一种高性能脂肪族聚酯弹性体及其制备方法
文献发布时间:2023-06-19 11:09:54
技术领域
本发明涉及热塑性弹性体技术领域,尤其涉及一种高性能脂肪族聚酯弹性体及其制备方法。
背景技术
热塑性弹性体具有良好的弹性、热加工和再循环的能力,因此受到了研究者的广泛关注。目前商业化的热塑性弹性体,大多由来自于化石能源的化学品合成,而由于石油资源的日益枯竭,以及此类弹性体在使用服役结束后不能降解往往造成严重的白色污染,因此普遍具有不可降解和不可再生的特性。而来源于可再生脂肪族环内酯单体的脂肪族聚酯弹性体由于其可降解和可再生的特点,很好的克服了上述石油基弹性体的缺点,近年来备受研究者的关注。
热塑性聚酯弹性体普遍具有ABA三嵌段的结构,其中A为末端硬段,一般采用具较有高玻璃化转变温度的无定形聚合物或者结晶度较高的聚合物,常见的有聚乳酸PLA、L型聚乳酸PLLA以及邻苯二甲酸酐和氧化环己烯的交替共聚物;B为中间软段,一般使用具有较低玻璃化温度的无定形聚合物或者低结晶度的聚合物,如:ε-己内酯(ε-CL)、δ-戊内酯(δ-VL)、menthide、1,3-trimethylene carbonate(1,3-碳酸三甲酯)、6-methyl-ε-caprolactone(6-甲基-ε-己内酯)、3-methyl-1,5-pentylene succinate(丁二酸3-甲基-1,5-戊烯酯)、1,4-二恶烷-2-酮、γ-甲基-ε-己内酯、β-甲基-δ-戊内酯等单体的均聚物或者共聚物。但是由以上这些单体组合所得到的聚酯弹性体一般断裂伸长率小于2100%,断裂拉伸强度普遍低于40MPa,且二者不可兼得,即只有弹性体的断裂拉伸率较低时才能得到较大的应力,具有较高断裂伸长率的弹性体,往往断裂拉伸强度较低,如目前已报道的断裂伸长率能达到2000%的弹性体的应力基本都小于13.5MPa。因此,断裂拉伸率和断裂拉伸强度相互制约,现有技术不能同时获得较大断裂拉伸率和断裂拉伸强度值的弹性体。而为了满足生产生活应用,普遍要求材料同时具有足够的强度和断裂伸长率,因此,催化合成序列结构可控的高性能聚酯弹性体,使其同时具有高强度和韧性亟待解决。
发明内容
本发明的目的在于提供一种高性能脂肪族聚酯弹性体及其制备方法,所述高性能脂肪族聚酯弹性体兼具高强度和高断裂伸长率。
为了实现上述发明目的,本发明提供以下技术方案:
本发明提供了一种高性能脂肪族聚酯弹性体,具有式I所示结构:
其中,n、z、y和x独立为20~400,且均为整数;
R
R同时为甲基或同时为氢。
优选的,所述高性能脂肪族聚酯弹性体包括
本发明提供了上述技术方案所述高性能脂肪族聚酯弹性体的制备方法,包括以下步骤:
将ε-己内酯、δ-戊内酯、催化剂、引发剂和有机溶剂混合,进行第一聚合反应,得到聚合产物;
将所述聚合产物与交酯单体混合,进行第二聚合反应,得到高性能脂肪族聚酯弹性体;
所述催化剂的结构为
所述引发剂包括二元醇,所述二元醇包括
所述交酯单体为L型丙交酯、D型丙交酯、外消旋丙交酯或乙交酯;
所述引发剂与催化剂的摩尔比为(0.5~10):1;所述δ-戊内酯与ε-己内酯的摩尔比为(0.5~10):1;所述交酯单体与ε-己内酯的摩尔比为(0.5~10):1;所述ε-己内酯与引发剂的摩尔比为(100~1000):1。
优选的,所述催化剂、引发剂、ε-己内酯、δ-戊内酯和交酯单体的摩尔比为1:1:200:228:50。
优选的,所述催化剂、引发剂、ε-己内酯、δ-戊内酯和交酯单体的摩尔比为1:1:300:342:50~150。
优选的,所述催化剂、引发剂、ε-己内酯、δ-戊内酯和交酯单体的摩尔比为1:1:400:456:50~150。
优选的,所述第一聚合反应的温度为室温~100℃,时间为10~180min。
优选的,所述第二聚合反应的温度为为室温~100℃,时间为10~90min。
优选的,完成所述第二聚合反应后,还包括将所得产物与石油醚混合,淬出聚合物,抽至恒重,得到高性能脂肪族聚酯弹性体。
本发明提供了一种高性能脂肪族聚酯弹性体,具有式I所示结构:
其中,n、z、y和x独立为20~400,且均为整数;
R
R同时为甲基或同时为氢。
本发明的高性能脂肪族聚酯弹性体,在聚合物的中间软段链段结构中,均聚连接占有优势,大于无规共聚连接,整体呈梯度渐变结构,同时无规链段中的两种单体可以同构结晶,在外力的作用下软段链段结构中存在的均聚链很容易有序排列取向结晶,所形成的晶体作为新的硬相填充到整个聚合物网络中,进行自增韧,即本发明提供的高性能脂肪族聚酯弹性体的梯度渐变类似于多嵌段的软段所形成的弹性网络在提供应变的时候通过受力诱导结晶增强材料的应力,从而实现了超韧热塑性聚酯弹性体的合成,得到了高强度和高断裂伸长率的弹性体。
本发明提供了所述高性能脂肪族聚酯弹性体的制备方法,本发明采用能够同构结晶的两种单体ε-己内酯和δ-戊内酯的无规共聚物作为软段,采用交酯单体开环聚合形成的均聚物作为硬段,ε-己内酯和δ-戊内酯单体的无规共聚能够降低结晶度,低的结晶度能够使弹性体具有更好的链段自由度,在拉伸的时候容易解缠结,有利于提供更高的断裂伸长率。
本发明采用具有高活性、高选择性和良好控制性的含Mg有机金属催化剂,同时结合二元醇引发剂,催化ε-己内酯和δ-戊内酯单体的无规共聚以及交酯单体的开环聚合(配位插入聚合),无酯交换副反应,能够很好地调控软硬段的分子量以及微结构,并达到调控聚合物机械性能的目的,从而使所合成的聚合物的特殊链段结构能够受力诱导结晶,实现自增韧,因而具有优异的机械性能。
本发明所用催化剂廉价易得,且对单体具有不同的选择性,从而能够实现一锅两步法合成高性能的脂肪族聚酯弹性体,无需分离纯化,简单快捷节能;而现有技术是在加热的条件下,先合成中间软段弹性体,而后将其分离提纯后,加入新的催化剂和单体进行嵌段共聚。
附图说明
图1为实施例1制备的
图2为实施例1制备的
图3为实施例1中弹性体合成及结构序列示意图;
图4为实施例2制备的
图5为实施例2制备的
图6为实施例3制备的
图7为实施例3制备的
图8为实施例4制备的
图9为实施例4制备的
图10为实施例5制备的
图11为实施例5制备的
图12为实施例6制备的
图13为实施例6制备的
图14为实施例7制备的
图15为实施例7制备的
图16为实施例3反应过程中碳谱随转化率的变化图;
图17为对比例1反应过程中碳谱随转化率的变化图;
图18为对比例1反应过程中监测产生的PCVL碳谱图;
图19为实施例1~3制备的弹性体软段样品的差示扫描量热分析图;
图20为实施例2、4和5制备的弹性体样品的差示扫描量热分析图;
图21为实施例3、6和7制备的弹性体样品的差示扫描量热分析图;
图22为室温下,含Mg催化剂和对苯二甲醇在甲苯和二氯甲烷混合溶剂中催化合成的
图23为实施例1~7制备的弹性体样品的应力应变曲线图;
图24为实施例1制备的
图25为实施例2制备的
图26为实施例3制备的
图27为实施例1制备的弹性体样品的WAXD曲线;
图28为实施例2制备的弹性体样品的WAXD曲线;
图29为实施例3制备的弹性体样品的WAXD曲线;
图30为实施例1~3制备的弹性体样品的结晶度定量分析曲线图。
具体实施方式
本发明提供了一种高性能脂肪族聚酯弹性体,具有式I所示结构:
其中,n、z、y和x独立为20~400,且均为整数;
R
R同时为甲基或同时为氢。
在本发明中,所述n、z、y和x独立优选为50~350,更优选为100~300,进一步优选为150~250。
在本发明中,所述高性能脂肪族聚酯弹性体包括
本发明提供了上述技术方案所述高性能脂肪族聚酯弹性体的制备方法,包括以下步骤:
将ε-己内酯、δ-戊内酯、催化剂、引发剂和有机溶剂混合,进行第一聚合反应,得到聚合产物;
将所述聚合产物与交酯单体混合,进行第二聚合反应,得到高性能脂肪族聚酯弹性体;
所述催化剂的结构为
所述引发剂包括二元醇,所述二元醇包括
所述交酯单体为L型丙交酯、D型丙交酯、外消旋丙交酯或乙交酯;
所述引发剂与催化剂的摩尔比为(0.5~10):1;所述δ-戊内酯与ε-己内酯的摩尔比为(0.5~10):1;所述交酯单体与ε-己内酯的摩尔比为(0.5~10):1;所述ε-己内酯与引发剂的摩尔比为(100~1000):1。
在本发明中,若无特殊说明,所需制备原料均为本领域技术人员熟知的市售商品。
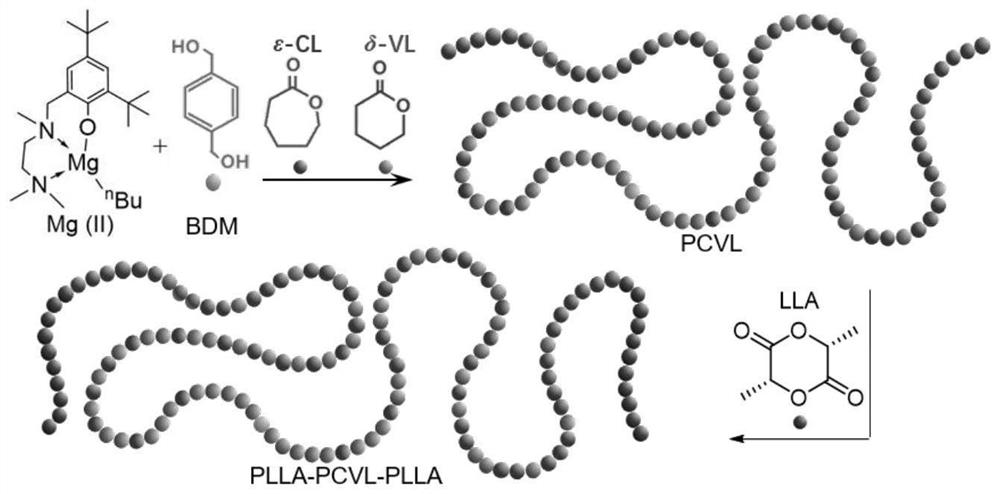
本发明将ε-己内酯、δ-戊内酯、催化剂、引发剂和有机溶剂混合,进行第一聚合反应,得到聚合产物。在本发明中,所述ε-己内酯和δ-戊内酯作为软段类单体,进行无规共聚,聚合产生的无规共聚物PCVL作为弹性体软段,所述ε-己内酯和δ-戊内酯的结构式分别为:
在本发明中,所述催化剂的结构为
在本发明中,所述催化剂的制备方法优选包括以下步骤:
将2,4-二叔丁基苯酚(15.6g,75.0mmol)、N,N,N’-三甲基-1,2-乙二胺(7.9g,75.0mmol)和多聚甲醛(3.1g,98.0mmol)溶于乙醇,加热至80℃回流24h,恢复至室温后,向所得产物中加入溴化氢水溶液(12.73mL,48%)反应生成季铵盐从而析出(Mannich反应),过滤收集沉淀,用乙醇洗涤沉淀至洗液呈无色,所得的白色固体溶于水中,用NaHCO
在本发明中,所述催化剂的制备过程如下式所示:
在本发明中,所述引发剂包括二元醇,所述二元醇包括
本发明利用引发剂与催化剂作为双组分催化剂,协同催化引发ε-己内酯和δ-戊内酯的无规共聚,聚合产生无规共聚物PCVL作为弹性体的软段,同时在不淬灭反应的情况下催化中间软段PCVL引发交酯单体聚合,产生PLLA作为硬段,从而得到具有PLLA-PCVL-PLLA结构的弹性体。
在本发明中,所述有机溶剂优选为无水甲苯。本发明对所述有机溶剂的用量没有特殊的限定,能够将物料充分溶解即可。
在本发明中,所述引发剂与催化剂的摩尔比为(0.5~10):1,优选为(2~8):1,进一步优选为(4~6):1;所述δ-戊内酯与ε-己内酯的摩尔比为(0.5~10):1,优选为(2~8):1,更优选为(4~6):1;所述交酯单体与ε-己内酯的摩尔比为(0.5~10):1,优选为(2~8):1,更优选为(4~6):1;所述ε-己内酯与引发剂的摩尔比为(100~1000):1,优选为(300~800):1,更优选为(500~700):1。
在本发明的实施例中,所述催化剂、引发剂、ε-己内酯、δ-戊内酯和交酯单体的摩尔比具体为1:1:200:228:50、1:1:300:342:(50~150)或1:1:400:456:(50~150)。
在本发明中,所述ε-己内酯、δ-戊内酯、催化剂、引发剂和有机溶剂混合的过程优选为将催化剂溶解于第一部分有机溶剂中,向所得溶液中加入引发剂,向所得溶液中加入第二部分有机溶剂,含Mg催化剂与引发剂反应1min(生成活性物种),向所得体系中加入ε-己内酯和δ-戊内酯。在本发明中,所述第一部分有机溶剂的用量优选少于第二部分有机溶剂,本发明对所述第一部分有机溶剂和第二部分有机溶剂的体积比没有特殊的限定,保证第一部分有机溶剂的用量明显少于第一部分有机溶剂即可。本发明首先加入少量第一部分有机溶剂,物料浓度高,能够保证含Mg催化剂与引发剂快速反应生成活性物种,便于后续聚合反应进行。
在本发明中,所述第一聚合反应的温度优选为室温~100℃,更优选为30~90℃,进一步优选为50~80℃,时间优选为10~180min,更优选为30~150min,进一步优选为50~100min;本发明优选根据不同单体比例确定反应时间,保证单体完全转化即可。
完成所述第一聚合反应后,本发明优选不进行任何后处理,直接向所得体系中加入交酯单体,进行第二聚合反应。在本发明中,所述交酯单体优选以溶液的形式使用,所述交酯单体的溶液所用溶剂优选为二氯甲烷,本发明对所述交酯单体的溶液的浓度没有特殊的限定,能够完全溶解交酯单体即可。在本发明中,所述交酯单体为L型丙交酯(LLA)、D型丙交酯(DLA)、外消旋丙交酯(rac-LA)或乙交酯(GA),其结构式分别为;
本发明利用交酯单体作为硬段单体,合成PGA或PLA作为弹性体的硬段,与ε-己内酯和δ-戊内酯单体共聚产生的PCVL软段构成具有PLLA-PCVL-PLLA结构的弹性体。
在本发明中,所述第二聚合反应的温度优选为室温~100℃,更优选为30~90℃,进一步优选为50~80℃;时间优选为10~90min,更优选为30~60min,进一步优选为40~50min。
完成所述第二聚合反应后,本发明优选还包括将所得产物与石油醚混合,淬出聚合物,抽至恒重,得到高性能脂肪族聚酯弹性体。本发明对所述石油醚的用量没有特殊的限定,能够完全淬出产物即可;本发明对所述抽至恒重的过程没有特殊的限定,按照本领域熟知的过程进行即可。
下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
以下实施例中,所用催化剂的制备方法为:
将2,4-二叔丁基苯酚(15.6g,75.0mmol)、N,N,N’-三甲基-1,2-乙二胺(7.9g,75.0mmol)和多聚甲醛(3.1g,98.0mmol)溶于乙醇,加热至80℃回流24h,恢复至室温后,加入溴化氢水溶液(12.73mL,48%)反应生成季铵盐从而析出,过滤收集沉淀,用乙醇洗涤沉淀至洗液呈无色,所得的白色固体溶于水中,用NaHCO
实施例1
本实施例中,催化剂:对苯二甲醇:ε-CL:δ-VL:LLA(L型丙交酯)的摩尔比为1:1:200:228:50,具体步骤为:
称量10mg(0.025mmol)催化剂溶解于500μL无水甲苯,称量3.5mg(0.025mmol)对苯二甲醇加入所得催化剂溶液中,再加入3.5mL无水甲苯,反应一分钟后,同时加入529μLε-CL(5.0mmol)和520μLδ-VL(5.7mmol),开始聚合反应35min,聚合完成后得到
表征及测试
1)对实施例1制备的弹性体样品进行核磁表征,结果见图1~2。
由核磁碳谱(图1)可知,中间软段PCVL存在四种比例不同的碳信号,其中ε-CL和δ-VL的均聚信号明显高于两种单体的无规共聚信号,由中间到两端首先是以δ-VL均聚为主导的无规共聚物,而后是ε-CL的均聚物,最后是LLA的均聚,PCVL与PLLA之间不存在酯交换,具体的序列结构分析见图3;同时核磁氢谱(图2)显示PCVL中的OCH
2)采用凝胶渗透色谱-激光光散射检测器GPC-MALLS联用测得弹性体样品的绝对分子量为47.4kg/mol,分子量分布为1.04。
实施例2
本实施例中,催化剂:对苯二甲醇:ε-CL:δ-VL:LLA(L型丙交酯)的摩尔比为1:1:300:342:50,具体步骤为:
称量6.7mg(0.0167mmol)催化剂溶解于500μL无水甲苯,称量2.3mg(0.0167mmo l)对苯二甲醇加入所得催化剂溶液中,再加入3.5mL无水甲苯,反应一分钟后,同时加入529μLε-CL(5.0mmol)和520μLδ-VL(5.7mmol),开始聚合反应60min,聚合完成后得到
表征及测试
1)对实施例2制备的弹性体样品进行核磁表征,结果见图4~5。
由核磁碳谱(图4)可知,中间软段PCVL存在四种比例不同的碳信号,其中ε-CL和δ-VL的均聚信号明显高于两种单体的无规共聚信号,由中间到两端首先是以δ-VL均聚为主导的无规共聚物,而后是ε-CL的均聚物,最后是LLA的均聚,PCVL与PLLA之间不存在酯交换。同时,核磁氢谱(图5)显示PCVL中的OCH
2)采用凝胶渗透色谱-激光光散射检测器GPC-MALLS联用测得弹性体样品的绝对分子量为67.2kg/mol,分子量分布为1.08。
实施例3
本实施例中,催化剂:对苯二甲醇:ε-CL:δ-VL:LLA(L型丙交酯)的摩尔比为1:1:400:456:50,具体步骤为:
称量5.0mg(0.0125mmol)催化剂溶解于500μL无水甲苯,称量1.7mg(0.0125mmo l)对苯二甲醇加入所得催化剂溶液中,再加入3.5mL无水甲苯,反应一分钟后,同时加入529μLε-CL(5.0mmol)和520μLδ-VL(5.7mmol),开始聚合反应75min,聚合完成后得到
表征及测试
1)对实施例3制备的弹性体样品进行核磁表征,结果见图6~7。
由核磁碳谱(图6)可知,中间软段PCVL存在四种比例不同的碳信号,其中ε-CL和δ-VL的均聚信号明显高于两种单体的无规共聚信号,由中间到两端首先是以δ-VL均聚为主导的无规共聚物,而后是ε-CL的均聚物,最后是LLA的均聚,PCVL与PLLA之间不存在酯交换。同时核磁氢谱(图7)显示PCVL中的OCH
2)采用凝胶渗透色谱-激光光散射检测器GPC-MALLS联用测得弹性体样品的绝对分子量为79.2kg/mol,分子量分布为1.09。
实施例4
本实施例中,催化剂:对苯二甲醇:ε-CL:δ-VL:LLA(L型丙交酯)的摩尔比为1:1:300:342:100,具体步骤为:
称量6.7mg(0.0167mmol)催化剂溶解于500μL无水甲苯,称量2.3mg(0.0167mm ol)对苯二甲醇加入所得催化剂溶液中,再加入3.5mL无水甲苯,反应一分钟后,同时加入529μLε-CL(5.0mmol)和520μLδ-VL(5.7mmol),开始聚合反应60min,聚合完成后得到
表征及测试
1)对实施例4制备的弹性体样品进行核磁表征,结果见图8~9。
由核磁碳谱(图8)可知,中间软段PCVL存在四种比例不同的碳信号,其中ε-CL和δ-VL的均聚信号明显高于两种单体的无规共聚信号,由中间到两端首先是以δ-VL均聚为主导的无规共聚物,而后是ε-CL的均聚物,最后是LLA的均聚,PCVL与PLLA之间不存在酯交换。同时,核磁氢谱(图9)显示PCVL中的OCH
2)采用凝胶渗透色谱-激光光散射检测器GPC-MALLS联用测得弹性体样品的绝对分子量为74.3kg/mol,分子量分布为1.09。
实施例5
本实施例中,催化剂:对苯二甲醇:ε-CL:δ-VL:LLA(L型丙交酯)的摩尔比为1:1:300:342:150,具体步骤为:
称量6.7mg(0.0167mmol)催化剂溶解于500μL无水甲苯,称量2.3mg(0.0167mmo l)对苯二甲醇加入所得催化剂溶液中,再加入3.5mL无水甲苯,反应一分钟后,同时加入529μLε-CL(5.0mmol)和520μLδ-VL(5.7mmol),开始聚合反应60min,聚合完成后得到
表征及测试
1)对实施例5制备的弹性体样品进行核磁表征,结果见图10~11。
由核磁碳谱(图10)可知,表明中间软段PCVL存在四种比例不同的碳信号,其中ε-CL和δ-VL的均聚信号明显高于两种单体的无规共聚信号,由中间到两端首先是以δ-V L均聚为主导的无规共聚物,而后是ε-CL的均聚物,最后是LLA的均聚,PCVL与PLL A之间不存在酯交换。同时,核磁氢谱(图11)显示PCVL中的OCH
2)采用凝胶渗透色谱-激光光散射检测器GPC-MALLS联用测得弹性体样品的绝对分子量为77.6kg/mol,分子量分布为1.07。
实施例6
本实施例中,催化剂:对苯二甲醇:ε-CL:δ-VL:LLA(L型丙交酯)的摩尔比为1:1:400:456:100,具体步骤为:
称量5.0mg(0.0125mmol)催化剂溶解于500μL无水甲苯,称量1.7mg(0.0125mmol)对苯二甲醇加入所得催化剂溶液中,再加入3.5mL无水甲苯,反应一分钟后,同时加入529μLε-CL(5.0mmol)和520μLδ-VL(5.7mmol),开始聚合反应75min,聚合完成后得到
表征及测试
1)对实施例6制备的弹性体样品进行核磁表征,结果见图12~13。
由核磁碳谱(图12)可知,聚合物碳谱表明中间软段PCVL存在四种比例不同的碳信号,其中ε-CL和δ-VL的均聚信号明显高于两种单体的无规共聚信号,由中间到两端首先是以δ-VL均聚为主导的无规共聚物,而后是ε-CL的均聚物,最后是LLA的均聚,PCVL与PLLA之间不存在酯交换。同时核磁氢谱(图13)显示PCVL中的OCH
2)采用凝胶渗透色谱-激光光散射检测器GPC-MALLS联用测得弹性体样品的绝对分子量为87.0kg/mol,分子量分布为1.07。
实施例7
本实施例中,催化剂:对苯二甲醇:ε-CL:δ-VL:LLA(L型丙交酯)的摩尔比为1:1:400:456:150,具体步骤为:
称量5.0mg(0.0125mmol)催化剂溶解于500μL无水甲苯,称量1.7mg(0.0125mmol)对苯二甲醇加入所得催化剂溶液中,再加入3.5mL无水甲苯,反应一分钟后,同时加入529μLε-CL(5.0mmol)和520μLδ-VL(5.7mmol),开始聚合反应75min,聚合完成后得到
表征及测试
1)对实施例7制备的弹性体样品进行核磁表征,结果见图14~15。
由核磁碳谱(图14)可知,中间软段PCVL存在四种比例不同的碳信号,其中ε-CL和δ-VL的均聚信号明显高于两种单体的无规共聚信号,由中间到两端首先是以δ-VL均聚为主导的无规共聚物,而后是ε-CL的均聚物,最后是LLA的均聚,PCVL与PLLA之间不存在酯交换。同时,核磁氢谱(图15)显示PCVL中的OCH
2)采用凝胶渗透色谱-激光光散射检测器GPC-MALLS联用测得弹性体样品的绝对分子量为102kg/mol,分子量分布为1.09。
对比例1
按照Sn(Oct)
对比例2
将Sn(Oct)
取所述PCVL 5.0g,加入80mL干燥甲苯,然后加入0.03gSn(Oct)
其他测试
1)在实施例3的聚合反应过程中监测产物的
2)在对比例1反应过程中监测产物,结果见图17;反应过程中产物的碳谱图见图18;由图17和18可知,在聚合反应中两种单体的速度差异不大,生成的PCVL的碳谱中四种酯基的信号比例基本一致,说明聚合物结构无规度较高,不能特异性的选择单体的插入顺序和速度,不能控制共聚物的序列结构,从而控制产物的性能。
3)分别对实施例1、2、3制备的软段
4)图22为室温下,含Mg催化剂和对苯二甲醇在甲苯和二氯甲烷混合溶剂中催化合成的
5)对实施例1~7制备的弹性体样品进行应力应变测试(即强度和断裂伸长率),具体方法为:
分别将实施例1~7制备的聚合物弹性体充分溶于适量的氯仿中,通过溶剂挥发制备测试所需的样品膜,并在真空烘箱中40℃抽7h,然后使用哑铃型裁刀裁成样品条,进行力学测试使用。
单轴拉伸测试使用长12mm、宽2mm的样品条在英特斯朗Instron UniversalTesting Machine(Model 5944)万能材料机进行测试,拉力传感器为2kN,拉伸速度15mm/min,具体测试结果见表1。
表1实施例1~7制备的弹性体样品的表征数据
对实施例1~7所制备的弹性体样品的断裂拉伸率曲线绘图,结果见图23,由图23和表1数据可知,通过调控样品软硬段的组成比例可以在大范围内调节弹性体的机械性能(13.6~71.5MPa,1290~2100MPa),得到同时兼具韧性与强度的聚酯弹性体(比如实施例3,2100%,46.3MPa),明显优于对比例2的弹性体(断裂拉伸率在700%左右,断裂拉伸强度在5.74MPa左右)。
6)分别对实施例1、2和3制备的弹性体样品进行广角X射线衍射WAXD测试,结果分别见图24~26;由图24~26研究其增韧机理在于:软段的微结构促使弹性体在拉伸的时候可以受力诱导结晶进而进行自增韧,达到兼具强度和韧性的目的。
7)分别对实施例1、2和3制备的弹性体样品绘制WAXD曲线,并进行结晶度定量分析,结果见图27~30;其中图27为实施例1制备的弹性体样品的WAXD曲线;图28为实施例2制备的弹性体样品的WAXD曲线;图29为实施例3制备的弹性体样品的WAXD曲线;图30为实施例1~3制备的弹性体样品的结晶度定量分析曲线图。由图27~30结合二维衍射图24~26可知,随着样品不断被拉伸样品出现取向结晶,样品的伸长率越大,取向结晶信号越明显(图24~26),在一维曲线图27~29)上也看出,峰越来越尖锐,晶体的衍射信号越来越明显表明结晶度越来越高,说明了受力诱导结晶的存在。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 一种高性能脂肪族聚酯弹性体及其制备方法
- 一种芳香族-脂肪族聚酯弹性体复合材料及制备方法