):1048.3。
S3)制备化合物1054
在25mL的干燥史莱克管中加入化合物3-5(350.3mg,0.3mmol),抽真空氮气置换三次后加入3mL叔丁基苯,冷却至-30℃,将正丁基锂的正己烷溶液(2.5M,252μL)逐滴加入到反应管内,在-30℃下保温30min后,恢复室温继续反应。通过硅胶薄层色谱监测反应完全后冷却至-30℃,逐滴加入三溴化硼(225.5mg,0.9mmol),自然恢复至室温继续反应1h。将混合物冷却至-30℃,逐滴加入N,N’-二异丙基乙胺(206.8mg,1.6mmol),在-30℃下保温30min后,加热到170℃回流反应8h后结束反应。用饱和食盐水和乙酸乙酯萃取,收集有机相用无水硫酸镁干燥,减压蒸馏除去有机溶剂,使用色谱柱纯化得到化合物1054,产率3%。MS(ASAP):908.5。
化合物1054的核磁数据如下:
1
发光光谱测试:图3为化合物1054的甲苯稀溶液(1*10-5M)在光致发光光谱图,如图3所示,化合物1054的甲苯稀溶液(1*10-5M)在光致发光测试中展现出了纯蓝色发光,化合物1054的发光峰值为459nm,且发光光谱FWHM仅33nm,具有高的发光效率与窄的发光光谱FWHM,具有实现高效蓝光显示的潜力。
实施例4
化合物1055的制备过程包括以下步骤:
S1)制备化合物4-3
在500mL的三口烧瓶中依次加入化合物4-1(7.5g,50.3mmol)、化合物4-2(23.6g,100mmol)、三(二亚苄基丙酮)二钯(0.5g,0.5mmol)、2-二环己基膦-2′,6′-二甲氧基-联苯(0.4g,1.0mmol),叔丁醇钠(9.7g,100.6mmol),抽真空氮气置换三次后加入300mL甲苯,加热到110℃回流反应,TLC跟踪反应。待反应完全,冷却至室温,旋蒸除去反应体系中的有机溶剂,用二氯甲烷和饱和氯化钠溶液萃取,合并有机相干燥、过滤浓缩后用二氯甲烷:正己烷做淋洗剂通过硅胶层析柱分离得到产品4-3,干燥后约9.1g,产率为60%。MS(ASAP)=304.2。
S2)制备化合物4-4
在500mL的三口烧瓶中依次加入化合物4-3(25.5g,84mmol)、邻二甲苯(250mL)、1,8-二氮杂二环[5.4.0]十一碳-7-烯(14.1g,92.4mmol)、醋酸钯(566mg,2mmol)抽真空氮气置换三次后加入三叔丁基膦的邻二甲苯溶液(25wt%,3mL),加热到140℃回流反应,TLC跟踪反应。待反应完全,冷却至室温,旋蒸除去反应体系中的有机溶剂,用二氯甲烷和饱和氯化钠溶液萃取,合并有机相干燥、过滤浓缩后用二氯甲烷:正己烷做淋洗剂通过硅胶层析柱分离得到产品4-4,干燥后约17.2g,产率为92%。MS(ASAP)=223.1。
S3)制备化合物4-5
在500mL的三口烧瓶中加入三氯化铁(17.2g,106.0mmol),抽真空氮气置换三次后逐滴加入溶有中间体4-4(4.73g,21.2mmol)的200mL二氯甲烷,室温反应,TLC跟踪反应。待反应完全,旋蒸除去反应体系中的有机溶剂,用二氯甲烷和饱和氯化钠溶液萃取,合并有机相干燥、过滤浓缩后用二氯甲烷:正己烷做淋洗剂通过硅胶层析柱分离得到产品4-5,干燥后约4.9g,产率为52%。MS(ASAP)=444.3。
S4)制备化合物4-7
化合物4-7的合成方法与化合物1-3一致,仅需将化合物1-2替换为等量的化合物4-6。MS(ASAP)=322.0。
S5)制备化合物4-8
化合物4-8的合成方法与化合物4-7一致,仅需将化合物4-6替换为等量的化合物4-5,化合物1-1替换为双倍量的化合物4-7。MS(ASAP)=1048.3。
S6)制备化合物1055
化合物1055的合成方法与化合物1一致,仅需将化合物1-5替换为等量的化合物4-8。MS(ASAP)=908.5。
化合物1055的核磁数据如下:
1H NMR(500MHz,Chloroform-d)δ8.50–8.40(m,2H),8.31(d,J=7.5Hz,1H),7.89(ddd,J=7.5,6.0,1.5Hz,1H),7.72(ddd,J=7.5,4.6,2.0Hz,1H),7.54–7.48(m,2H),7.41(ddd,J=9.4,7.5,1.7Hz,2H),6.97–6.90(m,2H),1.28(s,18H).
发光光谱测试:图4为化合物1055的甲苯稀溶液(1*10-5M)在光致发光光谱图,如图4所示,化合物1055的甲苯稀溶液(1*10-5M)在光致发光测试中展现出了纯蓝色发光,化合物1055的发光峰值为459nm,且发光光谱FWHM仅33nm,具有高的发光效率与窄的发光光谱FWHM,具有实现高效蓝光显示的潜力。
实施例5-化合物959
化合物959的制备过程包括以下步骤:
S1)制备化合物5-2
化合物5-2的合成方法与化合物4-7的合成方法一致,仅需将化合物4-6替换为等量的化合物5-1。MS(ASAP)=266.0。
S2)制备化合物5-3
化合物5-3的合成方法与化合物4-8的合成方法一致,仅需将化合物4-7替换为等量的化合物5-2。MS(ASAP)=936.2。
S3)制备化合物959
化合物959的合成方法与化合物4的合成方法一致,仅需将化合物4-7替换为等量的化合物5-3。MS(ASAP)=796.3。
化合物959的核磁数据如下:
1H NMR(500MHz,Chloroform-d)δ8.48(t,J=1.3Hz,1H),8.42(dd,J=5.9,1.6Hz,1H),8.31(d,J=7.5Hz,1H),7.90(ddd,J=7.5,5.1,1.5Hz,1H),7.76–7.68(m,2H),7.51(dd,J=3.3,1.5Hz,1H),7.42(td,J=7.4,1.6Hz,1H),7.33(td,J=7.5,2.0Hz,1H),7.11(td,J=7.5,2.0Hz,1H),7.02–6.91(m,2H),1.28(s,9H).
发光光谱测试:图5为化合物959的甲苯稀溶液(1*10-5M)在光致发光光谱图,如图5所示,化合物959的甲苯稀溶液(1*10-5M)在光致发光测试中展现出了纯蓝色发光,化合物959的发光峰值为460nm,且发光光谱FWHM仅29nm,具有高的发光效率与窄的发光光谱FWHM,具有实现高效蓝光显示的潜力。
实施例6
化合物1056的制备过程包括以下步骤:
S1)制备化合物6-2
化合物6-2的合成方法与化合物4-5的合成方法一致,仅需将化合物4-4替换为等量的化合物6-1。MS(ASAP)=332.1。
S2)制备化合物6-3
化合物6-3的合成方法与化合物5-3的合成方法一致,仅需将化合物5-2替换为等量的化合物4-7。MS(ASAP)=936.2。
S3)制备化合物1056
化合物1056的合成方法与化合物5的合成方法一致,仅需将化合物5-3替换为等量的化合物6-3。MS(ASAP)=796.3。
化合物1056的核磁数据如下:
1H NMR(500MHz,Chloroform-d)δ8.45–8.37(m,2H),8.31(d,J=7.5Hz,1H),7.90(dt,J=7.5,1.7Hz,1H),7.73(ddd,J=7.9,6.3,1.9Hz,1H),7.55–7.46(m,2H),7.41(tdd,J=7.2,5.6,2.4Hz,3H),6.98–6.90(m,2H),1.28(s,9H).
发光光谱测试:图6为化合物1056的甲苯稀溶液(1*10-5M)在光致发光光谱图,如图6所示,化合物1056的甲苯稀溶液(1*10-5M)在光致发光测试中展现出了纯蓝色发光,化合物1056的发光峰值为456nm,且发光光谱FWHM仅30nm,具有高的发光效率与窄的发光光谱FWHM,具有实现高效蓝光显示的潜力。
实施例7
化合物1057的制备过程包括以下步骤:
S1)制备化合物7-3
在500mL的三口烧瓶中依次加入化合物7-1(23.6g)、化合物7-2(41.4g)、四三苯基膦钯(1.2g)、碳酸钾(28g),抽真空氮气置换三次后加入甲苯(220mL)、乙醇(150mL)与去离子水(75mL),70℃下反应8小时。通过硅胶薄层色谱监测反应结束,用饱和食盐水和乙酸乙酯萃取,收集有机相用无水硫酸镁干燥,减压蒸馏除去有机溶剂,使用色谱柱纯化得到7-3。MS(ASAP):262.0。
S2)制备化合物7-4
化合物7-4的合成方法与化合物1-5的合成方法一致,仅需将化合物1-4替换为等量的化合物7-3。MS(ASAP)=1124.4。
S3)制备化合物7
化合物1057的合成方法与化合物1的合成方法一致,仅需将化合物1-5替换为等量的化合物7-4。MS(ASAP)=984.5。
化合物1057的核磁数据如下:
1H NMR(500MHz,Chloroform-d)δ8.44–8.36(m,2H),8.04(d,J=7.5Hz,1H),7.93–7.85(m,3H),7.71(dd,J=7.5,2.0Hz,1H),7.63(dd,J=7.5,2.0Hz,1H),7.51(dd,J=3.0,1.5Hz,1H),7.40(t,J=7.4Hz,1H),7.21–7.14(m,2H),6.95(dd,J=7.5,2.0Hz,1H),1.33(s,9H),1.28(s,9H).
实施例8
化合物1058的制备过程包括以下步骤:
S1)制备化合物8-2
化合物8-2的合成方法与化合物7-3的合成方法一致,仅需将化合物7-1替换为等量的化合物8-1。MS(ASAP)=252.0。
S2)制备化合物8-3
化合物8-3的合成方法与化合物1-5的合成方法一致,仅需将化合物1-4替换为等量的化合物8-2。MS(ASAP)=1114.3。
S3)制备化合物1058
化合物1058的合成方法与化合物1的合成方法一致,仅需将化合物1-5替换为等量的化合物8-3。MS(ASAP)=974.5。
化合物1058的核磁数据如下:
1H NMR(500MHz,Chloroform-d)δ8.47–8.37(m,4H),8.05(dd,J=7.5,3.5Hz,2H),7.71(ddt,J=6.7,4.2,2.1Hz,3H),7.62(d,J=1.5Hz,1H),7.54(d,J=1.5Hz,1H),7.41(td,J=7.5,2.0Hz,2H),7.25–7.13(m,4H),7.09–7.00(m,2H),6.96(td,J=7.9,2.0Hz,2H),1.33(s,18H),1.28(s,18H).
实施例9
化合物1059的制备过程包括以下步骤:
S1)制备化合物9-2
化合物9-2的合成方法与化合物7-3的合成方法一致,仅需将化合物7-1替换为等量的化合物9-1。MS(ASAP)=312.1。
S2)制备化合物9-3
化合物9-3的合成方法与化合物1-5的合成方法一致,仅需将化合物1-4替换为等量的化合物9-2。MS(ASAP)=1174.4。
S3)制备化合物1059
化合物1059的合成方法与化合物1的合成方法一致,仅需将化合物1-5替换为等量的化合物9-3。MS(ASAP)=1034.5。
化合物1059的核磁数据如下:
1H NMR(500MHz,Chloroform-d)δ8.44–8.36(m,2H),8.10(dt,J=7.3,1.5Hz,1H),8.04(d,J=7.5Hz,1H),7.99(dd,J=2.7,1.5Hz,1H),7.89(t,J=1.9Hz,1H),7.78(dt,J=7.6,1.4Hz,1H),7.72–7.62(m,2H),7.51(dd,J=6.4,1.6Hz,1H),7.40(t,J=7.5Hz,1H),7.21–7.12(m,2H),6.93(dd,J=7.5,2.0Hz,1H),1.33(s,9H),1.28(s,9H).
实施例10
化合物1060的制备过程包括以下步骤:
S1)制备化合物10-2
化合物10-2的合成方法与化合物7-3的合成方法一致,仅需将化合物7-1替换为等量的化合物10-1。MS(ASAP)=502.2。
S2)制备化合物10-3
化合物10-3的合成方法与化合物1-5的合成方法一致,仅需将化合物1-4替换为等量的化合物10-2。MS(ASAP)=1364.4。
S3)制备化合物1060
化合物1060的合成方法与化合物1的合成方法一致,仅需将化合物1-5替换为等量的化合物10-3。MS(ASAP)=1034.5。
化合物1060的核磁数据如下:
1H NMR(500MHz,Chloroform-d)δ8.66–8.60(m,2H),8.34(dt,J=7.4,1.5Hz,2H),8.20(t,J=1.8Hz,2H),8.12(td,J=6.8,6.3,2.3Hz,4H),7.98(d,J=2.0Hz,1H),7.95–7.82(m,3H),7.79–7.74(m,2H),7.68–7.56(m,3H),7.46(dd,J=7.5,2.0Hz,1H),7.35(t,J=7.4Hz,1H),7.30–7.19(m,6H),7.15(d,J=7.5Hz,1H),7.04(ddd,J=19.8,7.5,1.5Hz,2H),6.93(dd,J=7.5,2.0Hz,1H),6.85(dd,J=7.5,2.0Hz,1H),6.66(dd,J=7.5,2.0Hz,1H),6.50(d,J=7.5Hz,1H),6.41(t,J=7.5Hz,1H),6.14(dd,J=7.5,2.0Hz,1H),1.33(s,18H),1.28(s,18H).
实施例11
化合物1061的制备过程包括以下步骤:
S1)制备化合物11-3
在500mL的三口烧瓶中依次加入化合物1-1(23g)、化合物11-2(32g)、三(二亚苄基丙酮)二钯(1.8g)、三特丁基膦四氟硼酸盐(2.9g),叔丁醇钠(20g),抽真空氮气置换三次后加入300mL甲苯,加热到110℃回流反应,TLC跟踪反应。待反应完全,冷却至室温,旋蒸除去反应体系中的有机溶剂,用二氯甲烷和饱和氯化钠溶液萃取,合并有机相干燥、过滤浓缩后用二氯甲烷:正己烷做淋洗剂通过硅胶层析柱分离得到产品11-3。MS(ASAP)=305.2。
S2)制备化合物11-4
在500mL的三口烧瓶中加入化合物11-3(20g)与48%氢溴酸溶液(300mL),加热至回流,TLC跟踪反应。待反应完全,冷却至室温,用二氯甲烷和饱和氯化钠溶液萃取,合并有机相干燥、过滤浓缩后用二氯甲烷:正己烷做淋洗剂通过硅胶层析柱分离得到产品11-3。
S3)制备化合物11-5
化合物11-5的合成方法与化合物1-5的合成方法一致,仅需将化合物1-4替换为等量的化合物11-4。MS(ASAP)=1139.4。
S4)制备化合物1061
化合物1061的合成方法与化合物1的合成方法一致,仅需将化合物1-5替换为等量的化合物11-5。MS(ASAP)=999.5。
化合物1061的核磁数据如下:
1H NMR(500MHz,Chloroform-d)δ8.42–8.34(m,2H),8.04(t,J=7.7Hz,1H),7.71(ddd,J=17.4,7.4,1.9Hz,1H),7.46–7.33(m,3H),7.28–7.14(m,4H),7.11–7.04(m,1H),7.04–6.88(m,3H),1.33(s,9H),1.28(s,9H).
实施例12
化合物1062的制备过程包括以下步骤:
S1)制备化合物12-3
S2)制备化合物12-4
化合物12-4的合成方法与化合物11-4的合成方法一致,仅需将化合物11-4替换为等量的化合物12-4。MS(ASAP)=202.1。
S3)制备化合物12-5
化合物12-5的合成方法与化合物1-5的合成方法一致,仅需将化合物1-4替换为等量的化合物12-4。MS(ASAP)=1064.3。
S4)制备化合物1062
化合物1062的合成方法与化合物1的合成方法一致,仅需将化合物1-5替换为等量的化合物12-5。MS(ASAP)=924.4。
化合物1062的核磁数据如下:
1H NMR(500MHz,Chloroform-d)δ8.40(dd,J=16.7,1.6Hz,2H),8.04(d,J=7.5Hz,1H),7.70(dd,J=7.5,2.0Hz,1H),7.54(d,J=1.5Hz,1H),7.40(t,J=7.5Hz,1H),7.27(d,J=1.4Hz,1H),7.18(dd,J=7.5,1.5Hz,1H),7.01–6.90(m,3H),1.33(s,9H),1.28(s,9H).
对比例1
对比化合物1
对比例2
对比化合物2
分别测试不同实施例化合物和对比化合物的发光光谱以及荧光量子产率,测试结果列于表1。
表1
从表1中的测试数据可以看出,本申请化合物的具有更窄的半峰宽,可有效提高发光效率。
实施例7至实施例12的化合物的发光波长、弛豫能、振子强度的相关测试数据列于表2。
表2
从表2中的相关数据可知,本申请实施例的发光波长、弛豫能以及振子强度均得到改善,其中,对应化合物的弛豫能越小对应的半峰宽也越小,振子强度越大,其对应的化合物的PLQY也越大,由上述数据可以看出,本申请实施例的化合物能够满足高效、高显色指数的蓝光OLED显示技术的需求。
以上,仅为本申请的具体实施方式,但本申请的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本申请揭露的技术范围内,可轻易想到变化或替换,都应涵盖在本申请的保护范围之内。因此,本申请的保护范围应以权利要求的保护范围为准。