甲基甘氨酸二乙酸三碱金属盐的制备方法及其生产系统
文献发布时间:2023-06-19 18:35:48
技术领域
本发明涉及配合剂生产工艺技术领域,尤其涉及甲基甘氨酸二乙酸三碱金属盐的制备方法及其生产系统。
背景技术
现实生产中,一般洗涤用水都不是纯水,不同程度地含有各种金属离子。其中,构成水硬度的钙、镁等离子及铁、锰等离子对洗涤作用将产生严重的影响。如织物变黄、变灰、手感发硬等。为了消除水中金属离子对洗涤造成的影响,一般都要在洗涤剂中加入软水剂。其中的一种就是配合剂,配合剂的使用,可以节省活性物,并避免在被洗物上留下沉淀物,使被洗物色彩鲜艳。与常用配合剂,如氨基多磷酸盐、多羧酸盐或氨基多羧酸盐(例如乙二胺四乙酸,EDTA)相比,甲基甘氨酸二乙酸(MGDA)具有无毒且易于生物降解的优点,可用于生产清洁剂、抛光剂以及硬化基质的粘合剂。
甲基甘氨酸二乙酸,简称MGDA。是一种非毒性且容易生物降解的配合剂,用于生产清洁剂、生产抛光剂以及硬化基质的粘合剂,比常用配合剂EDTA更易生物降解。MGDA是德国BASF所开发生产的生物易分解性螫合剂,生物分解性试验时,28天以内的分解率达98%而被认定为生物易分解性,在螫合剂业界相当重视生物分解性型产品。
在现有技术中,甲基甘氨酸二乙酸通常采用Strecker反应合成,反应既可在碱性介质中进行,也可在酸性介质中进行,具体的合成路线可分为两大类:第一类合成路线是由丙氨腈或丙氨酸与氰化物和甲醛进行Strecker反应,再经水解获得产物。第二类合成路线是由亚氨基二乙腈或亚氨基二乙酸与氰化物和乙醛进行Strecker反应,再经水解获得产物。另外通过乙氧基化、催化脱氢制备MGDA的方法,采用氯乙酸作为原料的合成工艺也有相关专利报道。
专利WO9429421A首次公开丙氨酸、氢氰酸、甲醛为原料的Strecker反应路线。专利US5817864A采用丙氨酸、30%甲醛和33%氰化钠在pH值为10~12的碱性介质中反应,再经过水解脱氨获得MGDA·3Na。这类合成路线缺点是:由于是三组分反应体系,反应不容易控制,反应过程中副产物含量较多,MGDA·3Na的选择性较低,产品不容易提纯,NTA等杂质残留量较高。专利US5849950A分别采用丙氨酸、丙氨腈、亚氨基二乙腈和亚氨基二乙酸为原料,在酸性介质中与甲醛(或乙醛)和精制氢氰酸反应制备,与上述碱性介质中进行反应相比,该法可获得较高纯度的甲基甘氨酸二乙酸三钠盐产品,但对原料纯度要求较高,尤其是原料氢氰酸需达到99%,控制杂质NTA含量<0.3%,为了低成本的生产,对于各个步骤的合成收率和中间体的分离纯度要求很高。专利CN101171226B和US20080194873A采用甲基甘氨腈二乙腈(MGDN)进行碱性分阶段水解来制备甲基甘氨酸二乙酸三钠盐的方法,在每个阶段中水解温度逐渐提高,但是通过丙氨腈、氢氰酸和甲醛制备MGDN中间体时,仍需精制氢氰酸(纯度大于99%)为原料,为了降低副产杂质,需要将中间体MGDN进行结晶提纯,将高纯度的MGDN进行碱性水解,同时水解过程分成三段进行(低温30~40℃,中温50~80℃,高温110~200℃),虽然该法在一定程度上降低了有毒副产物次氮基三乙酸盐(NTA)的含量<0.3%,高纯度MGDN水解后最佳工况下,控制NTA的含量<0.1%,但是合成MGDA全过程中,存在不可避免的烷基甘氨腈-N,N-二乙腈的热稳定性水解问题,在碱性介质中存在其他离解产物,产生其他副产物如亚氨基二乙酸盐(IDA)、次氮基三乙酸盐(NTA)、碳酸盐、乙酸盐、甲酸盐、乙醇酸盐、乳酸盐、甘氨酸盐、丙氨酸盐、乙醛,总计含有0.1-10%不等的其他物质。专利US8802894B采用氢氧化钠溶液部分中和的98.5%以上丙氨酸、30%甲醛和99%以上氢氰酸,在pH值为9~12的碱性介质中反应制备丙氨酸二乙腈(ADAN),中间原料ADAN可以分离也可以不分离,后续进行两个阶段不同温度(低温45~50℃,高温95~102℃)直接水解来制备甲基甘氨酸二乙酸三钠盐溶液。该法在一定程度上降低了有毒副产物,控制副反应次氮基三乙腈,氢氰酸聚合物的生成,产品经过脱色后降低有色杂质,Hazen色号在150~650,产品中次氮基三乙酸盐的含量NTA<0.1%,最低可达到0.03%,但是制备ADAN过程仍需高纯98.5%以上的丙氨酸固体和精制99%以上氢氰酸为原料。专利CN102993034B介绍了一种利用粗氢氰酸和甲醛反应先合成羟基乙腈,在与氨气合成亚氨基二乙腈溶液,然后使其与粗氢氰酸与乙醛反应制备甲基甘氨腈二乙腈晶体,通入氮气与氢氧化钠回流水解制得甲基甘氨酸二乙酸三钠溶液,合成体系在酸性介质中,产品收率可达86%,但是未能完全解决转化率较低,副产物残留的问题。专利CN106928077B介绍了一种体系在酸性体系pH值为5~6.5以亚氨基二乙酸,乙醛和99%氢氰酸为原料获得甲基甘氨酸二乙腈,碱溶液水解制备甲基甘氨酸二乙酸,该方法采用工业级亚氨基二乙酸、乙醛和99%高纯氢氰酸进行三组分反应制备甲基甘氨酸二乙酸,反应收率可达到90%以上,杂质NTA<0.1%,由于是三组分反应体系,反应不容易控制,导致未能完全解决转化率较低,副产物残留的问题。专利CN107118114B介绍了L-丙氨酸和氯乙酸为原料,碱金属盐在催化剂存在下在一定温度和压力下与L-丙氨酸脱水生成甲基甘氨酸二乙酸(L-MGDA)对应的碱金属盐,不可避免该工艺副产含盐废水,分离困难,产品残留氯化物等。
从上述技术来看,专利US5817864单一采用氰化钠溶液在碱性条件下反应,无法避免氰化物的聚合和其他副产物产生,专利US5849950合成工艺采用高纯度的原料99%氢氰酸,合成的中间体的分离纯度至关重要,MGDA产品中NTA含量<0.3%,专利CN101171226B和专利US20080194873将中间体MGDN进行结晶纯化后,MGDN在三段不同温度下水解后,NTA的含量<0.3%,单一采用99%高浓度氢氰酸原料来源,或者采用原料中间体结晶提纯后,分段水解控制腈类中间体的方式,也只能达到NTA杂质的含量<0.3%,在碱性介质中存在其他离解产物。上述专利并未有完全解决如何更低控制副产杂质。专利US8802894B在前面专利基础上改进原料进料方式,采用高纯度丙氨酸与碱液部分预混合,与高纯度的氢氰酸反应,在9~12的碱性介质下合成中间体,后续采用两个阶段不同温度水解,从工艺上看一定程度的降低了NTA含量<0.1%,但是高纯度的原料使用,特别是高浓度的氢氰酸的使用,对于合成ADAN中间体的控制仍存在副反应和聚合的风险,对于安全和操作的水平要求极高。
综上来看,从上述专利资料和技术发展看来,制备MGDA的过程不可避免存在杂质残留的问题,工艺共性都存在需要控制一种或多种有毒杂质或活性杂质(如氰化物聚合物产生的有色杂质、氯化钠、次氮基三乙酸盐(NTA)、碳酸盐、乙酸盐、甲酸盐、乙醇酸盐、乳酸盐、甘氨酸盐、丙氨酸盐、醛类:甲醛、乙醛、多聚甲醛、醛类释放物等)。为了实现优化副产杂质的产生,不同的专利从不同的角度出发,工艺优化过程虽然某些方向控制了部分杂质的产生,单一采用部分中和高纯度99%丙氨酸原料工艺需要配合高浓度的99%氢氰酸原料,单一采用中间体MGDN的分三阶段水解不可或缺步骤是通过结晶工艺提纯MGDN,水解过程才能控制部分杂质产生。但是为了实现上述目的,采用高纯度99%氢氰酸或高纯99%丙氨酸固体溶液为原料,需另配置高纯度的反应原料精制设备,高纯度99%氢氰酸,生产成本高,安全控制水平较高,对设备密闭性要求高,局限了其在工业上的应用,中间体MGDN结晶提纯分离过程,安全环保成本高,能量消耗大,其他工艺存在温度和压力较高,副产含盐或含氰废水等问题,也未能完全解决其他副产物产生,控制副产物的量,限制了MGDA的合成工艺和杂质控制水平,未能实现连续化合成MGDA的工艺。
现有工业化技术中,以丙氨酸、甲醛、氰化氢或者丙氨酸与羟基乙腈为原料的生产工艺,由于甲醛和氰化氢或者羟基乙腈的过量,主要的副产物产生因素如下所示:
ADAN在碱性水解过程中离解反应,形成氨基酸类副产物(如甘氨酸钠),同时氢氰酸与过量的甲醛反应产生的羟基乙腈,羟基乙腈和氨存在的情况也能形成氨基乙腈,碱性条件下进一步水解生成甘氨酸钠,甘氨酸钠和甲醛在强碱性环境下40℃可进行羟甲基化形成副产物羟甲基甘氨酸钠。
羟甲基甘氨酸钠,它在pH3~12的范围内稳定性均良好,是极少数能在较高pH条件下保持活性的防腐剂,可用于多种洗涤用品,但在溶液中稳定性有差异限制,不同情况下可反应重新分解为原料甲醛和甘氨酸钠。
综合来看,对于甲基甘氨酸二乙酸的关键中间体丙氨酸-N,N-二乙腈(ADAN溶液)合成过程,使用高纯度的氢氰酸液体,生产成本较高,反应会放出大量热,反应过程中不容易控制,局部高浓度氢氰酸的加入,分布不均或者温度控制不好,聚合反应和副产物产生的量更多,并且有很大危险性,连续化生产实现比较困难,对设备的要求较高,高浓度的氢氰酸与甲醛溶液接触也容易形成副产物羟基乙腈,采用将氢氰酸液体气化后通入方法虽然控制反应放热,易于操作,但是这比较难于实现大规模连续化生产。而使用丙氨酸、甲醛和氰化氢混合气为原料,不可避免地产生大量的副产物,尤其氰化物的分解和聚合,以及有色杂质、氯化钠,亚氨基二乙酸盐(IDA)、氨三乙酸盐(NTA)、碳酸盐、乙酸盐、甲酸盐、乙醇酸盐、乳酸盐、甘氨酸盐、丙氨酸盐、醛类:甲醛、乙醛、多聚甲醛、醛类释放物等物质的产生给后续的纯化带来困难,造成产品的收率低、分离纯化困难,产生的废水量大等问题,造成产品的生产成本较高,市场竞争力低。使用亚氨基二乙酸、高纯度液体氰化氢和乙醛为原料,除了上述陈述的使用高纯度的氢氰酸液体存在的弊端,还存在最大的问题是不可避免产生氨三乙酸盐(NTA)杂质、有色杂质以及收率低,一锅法反应不容易控制,这为后续产品纯化带来了困难,再者,众所周知,工业级的亚氨基二乙酸主要用于生产除草剂草甘膦,其中主要杂质为氨三乙酸、甘氨酸、硫酸钠、乙醇酸、氯化钠等,这些杂质的带入会给后续的反应引入新的杂质,从而导致甲基甘氨酸二乙酸及其碱金属盐的分离纯化带来困难,导致产品收率降低。
发明内容
有鉴于此,本发明的目的是提供甲基甘氨酸二乙酸三碱金属盐的制备方法及其生产系统,制备方法反应时间短,操作简单,反应过程副产物很少,产生的废水极少,高效环保,适合大规模连续生产,且制备的甲基甘氨酸二乙酸三碱金属盐反应收率可达到98.5%以上,杂质极少,并且无副产物氨三乙酸产生。
本发明通过以下技术手段解决上述技术问题:
本发明的一方面在于提供了一种甲基甘氨酸二乙酸三碱金属盐的制备方法,其特征在于,包括以下步骤:
S1.乙醛在催化剂的作用下与脱氨后的氰化氢混合气在pH=5.0~6.0、温度35℃~70℃条件下氰化反应30~120min,制备得到2-羟基丙腈水溶液;
S2.2-羟基丙腈水溶液与亚氨基二乙酸一碱金属盐水溶液在温度60℃~110℃反应15~60min,制备得到N,N-二乙酸丙腈碱金属盐水溶液;
S3.将N,N-二乙酸丙腈碱金属盐水溶液和碱液在温度70℃~120℃条件下搅拌水解反应90~180min,经过脱氨、脱色和浓缩后得到甲基甘氨酸二乙酸三碱金属盐水溶液。
本发明的甲基甘氨酸二乙酸三碱金属盐的制备方法主要反应流程如下:
进一步,所述S1步骤中,乙醛以乙醛水溶液进行投料,所示乙醛水溶液的质量百分含量为40%;脱氨后的氰化氢混合气成分如下:HCN气体9.4%±2%,氢气1.6%±1%,氮气79.4%±2%,氧气1.7%±1%,一氧化碳5.8%±2%,二氧化碳1.5%±1%,甲烷0.6%±0.5%,水蒸气3.7%±2%。
进一步,所述催化剂为亚氨基二乙酸一碱金属盐水溶液。
进一步,所述亚氨基二乙酸一碱金属盐为亚氨基二乙酸一钠盐或者亚氨基二乙酸一钾盐,所述亚氨基二乙酸一碱金属盐水溶液的pH值为5.0~5.5,亚氨基二乙酸一碱金属盐质量百分含量为30%~40%。
进一步,所述2-羟基丙腈水溶液为无色透明液体,2-羟基丙腈质量百分含量为40%~60%,乙醛的残留量小于50ppm,氰化氢残留量小于500ppm。
进一步,所述S2步骤中,2-羟基丙腈与亚氨基二乙酸一碱金属盐投料摩尔比为1:0.99~1.0,在温度60℃~110℃条件下反应25~30min。
进一步,所述S3步骤中,按照N,N-二乙酸丙腈碱金属盐与碱的摩尔比为1:2投料N,N-二乙酸丙腈碱金属盐水溶液与碱液,所述碱液为氢氧化钠水溶液或者氢氧化钾水溶液,碱液的质量百分含量为30%~50%。
进一步,所述甲基甘氨酸二乙酸三碱金属盐水溶液经过脱氨和脱色后的质量百分含量为30%~40%,游离氨的含量小于20ppm,氨三乙酸未检出,Hazen色号20~200。
进一步,所述甲基甘氨酸二乙酸三碱金属盐的结构式如下:
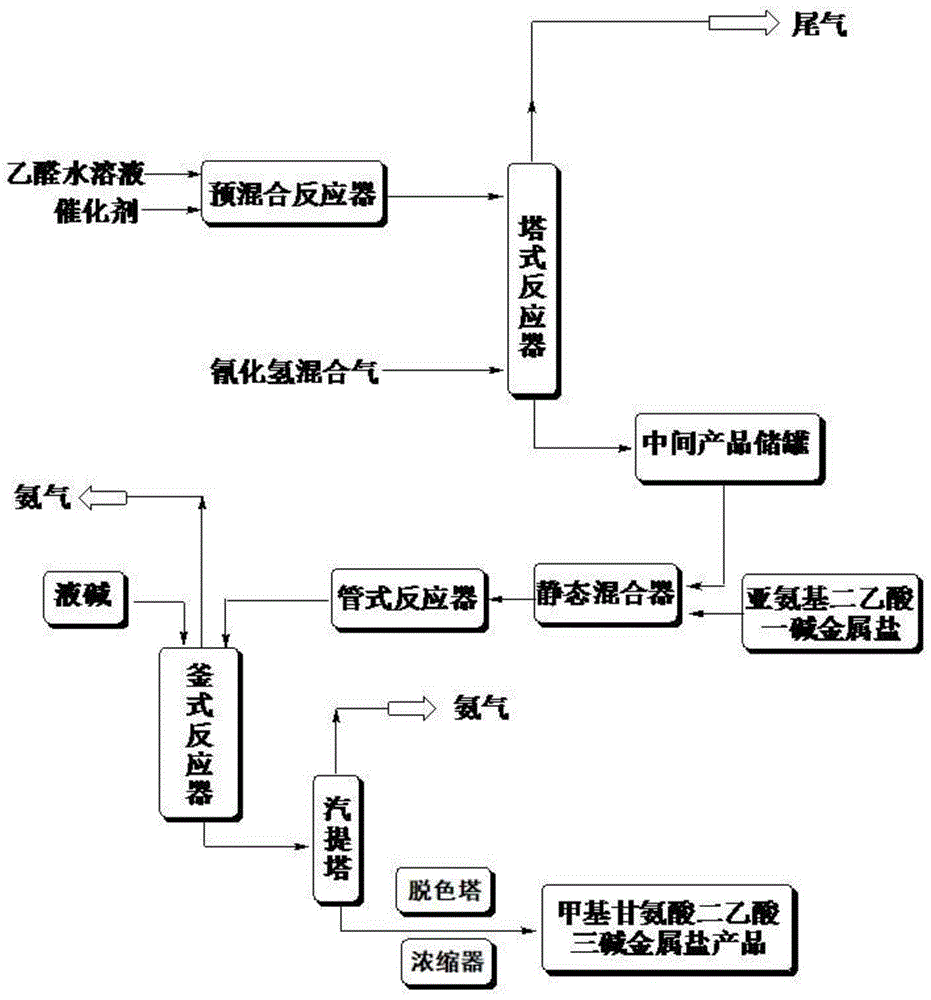
本发明的另一方面在于提供了执行上述制备方法的生产系统,所述生产系统包括:塔式反应器、管式反应器、釜式反应器、汽提塔、脱色塔和浓缩器,所述乙醛和脱氨后的氰化氢混合气在塔式反应器中进行氰化反应,所述2-羟基丙腈水溶液与亚氨基二乙酸一碱金属盐水溶液在管式反应器中反应,所述N,N-二乙酸丙腈碱金属盐水溶液和碱液在釜式反应器中进行水解反应;
所述塔式反应器和管式反应器之间还连接有中间产品储罐和静态混合器,所述塔式反应器的出口与中间产品储罐的入口连通,所述中间产品储罐的出口与静态混合器的入口连通,所述静态混合器的出口与管式反应器的入口连通,所述管式反应器的出口与釜式反应器的入口连通,所述釜式反应器的出口与汽提塔的入口连通,所述汽提塔的出口与脱色塔的入口连通,所述脱色塔的出口与浓缩器的入口连通;
所述釜式反应器设置有搅拌装置、加热器、温度传感器、液位计以及氨气排气阀口,所述釜式反应器的材质为304不锈钢或者316不锈钢。
所述汽提塔带有加热器、温度传感器、液位计以及氨气排气阀口,所述汽提塔的塔体材质为304不锈钢或者316不锈钢。
综合本发明的甲基甘氨酸二乙酸三碱金属盐(MGDA)制备方法全流程来看,从生产成本和安全环保,工艺优化看来,粗氢氰酸混合气的生产成本远远低于液体氢氰酸,同时也省去了气体氢氰酸多级吸收和精馏的投资,以及精馏制备99%的高浓度氢氰酸风险,粗氢氰酸气体在较低浓度情况下,也能快速参与反应,同时也能避免氢氰酸自身聚合所带来的后续颜色较深的问题;使用氢氰酸混合气与乙醛水溶液反应,先合成2-羟基丙腈,使用颜色二乙酸一碱金属盐作为催化剂,避免了其他物质的引入所带来的后续纯化问题,生成的2-羟基丙腈再与亚氨基二乙酸一碱金属盐水溶液反应,避免了三组分复杂反应体系,反应简单,易于控制;使用高纯度亚氨基二乙酸、氰化氢混合气和乙醛为原料,避免了传统生产工艺中副产物羟基乙腈、次氮基三乙腈等物质的生成,同时也将存在聚合趋势的氰化物、甲醛等残留物更充分的排出N,N-二乙酸丙腈一碱金属盐溶液体系,控制体系中氢氰酸的微过量状态以及严格控制乙醛的残留量,保证中间体2-羟基丙腈溶液的稳定性,防止副产物的产生,达到降低副反应和提高产品纯度和收率的目的。
本发明的甲基甘氨酸二乙酸三碱金属盐的制备方法,以高纯度亚氨基二乙酸、乙醛和氢氰酸混合气为原料,首先乙醛与氢氰酸混合气在催化剂的作用下,高效制备2-羟基丙腈水溶液,2-羟基丙腈再与亚氨基二乙酸一碱金属盐反应,制备N,N-二乙酸丙腈一碱金属盐溶液,将N,N-二乙酸丙腈一碱金属盐溶液滴入碱溶液水解,得到目标产物甲基甘氨酸二乙酸三碱金属盐,避免了传统生产工艺使用复杂的三组分反应体系,本发明两组分反应体系具有反应时间短,操作简单,反应易于控制,使用氰化氢混合气成本低,使用高纯度的亚氨基二乙酸,避免了原料中杂质的带入以及杂质与原料继续反应生成新的杂质,反应过程副产物很少(只有氨),无废水产生,高效环保,适合大规模连续生产。
本发明的甲基甘氨酸二乙酸三碱金属盐的制备方法制备得到的MGDA反应收率可达到98.5%以上,杂质极少,并且无副产物氨三乙酸(NTA)产生。
附图说明
图1是本发明的制备甲基甘氨酸二乙酸三碱金属盐的生产系统的装置流程图;
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
本发明的甲基甘氨酸二乙酸三碱金属盐的制备方法,首先以乙醛和脱氨后的氢氰酸混合气为原料制备2-羟基丙腈,2-羟基丙腈在与亚氨基二乙酸一碱金属盐制备N,N-二乙酸丙腈碱金属盐水溶液,该水溶液再经过碱液水解得到甲基甘氨酸二乙酸三碱金属盐水溶液。其主要反应流程如下:
制备得到的甲基甘氨酸二乙酸三碱金属盐的分子式如下所示:
上述的氰化氢混合气来源于现有技术甲烷氨氧化法,氰化氢混合气采用以下方法制备:以甲烷、氨气和氧气为原料,通过氰化氢合成塔,氰化氢合成塔产物通入酸洗塔进行脱氨处理,得到氰化氢混合气,氰化氢混合气成分如下:HCN气体9.4%±2%,氢气1.6%±1%,氮气79.4%±2%,氧气1.7%±1%,一氧化碳5.8%±2%,二氧化碳1.5%±1%,甲烷0.6%±0.5%,水蒸气3.7%±2%。
上述的亚氨基二乙酸一碱金属盐为亚氨基二乙酸一钠盐或者亚氨基二乙酸一钾盐,其中亚氨基二乙酸的纯度为≥99%;亚氨基二乙酸一碱金属盐水溶液采用以下方法制备:以高纯度亚氨基二乙酸与氢氧化钠或者氢氧化钾的水溶液反应,投料摩尔比为1:1,得到的亚氨基二乙酸一碱金属盐水溶液的pH值为5.0~5.5,亚氨基二乙酸一碱金属盐质量百分含量为30%~40%。
本发明还公开了用于执行上述制备方法的生产系统,如图1所示,该生产系统包括:塔式反应器、管式反应器、釜式反应器、汽提塔、脱色塔和浓缩器,乙醛和脱氨后的氰化氢混合气在塔式反应器中进行氰化反应,2-羟基丙腈水溶液与亚氨基二乙酸一碱金属盐水溶液在管式反应器中反应,N,N-二乙酸丙腈碱金属盐水溶液和碱液在釜式反应器中进行水解反应。
塔式反应器和管式反应器之间还连接有中间产品储罐和静态混合器,塔式反应器的出口与中间产品储罐的入口连通,中间产品储罐的出口与静态混合器的入口连通,静态混合器的出口与管式反应器的入口连通,管式反应器的出口与釜式反应器的入口连通,釜式反应器的出口与汽提塔的入口连通,汽提塔的出口与脱色塔的入口连通,脱色塔的出口与浓缩器的入口连通。
塔式反应器包括预混合反应器、填料塔、循环泵、塔中料口、尾气净化系统、换热器,填料塔上端一侧设置有预混合反应器,预混合反应器用于乙醛和催化剂亚氨基二乙酸一碱金属盐水溶液的混合,填料塔下端一侧设置有HCN气体入口,填料塔底端通过管道经循环泵与换热器底端连接,换热器的顶端分为两路,一路通过中部回路通道与塔中料口连接,另一路通过顶部回路通道与填料塔上端连接,填料塔的顶端与尾气净化系统连接。
釜式反应器设置有搅拌装置、加热器、温度传感器、液位计以及氨气排气阀口,釜式反应器的材质为304不锈钢或者316不锈钢。
汽提塔带有加热器、温度传感器、液位计以及氨气排气阀口,汽提塔的塔体材质为304不锈钢或者316不锈钢。
为了更好的理解本发明的甲基甘氨酸二乙酸三碱金属盐的制备方法,进行了实施例1-4和对比实施例1和2,以下实施例中采用的分析方法如下:
2-羟基丙腈和N,N-二乙酸丙腈一碱金属盐水溶液反应液中游离氰根含量:硝酸银电位滴定法。
2-羟基丙腈和N,N-二乙酸丙腈一碱金属盐反应液:液相色谱面积归一法,色谱柱5μm,250×4.6mm(5μm多孔球形,以硅胶为基质,表面键合C18的反相色谱柱),流动相:水:乙腈=45:55,流速:1mL/min,检测波长:210nm,进样体积:20μL,柱温30℃。
MGDA·3M含量检测方法:三氯化铁络合电位滴定法。
Hazen色号:液体化学产品颜色测定法(铂-钴色号)。
游离氨:采用国标通用检测氨氮的化学滴定法。
NTA·3M含量:离子色谱法或液相色谱法定量分析。
实施例1
原料预处理
(1)氰化氢混合气脱氨
来自氢氰酸合成车间安氏法粗HCN气体经过10~98%硫酸两级吸收脱氨后得道粗氢氰酸气体成分如下:HCN气体9.4%±2%,氢气1.6%±1%,氮气79.4%±2%,氧气1.7%±1%,一氧化碳5.8%±2%,二氧化碳1.5%±1%,甲烷0.6%±0.5%,水蒸气3.7%±2%。
(2)亚氨基二乙酸一钠盐水溶液制备
在1000升反应釜中加入质量百分含量为99.5%的亚氨基二乙酸固体133.77kg、软水
280kg,在搅拌状态下,慢慢滴加50%氢氧化钠水溶液80kg。加完氢氧化钠水溶液,得到无色透明的亚氨基二乙酸一钠盐水溶液493.77kg,其中亚氨基二乙酸一钠盐的质量百分含量为31.41%,水溶液的pH值为5.5。
2-羟基丙腈水溶液制备
开启填料塔换热器,控制塔体内部温度在35~70℃,在预混合反应器中加入40%的乙醛水溶液110.13kg和质量百分含量为31.41%的亚氨基二乙酸一钠盐水溶液130g。将预混合反应器得到的混合液从塔式反应器的顶部打入塔内,流速1~35L/min,来自脱氨安氏法粗HCN气体由填料塔底部进入,气速2~40L/min;塔内物料的pH保持在5.5左右,温度为65℃左右,二者在填料塔内逆流发生反应,在填料塔底部得到含1~8%的乙醛的粗2-羟基丙腈,粗2-羟基丙腈由填料塔底部循环泵打至塔顶循环口,进行全塔循环继续反应至乙醛完全转化为2-羟基丙腈(乙醛残留30ppm,氰化氢残留200ppm),得到的2-羟基丙腈水溶液为无色透明液体150.53kg,其中2-羟基丙腈含量为47.22%,2-羟基丙腈的收率≥99.9%(以乙醛计)。
N,N-二乙酸丙腈一钠盐水溶液制备
将上述得到的150.53kg质量百分含量为47.22%的2-羟基丙腈水溶液和质量百分含量为31.41%的亚氨基二乙酸一钠盐水溶液493.70kg通过静态混合器充分混合后打入管式反应器中,物料在管式反应器中停留时间为25min,管式反应器中温度为110℃,得到无色透明液体N,N-二乙酸丙腈一钠盐水溶液644.23kg,其中N,N-二乙酸丙腈一钠盐质量百分含量为32.27%,收率为99.9%(以2-羟基丙腈计)。
甲基甘氨酸二乙酸三钠盐水溶液制备
在3000升材质为304的反应釜中加入160kg质量百分含量为50%的氢氧化钠水溶液,加热至70℃,在搅拌状态下将上述得到的644.23kg质量百分含量为32.27%的N,N-二乙酸丙腈一钠盐水溶液流加至氢氧化钠水溶液中,随着N,N-二乙酸丙腈一钠盐水溶液加入,反应釜中物料温度上升至85℃,加完N,N-二乙酸丙腈一钠盐水溶液后,升温至110℃,继续搅拌反应90min,得到淡黄色的甲基甘氨酸二乙酸三钠盐水溶液,其中氨的含量为1.02%。将得到的甲基甘氨酸二乙酸三钠盐水溶液转入汽提塔中,汽提塔底温度控制在105℃,塔顶温度控制在95℃,经过汽提后的甲基甘氨酸二乙酸三钠盐水溶液中游离氨的含量为15ppm,氨三乙酸(NTA)未检测出,得到甲基甘氨酸二乙酸三钠盐水溶液767.23kg,其中甲基甘氨酸二乙酸三钠盐质量百分含量为35.0%,收率为99.9%(以N,N-二乙酸丙腈一钠盐计)。在甲基甘氨酸二乙酸三钠盐水溶液中加入7.0kg的活性炭,搅拌进行脱色处理,脱色温度为45℃,过滤活性炭后,得到无色透明的甲基甘氨酸二乙酸三钠盐水溶液766.85kg,进行减压浓缩至甲基甘氨酸二乙酸三钠盐质量百分含量为40%,水溶液的Hazen色号80,未检测出氨三乙酸(NTA),甲基甘氨酸二乙酸三钠盐的总收率为99%(以乙醛计)。
实施例2
原料预处理
(1)氰化氢混合气脱氨
来自氢氰酸合成车间安氏法粗HCN气体经过10~98%硫酸两级吸收脱氨后得道粗氢氰酸气体成分如下:HCN气体9.4%±2%,氢气1.6%±1%,氮气79.4%±2%,氧气1.7%±1%,一氧化碳5.8%±2%,二氧化碳1.5%±1%,甲烷0.6%±0.5%,水蒸气3.7%±2%。
(2)亚氨基二乙酸一钾盐水溶液制备
在1000升反应釜中加入质量百分含量为99.5%的亚氨基二乙酸固体133.77kg、软水204.51kg,在搅拌状态下,慢慢滴加50%氢氧化钾水溶液112.22kg。加完氢氧化钾水溶液,得到无色透明的亚氨基二乙酸一钾盐水溶液450.50kg,其中亚氨基二乙酸一钾盐的质量百分含量为38.0%,水溶液的pH值为5.5。
2-羟基丙腈水溶液制备
开启填料塔换热器,控制塔体内部温度在35~70℃,在预混合反应器中加入40%的乙醛水溶液110.13kg和质量百分含量为38.0%的亚氨基二乙酸一钾盐水溶液300g。将预混合反应器得到的混合液从塔式反应器的顶部打入塔内,流速1~35L/min,来自脱氨安氏法粗HCN气体由填料塔底部进入,气速2~40L/min;塔内物料的pH保持在5.5左右,温度为40℃左右,二者在填料塔内逆流发生反应,在填料塔底部得到含1~8%的乙醛的粗2-羟基丙腈,粗2-羟基丙腈由填料塔底部循环泵打至塔顶循环口,进行全塔循环继续反应至乙醛完全转化为2-羟基丙腈(乙醛残留20ppm,氰化氢残留300ppm),得到的2-羟基丙腈水溶液为无色透明液体177.70kg,其中2-羟基丙腈含量为40.0%,2-羟基丙腈的收率≥99.9%(以乙醛计)。
N,N-二乙酸丙腈一钾盐水溶液制备
将上述得到的177.70kg质量百分含量为40.0%的2-羟基丙腈水溶液和质量百分含量为38.0%的亚氨基二乙酸一钾盐水溶液450.50kg通过静态混合器充分混合后打入管式反应器中,物料在管式反应器中停留时间为30min,管式反应器中温度为100℃,得到无色透明液体N,N-二乙酸丙腈一钾盐水溶液628.20kg,其中N,N-二乙酸丙腈一钾盐质量百分含量为35.69%,收率为99.9%(以2-羟基丙腈计)。
甲基甘氨酸二乙酸三钾盐水溶液制备
在3000升材质为304的反应釜中加入224.44kg质量百分含量为50%的氢氧化钾水溶液,加热至70℃,在搅拌状态下将上述得到的628.20kg质量百分含量为35.69%的N,N-二乙酸丙腈一钾盐水溶液流加至氢氧化钾水溶液中,随着N,N-二乙酸丙腈一钾盐水溶液加入,反应釜中物料温度上升至90℃,加完N,N-二乙酸丙腈一钾盐水溶液后,升温至100℃,继续搅拌反应180min,得到淡黄色的甲基甘氨酸二乙酸三钾盐水溶液,其中氨的含量为1.52%。将得到的甲基甘氨酸二乙酸三钾盐水溶液转入汽提塔中,汽提塔底温度控制在110℃,塔顶温度控制在98℃,经过汽提后的甲基甘氨酸二乙酸三钾盐水溶液中游离氨的含量为10ppm,氨三乙酸(NTA)未检测出,得到甲基甘氨酸二乙酸三钾盐水溶液825.64kg,其中甲基甘氨酸二乙酸三钾盐质量百分含量为38.68%,收率为99.9%(以N,N-二乙酸丙腈一钾盐计)。在甲基甘氨酸二乙酸三钾盐水溶液中加入10.0kg的活性炭,搅拌进行脱色处理,脱色温度为50℃,过滤活性炭后,得到无色透明的甲基甘氨酸二乙酸三钾盐水溶液821.55kg,水溶液的Hazen色号120,未检测出氨三乙酸(NTA),甲基甘氨酸二乙酸三钾盐的总收率为99%(以乙醛计)。
实施例3
原料预处理
(1)氰化氢混合气脱氨
来自氢氰酸合成车间安氏法粗HCN气体经过10~98%硫酸两级吸收脱氨后得道粗氢氰酸气体成分如下:HCN气体9.4%±2%,氢气1.6%±1%,氮气79.4%±2%,氧气1.7%±1%,一氧化碳5.8%±2%,二氧化碳1.5%±1%,甲烷0.6%±0.5%,水蒸气3.7%±2%。
(2)亚氨基二乙酸一钠盐水溶液制备
在1000升反应釜中加入质量百分含量为99.5%的亚氨基二乙酸固体133.77kg、软水240kg,在搅拌状态下,慢慢滴加30%氢氧化钠水溶液133.33kg。加完氢氧化钠水溶液,得到无色透明的亚氨基二乙酸一钠盐水溶液507.10kg,其中亚氨基二乙酸一钠盐的质量百分含量为30.58%,水溶液的pH值为5.3。
2-羟基丙腈水溶液制备
开启填料塔换热器,控制塔体内部温度在35~70℃,在预混合反应器中加入40%的乙醛水溶液110.13kg和质量百分含量为30.58%的亚氨基二乙酸一钠盐水溶液500g。将预混合反应器得到的混合液从塔式反应器的顶部打入塔内,流速1~35L/min,来自脱氨安氏法粗HCN气体由填料塔底部进入,气速2~40L/min;塔内物料的pH保持在5.3左右,温度为60℃左右,二者在填料塔内逆流发生反应,在填料塔底部得到含1~8%的乙醛的粗2-羟基丙腈,粗2-羟基丙腈由填料塔底部循环泵打至塔顶循环口,进行全塔循环继续反应至乙醛完全转化为2-羟基丙腈(乙醛残留30ppm,氰化氢残留200ppm),得到的2-羟基丙腈水溶液为无色透明液体122.55kg,其中2-羟基丙腈含量为58.10%,2-羟基丙腈的收率≥99.9%(以乙醛计)。
N,N-二乙酸丙腈一钠盐水溶液制备
将上述得到的122.55kg质量百分含量为58.10%的2-羟基丙腈水溶液和质量百分含量为30.58%的亚氨基二乙酸一钠盐水溶液506.60kg通过静态混合器充分混合后打入管式反应器中,物料在管式反应器中停留时间为30min,管式反应器中温度为65℃,得到无色透明液体N,N-二乙酸丙腈一钠盐水溶液629.15kg,其中N,N-二乙酸丙腈一钠盐质量百分含量为33.08%,收率为99.9%(以2-羟基丙腈计)。
甲基甘氨酸二乙酸三钠盐水溶液制备
在3000升材质为316的反应釜中加入266.67kg质量百分含量为30%的氢氧化钠水溶液,加热至70℃,在搅拌状态下将上述得到的629.15kg质量百分含量为33.08%的N,N-二乙酸丙腈一钠盐水溶液流加至氢氧化钠水溶液中,随着N,N-二乙酸丙腈一钠盐水溶液加入,反应釜中物料温度上升至75℃,加完N,N-二乙酸丙腈一钠盐水溶液后,升温至120℃,继续搅拌反应90min,得到淡黄色的甲基甘氨酸二乙酸三钠盐水溶液,其中氨的含量为0.90%。将得到的甲基甘氨酸二乙酸三钠盐水溶液转入汽提塔中,汽提塔底温度控制在110℃,塔顶温度控制在98℃,经过汽提后的甲基甘氨酸二乙酸三钠盐水溶液中游离氨的含量为10ppm,氨三乙酸(NTA)未检测出,得到甲基甘氨酸二乙酸三钠盐水溶液860.82kg,其中甲基甘氨酸二乙酸三钠盐质量百分含量为31.18%,收率为99.9%(以N,N-二乙酸丙腈一钠盐计)。在甲基甘氨酸二乙酸三钠盐水溶液中加入10.0kg的活性炭,搅拌进行脱色处理,脱色温度为50℃,过滤活性炭后,得到无色透明的甲基甘氨酸二乙酸三钠盐水溶液898.55kg,进行减压浓缩至甲基甘氨酸二乙酸三钠盐质量百分含量为40%,水溶液的Hazen色号100,未检测出氨三乙酸(NTA),甲基甘氨酸二乙酸三钠盐的总收率为99%(以乙醛计)。
实施例4
原料预处理
(1)氰化氢混合气脱氨
来自氢氰酸合成车间安氏法粗HCN气体经过10~98%硫酸两级吸收脱氨后得道粗氢氰酸气体成分如下:HCN气体9.4%±2%,氢气1.6%±1%,氮气79.4%±2%,氧气1.7%±1%,一氧化碳5.8%±2%,二氧化碳1.5%±1%,甲烷0.6%±0.5%,水蒸气3.7%±2%。
(2)亚氨基二乙酸一钾盐水溶液制备
在1000升反应釜中加入质量百分含量为99.5%的亚氨基二乙酸固体133.77kg、软水230kg,在搅拌状态下,慢慢滴加30%氢氧化钾水溶液187.04kg。加完氢氧化钾水溶液,得到无色透明的亚氨基二乙酸一钾盐水溶液550.80kg,其中亚氨基二乙酸一钾盐的质量百分含量为31.08%,水溶液的pH值为5.5。
2-羟基丙腈水溶液制备
开启填料塔换热器,控制塔体内部温度在35~70℃,在预混合反应器中加入40%的乙醛水溶液110.13kg和质量百分含量为31.08%的亚氨基二乙酸一钾盐水溶液200g。将预混合反应器得到的混合液从塔式反应器的顶部打入塔内,流速1~35L/min,来自脱氨安氏法粗HCN气体由填料塔底部进入,气速2~40L/min;塔内物料的pH保持在5.5左右,温度为70℃左右,二者在填料塔内逆流发生反应,在填料塔底部得到含1~8%的乙醛的粗2-羟基丙腈,粗2-羟基丙腈由填料塔底部循环泵打至塔顶循环口,进行全塔循环继续反应至乙醛完全转化为2-羟基丙腈(乙醛残留15ppm,氰化氢残留300ppm),得到的2-羟基丙腈水溶液为无色透明液体118.50kg,其中2-羟基丙腈含量为60.0%,2-羟基丙腈的收率≥99.9%(以乙醛计)。
N,N-二乙酸丙腈一钾盐水溶液制备
将上述得到的118.50kg质量百分含量为60.0%的2-羟基丙腈水溶液和质量百分含量为31.08%的亚氨基二乙酸一钾盐水溶液550.60kg通过静态混合器充分混合后打入管式反应器中,物料在管式反应器中停留时间为25min,管式反应器中温度为65℃,得到无色透明液体N,N-二乙酸丙腈一钾盐水溶液669.10kg,其中N,N-二乙酸丙腈一钾盐质量百分含量为33.51%,收率为99.9%(以2-羟基丙腈计)。
甲基甘氨酸二乙酸三钾盐水溶液制备
在3000升材质为316的反应釜中加入374.07kg质量百分含量为30%的氢氧化钾水溶液,加热至70℃,在搅拌状态下将上述得到的669.10kg质量百分含量为33.51%的N,N-二乙酸丙腈一钾盐水溶液流加至氢氧化钾水溶液中,随着N,N-二乙酸丙腈一钾盐水溶液加入,反应釜中物料温度上升至80℃,加完N,N-二乙酸丙腈一钾盐水溶液后,升温至120℃,继续搅拌反应90min,得到淡黄色的甲基甘氨酸二乙酸三钾盐水溶液,其中氨的含量为0.89%。将得到的甲基甘氨酸二乙酸三钾盐水溶液转入汽提塔中,汽提塔底温度控制在110℃,塔顶温度控制在98℃,经过汽提后的甲基甘氨酸二乙酸三钾盐水溶液中游离氨的含量为10ppm,氨三乙酸(NTA)未检测出,得到甲基甘氨酸二乙酸三钾盐水溶液1020.17kg,其中甲基甘氨酸二乙酸三钾盐质量百分含量为31.31%,收率为99.9%(以N,N-二乙酸丙腈一钾盐计)。在甲基甘氨酸二乙酸三钾盐水溶液中加入15.0kg的活性炭,搅拌进行脱色处理,脱色温度为50℃,过滤活性炭后,得到无色透明的甲基甘氨酸二乙酸三钾盐水溶液1015.88kg,进行减压浓缩至甲基甘氨酸二乙酸三钾盐质量百分含量为40%,水溶液的Hazen色号110,未检测出氨三乙酸(NTA),甲基甘氨酸二乙酸三钾盐的总收率为99%(以乙醛计)。
对比实施例1
在烧瓶中加入135.8kg亚氨基二乙酸(98%),270kg水,50%NaOH 80kg,搅拌溶解,PH约5.5,控制温度30℃,同时滴加氢氰酸28.6kg(99%)和乙醛110kg(40%),1~2h滴完,升温至70℃,保温30min,然后将反应液滴加到287kg 30% NaOH中,控制滴加温度30~35℃,约2~3h滴完,滴完后,30℃保温3min,然后升温至沸腾,回流约4h,直至回流管尾气pH 7~8结束,降温至80℃,加入2kg双氧水,反应30min,再加入活性炭2kg,保温30min,过滤得含量30%的甲基甘氨酸二乙酸三钠盐溶液842kg,可继续浓缩制得40%的甲基甘氨酸二乙酸三钠盐溶液,水溶液的Hazen色号560,收率86%,NTA含量0.5%,亚氨基二乙酸含量1.05%,甘氨酸含量为0.1%,丙氨酸0.3%,甲酸含量0.2%。
对比实施例2
在烧瓶中加入135.8kg亚氨基二乙酸(98%),270kg水,50%KOH 112.22kg,搅拌溶解,PH约5.5,控制温度30℃,同时滴加氢氰酸28.6kg(99%)和乙醛110kg(40%),1~2h滴完,升温至70℃,保温30min,然后将反应液滴加到542.40kg 30%KOH中,控制滴加温度30~35℃,约2~3h滴完,滴完后,30℃保温3min,然后升温至沸腾,回流约4h,直至回流管尾气pH7~8结束,降温至80℃,加入2kg双氧水,反应30min,再加入活性炭2kg,保温30min,过滤得含量26.83%的甲基甘氨酸二乙酸三钠盐溶液1000.20kg,可继续浓缩制得40%的甲基甘氨酸二乙酸三钠盐溶液,水溶液的Hazen色号840,收率84%,NTA含量0.6%,亚氨基二乙酸含量1.25%,甘氨酸含量为0.1%,丙氨酸0.5%,甲酸含量0.3%。
以上实施例仅用以说明本发明的技术方案而非限制,尽管参照较佳实施例对本发明进行了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的宗旨和范围,其均应涵盖在本发明的权利要求范围当中。本发明未详细描述的技术、形状、构造部分均为公知技术。