基于高阶接近性和矩阵补全算法的lncRNA-疾病关联预测方法
文献发布时间:2023-06-19 11:57:35
技术领域
本发明涉及机器学习与生物基因结合领域,更具体地,涉及一种基于高阶接近性和矩阵补全算法的lncRNA-疾病关联预测方法。
背景技术
LncRNA是一类超过200个核苷酸的非编码RNA。大量研究表明,lncRNA在许多重要的生物学过程中发挥着关键作用,包括翻译、剪接、分化、表观遗传调控和免疫应答。近年来,科学家发现lncRNA过表达或失调与各种癌症等复杂疾病密切相关,如肝癌(HCC)、胃癌、乳腺癌、膀胱癌、帕金森病(PD)等。因此,开发计算方法来推断潜在的疾病-lncRNA之间的关联,不仅可以加快疾病的诊断和治疗,还可以从分子水平上理解疾病的机制。此外,发展计算方法可以减少时间成本,为生物研究提供有效的实验方向。因此,识别与疾病相关的潜在lncRNAs对发现疾病生物标志物以及治疗、诊断和预防人类复杂疾病具有重要意义。鉴于传统的实验费时费力,计算模型可以作为识别lncRNA疾病关联的有效辅助工具。
多年来,许多计算方法已被开发出来,以推断潜在的lncRNA-疾病关联。现有的计算方法大致可以分为三类:(1)基于机器学习的方法(2)基于网络的方法(3)基于矩阵补全的方法。
基于机器学习的方法通常假设功能上相似的lncRNA会与相同的疾病或密切相关的疾病相关。但不幸的是,大多数机器学习方法严重依赖于已知的标签样本,这导致基于机器学习的算法在对阴性样本进行分类时总是面临困境,因为通常只有阳性lncRNA与疾病相关的报道,而阴性样本在实际情况下很难获得。而使用大量未知样本作为阴性样本可能会将潜在的lncRNA-疾病关联错误地划分为阴性样本,这将影响该方法的预测准确性。
为了减轻阴性样本的负担,最近基于网络的lncRNA-疾病关联预测方法也是一个热门研究方向。如公开日为2019年01月18日,公开号为CN109243538A的中国专利公开了一种预测疾病与LncRNA关联关系的方法和系统,包括:从已知数据库获取LncRNA-miRNA关联关系和miRNA-疾病关联关系,根据二者构建LncRNA-miRNA-疾病相互作用网络;基于LncRNA-miRNA-疾病相互作用网络,构建疾病超级表达谱和LncRNA的超级表达谱;根据疾病超级表达谱和LncRNA的超级表达谱,采用基于RBF神经网络的LncRNA相似性计算和疾病相似性计算,训练疾病与LncRNA关联关系的预测模型;使用预测模型对候选样本的LncRNA-疾病关联对进行预测。然而,由于实验验证的lncRNA-疾病相关性仍然不足,因此一些lncRNA节点与一些疾病节点可能会存在关联路径这种情况依然会影响这类方法的预测精度。另外当引入新的疾病或者lncRNA节点时,基于网络的方法会面临冷启动问题,也就是无法预测该节点,从而导致了基于网络的方法往往需要考虑单个节点的预测或整合额外的生物学信息。同时,整合其他生物信息虽然可以提高预测性能,如基因与疾病的联系、miRNA与疾病的联系等,但其他生物信息的一些交互作用可能会包含一些干扰预测结果的噪声。
第三种方法使用矩阵补全法来挖掘lncRNA与疾病的关联。主要思想是更新LncRNA-disease邻接矩阵,恢复其缺失项,假设最终迭代结果中的元素与原始邻接矩阵中的元素尽可能接近。与其他两种方法相比,矩阵补全法能够捕捉lncRNA-疾病关联的整体模式,降低假阳性率,且不需要负样本。然而,现存的矩阵补全方法都融合了疾病与lncRNA的相似信息来辅助关联预测,但都侧重于利用显性相似信息来预测lncRNA与疾病的相关性,如lncRNA功能相似度、疾病语义相似度等,而忽略了lncRNA与疾病的高阶隐式相似度。
潜在的lncRNA-疾病关联的发现无疑对了解疾病发病机制和开发人类疾病的治疗方法的研究有很大的帮助。由于传统的生物实验费时费力,迫切需要高效可靠的计算预测方法。因此发展计算方法来揭示lncRNA与疾病的未知关联,不仅有利于了解lncRNA在人类疾病的病理和分子变化中的主要功能,也有助于复杂疾病的预后、治疗和预防。
但是,到目前为止的所有应用于lncRNA-疾病关联预测的方法尽管都有使用疾病与lncRNA的相似信息来辅助关联预测,然而都侧重于关注线性原始的lncRNA和疾病相似度的信息。同时由于在实际情况下阴性的lncRNA-疾病关联样本难以获得,这导致许多需要阴性样本的计算方法的预测精度被影响。另外在矩阵补全算法中,lncRNA-疾病关联矩阵中的1表示已知药物疾病关联,而0表示未知。合理的预测值应该在[0,1]的范围内,表示预测关联的可能性。然而,现在大部分矩阵补全方法都无法避免预测值超出[0,1]范围的情况,给生物学解释带来困难。
发明内容
本发明提供一种基于高阶接近性和矩阵补全算法的lncRNA-疾病关联预测方法,更好地预测lncRNA-疾病的关联。
为解决上述技术问题,本发明的技术方案如下:
一种基于高阶接近性和矩阵补全算法的lncRNA-疾病关联预测方法,包括以下步骤:
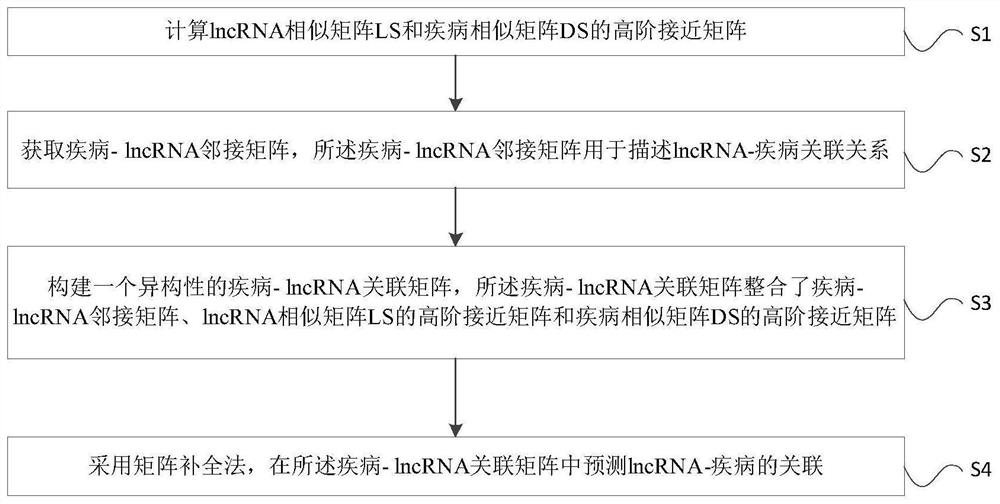
S1:计算lncRNA相似矩阵LS和疾病相似矩阵DS的高阶接近矩阵;
S2:获取疾病-lncRNA邻接矩阵DL,所述疾病-lncRNA邻接矩阵用于描述lncRNA-疾病关联关系;
S3:构建一个异构性的疾病-lncRNA关联矩阵,所述疾病-lncRNA关联矩阵整合了疾病-lncRNA邻接矩阵DL、lncRNA相似矩阵LS的高阶接近矩阵和疾病相似矩阵DS的高阶接近矩阵;
S4:采用矩阵补全法,在所述疾病-lncRNA关联矩阵中预测lncRNA-疾病的关联。
优选地,步骤S1中lncRNA相似矩阵LS,具体为:
从ArrayExpress下载lncRNA表达谱,通过RNA-Seq技术生成;在前期研究的基础上,通过计算每个lncRNA对表达谱之间的spearman相关系数来表示lncRNA的表达相似性,用矩阵LS(li,lj)来描述lncRNA li和lncRNA lj的表达相似性,相似性在0到1之间;lncRNAli与lncRNA lj的表达相似性越高,分值越高。
优选地,步骤S1中疾病相似矩阵DS,具体为:
从美国国家医学图书馆MeSH下载网格描述后,引入一个基于有向无环图DAG的模型来描述疾病之间的语义相似性,有向无环图(DAG)可用来描述疾病d,即DAG(d)=(d,T(d),E(d)),其中T(d)为节点集,E(d)为边集,对于给定特定疾病d,其祖先节点q在DAG(d)中的贡献值定义如下具体为:
结合其祖先节点在DAG(d)中的贡献值,可以将疾病d的语义值描述为:
如果两种疾病在DAG中有更多的共享节点,则可以认为两种疾病之间的语义相似性更高,使用语义相似矩阵DS(di,dj)来表示疾病di和疾病dj之间的语义相似度,定义为:
优选地,步骤S1中计算疾病相似矩阵DS的高阶接近矩阵,具体为:
在疾病相似矩阵DS的基础上构造q阶接近矩阵HD,以保持疾病语义相似矩阵的不同顺序邻近信息如下:
其中DS
采用奇异值分解技术来提高数据质量:
HD=UΣV
其中U∈R
然后通过保持k个最大奇异值来重建高阶邻近矩阵HD:
其中Σ
优选地,所述lncRNA相似矩阵LS的高阶接近矩阵HL的计算方法与疾病相似矩阵DS的高阶接近矩阵HD的计算方法相同。
优选地,步骤S2中获取疾病-lncRNA邻接矩阵DL,具体为:
从LncRNADisease数据库下载lncRNA-疾病联系数据集,删除其中重复的lncRNA、疾病和非人类数据,用疾病-lncRNA邻接矩阵DL∈R
优选地,步骤S3中构建一个异构性的疾病-lncRNA关联矩阵,具体定义为:
优选地,步骤S4中矩阵补全法用于将疾病-lncRNA关联矩阵T中DL值为0的元素。
优选地,步骤S4中采用矩阵补全法,预测lncRNA-疾病的关联,具体为:
设Ω为观测项X∈R
采用低秩矩阵补全算法,通过假设低秩矩阵X来推断缺失值,模型描述如下:
s.t 0≤X≤1
其中ω,α是平衡迹范数和核范数的非负参数,使用0≤X≤1的约束来确保恢复的矩阵元素的值在0到1之间。
优选地,步骤S4中使用交替方向的乘数法来转换模型进入待优化问题,引入变量矩阵Y,该模型可以优化为:
s.t X=Y,0≤Y≤1
相应地,对应于该方程的增广拉格朗日函数为:
其中Z是标准迹内积,β>0是自适应罚参数,在第k次迭代中,要求交替更新Y
计算Y
计算X
基于奇异值阈值算法,X
其中S
计算Z
Z
其中γ是非负学习率。
与现有技术相比,本发明技术方案的有益效果是:
本发明引入高阶接近性来重构lncRNA和疾病的相似度矩阵,建立一个更好的度量标准来精确描述药物或疾病之间的相似关系,采用构建异构矩阵来利用lncRNA和疾病的相似度信息来辅助预测,并设计了一种预测值受限制的矩阵补全算法预测lncRNA和疾病关联可能性,实现了更加准确的lncRNA-疾病的关联预测。
附图说明
图1为本发明的方法流程图。
图2为实施例中基于留一交叉验证实现了HOPMC,GMCLDA,DSCMF,SIMCLDA,BRWLDA和RWRlncD的AUCs示意图。
图3为实施例中基于5折交叉验证,利用HOPMC,GMCLDA,DSCMF,SIMCLDA,BRWLDA和RWRlncD实现的AUCs示意图。
具体实施方式
附图仅用于示例性说明,不能理解为对本专利的限制;
为了更好说明本实施例,附图某些部件会有省略、放大或缩小,并不代表实际产品的尺寸;
对于本领域技术人员来说,附图中某些公知结构及其说明可能省略是可以理解的。
下面结合附图和实施例对本发明的技术方案做进一步的说明。
实施例1
本实施例提供一种基于高阶接近性和矩阵补全算法的lncRNA-疾病关联预测方法,如图1所示,包括以下步骤:
S1:计算lncRNA相似矩阵LS和疾病相似矩阵DS的高阶接近矩阵;
S2:获取疾病-lncRNA邻接矩阵DL,所述疾病-lncRNA邻接矩阵用于描述lncRNA-疾病关联关系;
S3:构建一个异构性的疾病-lncRNA关联矩阵,所述疾病-lncRNA关联矩阵整合了疾病-lncRNA邻接矩阵DL、lncRNA相似矩阵LS的高阶接近矩阵和疾病相似矩阵DS的高阶接近矩阵;
S4:采用矩阵补全法,在所述疾病-lncRNA关联矩阵中预测lncRNA-疾病的关联。
步骤S1中lncRNA相似矩阵LS,具体为:
从ArrayExpress下载lncRNA表达谱,通过RNA-Seq技术生成;在前期研究的基础上,通过计算每个lncRNA对表达谱之间的spearman相关系数来表示lncRNA的表达相似性,用矩阵LS(li,lj)来描述lncRNA li和lncRNA lj的表达相似性,相似性在0到1之间;lncRNAli与lncRNA lj的表达相似性越高,分值越高。
步骤S1中疾病相似矩阵DS,具体为:
从美国国家医学图书馆MeSH下载网格描述后,引入一个基于有向无环图DAG的模型来描述疾病之间的语义相似性,有向无环图(DAG)可用来描述疾病d,即DAG(d)=(d,T(d),E(d)),其中T(d)为节点集,E(d)为边集,对于给定特定疾病d,其祖先节点q在DAG(d)中的贡献值定义如下具体为:
结合其祖先节点在DAG(d)中的贡献值,可以将疾病d的语义值描述为:
如果两种疾病在DAG中有更多的共享节点,则可以认为两种疾病之间的语义相似性更高,使用语义相似矩阵DS(di,dj)来表示疾病di和疾病dj之间的语义相似度,定义为:
步骤S1中计算疾病相似矩阵DS的高阶接近矩阵,具体为:
根据生物学实验观察,lncRNA与疾病预测的一个基本假设是,功能相似的lncRNA往往与表型相似的疾病相关,反之亦然。因此,lncRNA与疾病的相似性测度是预测lncRNA与疾病相关性的关键。高阶相似度可以描述矩阵元素之间的间接相似信息,不同于显式的成对相似。例如,在一个网络中,如果vi和vj有较多的共同邻居和丰富的路径信息,那么vi通过2步随机漫步到达节点vj的可能性就会很大,这就意味着两个节点的二阶接近值会太高。因此,通过考虑高阶邻近性来推断lncRNA与疾病的相似度度量,这将有助于我们更有效地表达lncRNA与疾病的相似信息。在疾病相似矩阵DS的基础上构造q阶接近矩阵HD,以保持疾病语义相似矩阵的不同顺序邻近信息如下:
其中DS
然而,由于矩阵的高维数,在矩阵HD中可能存在噪声,采用奇异值分解技术来提高数据质量,SVD的细节如下:
HD=UΣV
其中U∈R
然后通过保持k个最大奇异值来重建高阶邻近矩阵HD:
其中Σ
所述lncRNA相似矩阵LS的高阶接近矩阵HL的计算方法与疾病相似矩阵DS的高阶接近矩阵HD的计算方法相同。
步骤S2中获取疾病-lncRNA邻接矩阵DL,具体为:
从LncRNADisease数据库下载lncRNA-疾病联系数据集,其中包括687个经实验验证的lncRNA-疾病联系,删除其中重复的lncRNA、疾病和非人类数据,最后,在115个独特的lncRNA和178个独特的疾病之间得到了540个独特的经实验验证的lncRNA-疾病关联,用疾病-lncRNA邻接矩阵DL∈R
步骤S3中构建一个异构性的疾病-lncRNA关联矩阵,具体定义为:
步骤S4中矩阵补全法用于将疾病-lncRNA关联矩阵T中DL值为0的元素。
步骤S4中采用矩阵补全法,预测lncRNA-疾病的关联,具体为:
基于功能相似的lncRNA倾向于涉及相似疾病的lncRNA和疾病预测的假设,因此决定lncRNA与疾病关联可能性的潜在因素往往是高度相关的,这导致相应的数据矩阵中存在相关性。因此,在疾病-lncRNA关联的异构相邻矩阵T中,lncRNA与疾病相互作用的独立因素数量有限,这导致异构相邻矩阵T形成低秩结构。因此,我们使用矩阵完成来预测潜在的疾病-lncRNA关联。
设Ω为观测项X∈R
采用低秩矩阵补全算法,通过假设低秩矩阵X来推断缺失值,模型描述如下:
s.t 0≤X≤1
其中ω,α是平衡迹范数和核范数的非负参数,使用0≤X≤1的约束来确保恢复的矩阵元素的值在0到1之间,这使得结果在生物学上更容易解释。
步骤S4中使用交替方向的乘数法来转换模型进入待优化问题,引入变量矩阵Y,该模型可以优化为:
s.t X=Y,0≤Y≤1
相应地,对应于该方程的增广拉格朗日函数为:
其中Z是标准迹内积,β>0是自适应罚参数,在第k次迭代中,要求交替更新Y
计算Y
计算X
基于奇异值阈值算法,X
其中S
计算Z
Z
其中γ是非负学习率,在本实施例中可设置为1。
为了检验本实施例的方法(HOPMC)的预测精度,将HOPMC与GMCLDA、SIMCLDA、DSCMF、BRWLDA和RWRlncD5种先进方法进行比较。从图2可以看出,在留一交叉验证框架下,HOPMC曲线AUC下面积为0.8757,大于其他计算方法(GMCLDA 0.8501,SIMCLDA 0.8237,DSCMF 0.8176,BRWLDA0.7969,RWRlncD 0.6540),说明HOPMC的性能优于其他计算方法。为了进一步验证HOPMC的预测性能,采用5折交叉验证框架进行验证。从图3可以看出,HOPMC可以得到可靠的AUC为0.8353±0.0045,远远超过的AUC值0.7894±0.0040、0.7839±0.0045、0.7734±0.0045、0.7659±0.0045和0.6179±0.0045。这意味着HOPMC在5折交叉验证框架下比其他方法更有效。以上结果充分说明HOPMC方法优于所比较的其它方法,更有利于lncRNA-disease相关性的预测。
同时也使用HOPMC来预测已知的lncRNA在实际lncRNA-疾病预测中的应用。在预测新的lncRNA-疾病关联过程中,我们将已知的lncRNA-疾病相关性作为HOPMC的训练数据集,然后计算每个未知lncRNA-疾病对的预测得分并进行排序。我们选择骨肉瘤、胃癌和肝细胞癌作为病例研究。在第三方数据库(Lnc2Cancer和MNDR)中验证了排名前10位的癌症lncRNA。结果如表一,表二和表三表明,预测的lncRNA分别有100%、90%和90%与癌症相关
此外,HOPMC所预测一些没被证明lncRNA-疾病,包括:MINA与骨肉瘤,PCA3与肝细胞癌等。这些预测的关联尚未在目前的文献中报道,但有较大的可能性存在,有待医学研究人员去研究和验证这些关联。
表一HOPMC预测的与胃癌相关的前10位潜在lncRNA
表二HOPMC预测的与骨肉瘤相关的前10位潜在lncRNA
表三HOPMC预测的与肝细胞癌相关的前10位潜在lncRNA
相同或相似的标号对应相同或相似的部件;
附图中描述位置关系的用语仅用于示例性说明,不能理解为对本专利的限制;
显然,本发明的上述实施例仅仅是为清楚地说明本发明所作的举例,而并非是对本发明的实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明权利要求的保护范围之内。
- 基于高阶接近性和矩阵补全算法的lncRNA-疾病关联预测方法
- 基于拉普拉斯正则化最小二乘和网络投影的lncRNA-疾病关联预测方法和系统