一种2-氯-4-(4-氯苯氧基)-苯乙酮的合成方法
文献发布时间:2023-06-19 13:48:08
技术领域
本发明涉及一种合成方法,特别涉及一种2-氯-4-(4-氯苯氧基)-苯乙酮的合成方法。
背景技术
2-氯-4-(4-氯苯氧基)-苯乙酮(以下简称“二苯醚酮”)是合成农用苯醚甲环唑的中间体。苯醚甲环唑是一种超高效、光谱、内吸性三唑类杀菌剂,对植物病原菌的孢子形成强烈的抑制作用,并在防治病害的过程中,表现出预防、治疗、铲除的三大功效。因此其应用技术已成为农药企业的研究重点,潜在应用市场非常广阔。目前我国三氮唑类杀菌剂使用量逐年增高,因此加紧对其中间体二苯醚酮的研制开发并使之大规模生产,可进一步提高企业的经济效益。
2007年,中国中化集团公司申请了《一种制备2-氯-4-(4-氯苯氧基)-苯乙酮的方法》的专利(CN101434528A),该专利采用了二苯醚、三氯化铝、乙酸酐为原料,二氯甲烷为溶剂,经过合成、水解、萃取、脱溶、重结晶等工序获得二苯醚酮。该工艺主要采用先水解、后处理溶剂的方式生产二苯醚酮,溶剂回收率低,且操作复杂。重结晶工序中采用甲醇作为重结晶溶剂,由于甲醇低温下对二苯醚酮仍有一定的溶解度,造成二苯醚酮回收率低。多批次平均收率约为90%,仍有一定的提升空间。
发明内容
本发明为了解决上述问题,提出了一种2-氯-4-(4-氯苯氧基)-苯乙酮的合成方法,包括原料:原料二苯醚、路易斯酸和乙酰氯溶液,反应化学方程式如下:
制作如下步骤:
(1)将二苯醚和路易斯酸加入溶剂中,采用计量泵将乙酰氯溶液也打入溶剂中,温度为-15~50℃合成反应生成二苯醚酮合成液;
(2)将二苯醚酮合成液升温脱除溶剂,析出固体,脱除的溶剂回用至步骤(1)中,产生的固体加入去离子水水解,分层获得油层与水层;
(3)将步骤(2)中油层用去离子水洗涤一次,水洗后的水与步骤(2)中水解后的水层合并后放于釜中,在温度为5~50℃下用溶剂萃取,萃取后油层在温度为50~100℃蒸发回收溶剂,溶剂干燥后回到步骤(1)中重复利用,蒸发后所得釜余与步骤(3)中水洗后油层合加入石油醚重结晶,所述的重结晶升温溶解终点温度为30~105℃,结晶降温终点温度为0~25℃,之后抽滤得到精品二苯醚酮。
进一步的,所述的溶剂为二氯乙烷、邻二氯苯、氯苯、甲苯、二硫化碳、四氯化碳、硝基苯或四氢呋喃。
进一步的,步骤(1)中所述的路易斯酸是三氯化铝、硝酸镧、氯化锡或氯化铈。
进一步的,步骤(1)中所述的合成反应温度为-15~25℃。
进一步的,步骤(2)中所述的升温脱除溶剂温度为25~85℃,所述的去离子水量为析出固体质量的2.5~3倍。
进一步的,步骤(3)中所述的水解过程温度为60~80℃,
进一步的,步骤(3)中所述的洗涤温度为5~50℃。
进一步的,所述的萃取温度为5~15℃,所述的萃取用溶剂量为所述水层质量的0.5~1倍。
进一步的,步骤(3)中所述的萃取油层的温度为50~78℃。
进一步的,步骤(3)中所述的石油醚的质量为蒸发釜余和水洗后油层总量的1.5~2倍。
与现有技术相比,本发明的有益效果是:
本发明一种2-氯-4-(4-氯苯氧基)-苯乙酮的合成方法,提供了以二苯醚、路易斯酸、乙酰氯等原料经过先脱溶后水解的方式合成二苯醚酮,同时本申请也提供了一种二苯醚酮重结晶方法。本发明简化了生产工艺,产品收率高,原料消耗低,易于工业化实施。
附图说明
为了更清楚的说明本发明的技术方案,下面将对实施例中所需要使用的附图作简要的介绍,显而易见地,对于本领域普通技术人员而言,在不付出创造性的前提下,还可以根据这些附图获得其他的附图。
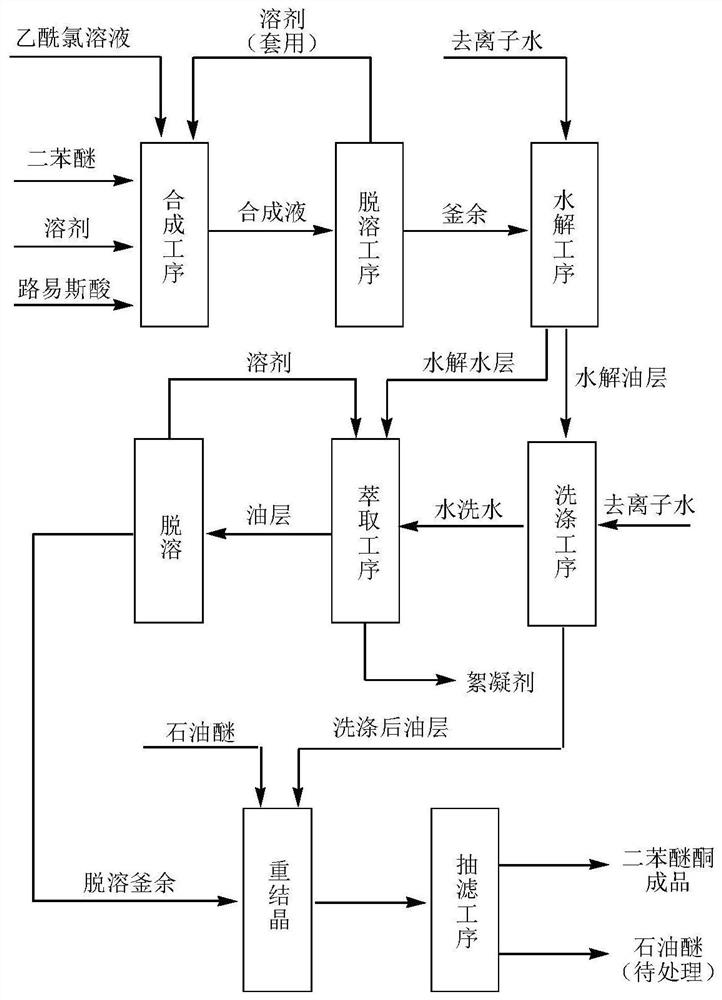
图1为本发明工艺流程图。
具体实施方式
为了使本技术领域的人员更好地理解本申请中的技术方案,下面将结合附图,对本申请实施例中的技术方案进行清楚完整的描述。为了使本领域技术人员更好地理解本方案,下面以具体实施例对本发明进行详细说明。下面的实施例是对本发明进行更详细的阐述,而不是对本发明的进一步限定。除纯度为百分比外,除非另有说明,其中的“%”均为“质量%”。
如图1所示:
实验实例1
(1)在1000ml四口烧瓶中投入二苯醚(98.5%)75.0g,加入溶剂二氯乙烷90.2g,催化剂三氯化铝49.2g,降温至5℃。将30.0g乙酰氯与90.0g二氯乙烷配置成均匀溶液,使用计量泵打入反应体系,控制反应滴加时间2h,保温时间4h。保温结束后,取少量反应液水解得到油层,归一气谱检测,二苯醚含量0.005%,二苯醚酮含量95.2%。改合成装置为蒸馏装置,负压条件下脱除171.3g二氯乙烷(95%馏分),釜余为黑色固体颗粒物132.7g。
(2)准备1000mL四口烧瓶一个,加入去离子水380g,20g试剂盐酸,降温至5℃,缓慢将上述132.7g固体放入水中,待水解彻底结束后,70℃保温0.5h,趁热分层。得到油层92.0g。水层418.7g
(3)将(2)中油层用50g去离子水室温洗涤一次,得水洗后油层90.2g。将所得水洗水51.5g与(2)中水解后水层合并后用240g二氯乙烷萃取一次,萃取后油层蒸发回收溶剂,干燥后重复利用。蒸发后所得釜余3.5g与(3)中水洗后油层合并。
(4)将(3)中93.7g合并油层溶于165g石油醚中,升温至72℃,保温15min后,降温结晶,10℃抽滤得到精制二苯醚酮固体81.87g。
结果由气谱归一检测,成品归一结果为99.92%,总收率为94.2%。
实验实例2
(1)在1000ml四口烧瓶中投入二苯醚(98.5%)75.0g(0.3mol),加入溶剂邻二氯苯132g(0.9mol),催化剂三氯化铝49.2g,降温至5℃。将30.0g乙酰氯与143g邻二氯苯配置成均匀溶液,使用计量泵打入反应体系,控制反应滴加时间2h,保温时间4h。保温结束后,取少量反应液水解获得油层,归一气谱检测,二苯醚含量0.003%,二苯醚酮含量95.9%。改合成装置为蒸馏装置,负压条件下脱除264g邻二氯苯(96%馏分),釜余为黑色固体颗粒物136.3g。
(2)向1000mL四口烧瓶中加入去离子水380g,20g试剂盐酸,降温至5℃,缓慢将上述136.3g固体放入水中,待水解彻底结束后,60℃保温0.5h,趁热分层。得到油层92.4g。水层421.3g
(3)将(2)中油层用50g去离子水室温洗涤一次,得水洗后油层90.8g。将所得水洗水50.9g与(2)中水解后水层合并后用240g邻二氯苯萃取一次,萃取后油层蒸发回收溶剂,干燥后重复利用。蒸发后所得釜余3.4g与(3)中水洗后油层合并。
(4)将(3)中94.2g合并油层溶于165g石油醚中,升温至70℃,保温15min后,降温结晶,10℃抽滤得到精制二苯醚酮固体82.8g。
结果由气谱归一检测,成品归一结果为99.1%,总收率为94.5%。
实验实例3
(1)在1000ml四口烧瓶中投入二苯醚(98.5%)75.0g(0.3mol),加入溶剂甲苯83.2g(0.90mol),催化剂氯化锡89.9g,降温至5℃。将30.0g乙酰氯与82.7g甲苯配置成均匀溶液,使用计量泵打入反应体系,控制反应滴加时间2h,保温时间4h。保温结束后,取少量反应液水解获得油层,归一气谱检测,二苯醚含量0.008%,二苯醚酮含量94.9%。改合成装置为蒸馏装置,负压条件下脱除157.6g甲苯(95%馏分),釜余为黑色固体颗粒物174.3g。
(2)向1000mL四口烧瓶中加入去离子水380g,20g试剂盐酸,降温至5℃,缓慢将上述174.3g固体放入水中,待水解彻底结束后,60℃保温0.5h,趁热分层。得到油层91.8g。水层459.8g
(3)将(2)中油层用50g去离子水室温洗涤一次,得水洗后油层90.5g。将所得水洗水50.8g与(2)中水解后水层合并后用240g甲苯萃取一次,萃取后油层蒸发回收溶剂,干燥后重复利用。蒸发后所得釜余3.2g与(3)中水洗后油层合并。
(4)将(3)中93.7g合并油层溶于165g石油醚中,升温至70℃,保温15min后,降温结晶,10℃抽滤得到精制二苯醚酮固体82.32g。
结果由气谱归一检测,成品归一结果为99.2%,总收率为94%。
在实验实例4-8选用三氯化铝为催化剂,考察不同合成温度下,对催化反应合成二苯醚酮的影响:
实验实例4
(1)在1000ml四口烧瓶中投入二苯醚(98.5%)75.0g,加入溶剂二氯乙烷90.2g,催化剂三氯化铝49.2g,降温至-15℃。将30.0g乙酰氯与90.0g二氯乙烷配置成均匀溶液,使用计量泵打入反应体系,控制反应滴加时间2h,保温时间4h。保温结束后。取少量反应液水解得到油层,归一气谱检测,当二苯醚含量<1%时,改合成装置为蒸馏装置,负压条件下脱除171g二氯乙烷(95%馏分),釜余为黑色固体颗粒物134.5g。
(2)向1000mL四口烧瓶中加入去离子水380g,20g试剂盐酸,降温至5℃,缓慢将上述134.5g固体放入水中,待水解彻底结束后,70℃保温0.5h,趁热分层。得到油层91.8g。水层420.5g
(3)将(2)中油层用50g去离子水室温洗涤一次,得水洗后油层90g。将所得水洗水51.4g与(2)中水解后水层合并后用240g二氯乙烷萃取一次,萃取后油层蒸发回收溶剂,干燥后重复利用。蒸发后所得釜余3.3g与(3)中水洗后油层合并。
(4)将(3)中93.3g合并油层溶于165g石油醚中,升温至70℃,保温15min后,降温结晶,10℃抽滤得到精制二苯醚酮固体82.68g。
结果由气谱归一检测,成品归一结果为99.5%,总收率为94.7%。
实验实例5
(1)在1000ml四口烧瓶中投入二苯醚(98.5%)75.0g,加入溶剂二氯乙烷90.2g,催化剂三氯化铝49.2g,降温至-5℃。将30.0g乙酰氯与90.0g二氯乙烷配置成均匀溶液,使用计量泵打入反应体系,控制反应滴加时间2h,保温时间4h。保温结束后。取少量反应液水解得到油层,归一气谱检测,当二苯醚含量<1%时,改合成装置为蒸馏装置,负压条件下脱除170.8g二氯乙烷(95%馏分),釜余为黑色固体颗粒物135.2g。
(2)向1000mL四口烧瓶中加入去离子水380g,20g试剂盐酸,降温至5℃,缓慢将上述135.2g固体放入水中,待水解彻底结束后,70℃保温0.5h,趁热分层。得到油层92.4g。水层420.1g
(3)将(2)中油层用50g去离子水室温洗涤一次,得水洗后油层90.7g。将所得水洗水51.3g与(2)中水解后水层合并后用240g二氯乙烷萃取一次,萃取后油层蒸发回收溶剂,干燥后重复利用。蒸发后所得釜余3.8g与(3)中水洗后油层合并。
(4)将(3)中94.5g合并油层溶于165g石油醚中,升温至70℃,保温15min后,降温结晶,10℃抽滤得到精制二苯醚酮固体82.8g。
结果由气谱归一检测,成品归一结果为99.3%,总收率为94.65%。
实验实例6
(1)在1000ml四口烧瓶中投入二苯醚(98.5%)75.0g,加入溶剂二氯乙烷90.2g,催化剂三氯化铝49.2g,降温至5℃。将30.0g乙酰氯与90.0g二氯乙烷配置成均匀溶液,使用计量泵打入反应体系,控制反应滴加时间2h,保温时间4h。保温结束后。取少量反应液水解得到油层,归一气谱检测,当二苯醚含量<1%时,改合成装置为蒸馏装置,负压条件下脱除171.5g二氯乙烷(95%馏分),釜余为黑色固体颗粒物135.5g。
(2)向1000mL四口烧瓶中加入去离子水380g,20g试剂盐酸,降温至5℃,缓慢将上述135.5g固体放入水中,待水解彻底结束后,70℃保温0.5h,趁热分层。得到油层92.7g。水层420.8g
(3)将(2)中油层用50g去离子水室温洗涤一次,得水洗后油层90.5g。将所得水洗水51.6g与(2)中水解后水层合并后用240g二氯乙烷萃取一次,萃取后油层蒸发回收溶剂,干燥后重复利用。蒸发后所得釜余3.3g与(3)中水洗后油层合并。
(4)将(3)中93.8g合并油层溶于165g石油醚中,升温至70℃,保温15min后,降温结晶,10℃抽滤得到精制二苯醚酮固体82.75g。
结果由气谱归一检测,成品归一结果为99.2%,总收率为94.5%。
实验实例7
(1)在1000ml四口烧瓶中投入二苯醚(98.5%)75.0g,加入溶剂二氯乙烷90.2g,催化剂三氯化铝49.2g,降温至15℃。将30.0g乙酰氯与90.0g二氯乙烷配置成均匀溶液,使用计量泵打入反应体系,控制反应滴加时间2h,保温时间4h。保温结束后。取少量反应液水解得到油层,归一气谱检测,当二苯醚含量<1%时,改合成装置为蒸馏装置,负压条件下脱除171.2g二氯乙烷(95%馏分),釜余为黑色固体颗粒物135.3g。
(2)向1000mL四口烧瓶中加入去离子水380g,20g试剂盐酸,降温至5℃,缓慢将上述135.3g固体放入水中,待水解彻底结束后,70℃保温0.5h,趁热分层。得到油层92.4g。水层420.1g
(3)将(2)中油层用50g去离子水室温洗涤一次,得水洗后油层90.8g。将所得水洗水51g与(2)中水解后水层合并后用240g二氯乙烷萃取一次,萃取后油层蒸发回收溶剂,干燥后重复利用。蒸发后所得釜余3.6g与(3)中水洗后油层合并。
(4)将(3)中94.4g合并油层溶于165g石油醚中,升温至70℃,保温15min后,降温结晶,10℃抽滤得到精制二苯醚酮固体82.43g。
结果由气谱归一检测,成品归一结果为99.3%,总收率为94.23%。
实验实例8
(1)在1000ml四口烧瓶中投入二苯醚(98.5%)75.0g,加入溶剂二氯乙烷90.2g,催化剂三氯化铝49.2g,降温至25℃。将30.0g乙酰氯与90.0g二氯乙烷配置成均匀溶液,使用计量泵打入反应体系,控制反应滴加时间2h,保温时间4h。保温结束后。取少量反应液水解得到油层,归一气谱检测,当二苯醚含量<1%时,改合成装置为蒸馏装置,负压条件下脱除172.0g二氯乙烷(95.4%馏分),釜余为黑色固体颗粒物134.3g。
(2)向1000mL四口烧瓶中加入去离子水380g,20g试剂盐酸,降温至5℃,缓慢将上述134.3g固体放入水中,待水解彻底结束后,70℃保温0.5h,趁热分层。得到油层91.8g。水层420.8g
(3)将(2)中油层用50g去离子水室温洗涤一次,得水洗后油层90.2g。将所得水洗水51.1g与(2)中水解后水层合并后用240g二氯乙烷萃取一次,萃取后油层蒸发回收溶剂,干燥后重复利用。蒸发后所得釜余3.2g与(3)中水洗后油层合并。
(4)将(3)中93.4g合并油层溶于165g石油醚中,升温至70℃,保温15min后,降温结晶,10℃抽滤得到精制二苯醚酮固体82.23g。
结果由气谱归一检测,成品归一结果为99.4%,总收率为94.1%。
改变合成温度,考察对合成二苯醚酮的影响,相应结果统计如下:
由以上实验数据可以得出,在其他物质的量不变的情况下,只改变温度,在-15℃时总收率最高,-15℃为最佳反应温度。
本领域技术人员在考虑说明书及实践这里公开的申请后,将容易想到本申请的其他实施方案。本申请旨在涵盖本申请的任何变型、用途或者适应性变化,这些变型、用途或者适应性变化遵循本申请的一般性原理并包含本申请公开的本技术领域中的公知常识或惯用技术手段。说明书和实施例仅被视为实验实例性的,本申请的真正范围由权利要求指出。
应当理解的是,本申请并不局限于上面已经描述并在附图中示出的精确结构,并且可以在不脱离其范围进行各种修改和改变。以上所述的本申请实施方式并不构成对本申请保护范围的限定。