一种苯并咪唑类药物中间体3-硝基-4-乙基氨基苯甲醇的制备方法
文献发布时间:2024-04-18 19:53:33
技术领域
本发明涉及有机合成及药物技术领域,具体涉及苯并咪唑类药物中间体3-硝基-4-乙基氨基苯甲醇的制备方法。
背景技术
苯并咪唑类是一类重要的化合物,在药物合成方面应用广泛。比如其可用于合成抗菌药多菌灵、阿苯达唑、芬苯达唑、奥苯达唑等;质子泵抑制剂药物奥美拉唑、兰索拉唑、泮托拉唑、雷贝拉唑等;还可用于溴结构域抑制剂(比特R.A.等人, WO2016146738A1,CN107635989B),以及鞘氨醇1-磷酸受体1拮抗剂药物(Hennessy E.J.等人,
苯并咪唑类衍生物的合成方法之一是2-硝基苯胺类化合物与羧酸类化合物缩合。本发明所涉及的3-硝基-4-乙基氨基苯甲醇即属于2-硝基苯胺类化合物,可用于合成以苯并咪唑为母核的磺胺类或2-吡啶酮类的药物合成。
现有技术公开了一种3-硝基-4-乙基氨基苯甲醇的制备方法:利用微波反应,在四氢呋喃溶剂中,加入过量的二异丙基乙基胺,以3-硝基-4-氟苯甲醇为原料,与等当量的70%乙胺水溶液在120℃反应3小时得到3-硝基-4-乙基氨基苯甲醇。该方法反应条件苛刻,需利用微波反应,温度较高,原料成本高。因此针对上述问题有必要研究一种新的制备方法来合成3-硝基-4-乙基氨基苯甲醇。
发明内容
为解决上述问题,本发明提供一种苯并咪唑类药物中间体3-硝基-4-乙基氨基苯甲醇的制备方法,该方法一步反应实现两种官能团的化学转化,反应条件比较温和,设备简单,易于控制,降低了原料成本,产物收率较高。
本发明的目的是以下述方式实现的:一种苯并咪唑类药物中间体3-硝基-4-乙基氨基苯甲醇的制备方法,以3-硝基-4-乙酰氨基苯甲酸为原料,在有机溶剂中与还原剂发生还原反应,合成出3-硝基-4-乙基氨基苯甲醇,其中还原剂为硼烷,二异丁基氢化铝或者红铝,或者是还原性含锂、含钠或含钾的化合物与三氟化硼/乙醚为还原剂。
1、硼烷作为还原剂的具体制备方法
(1)以3-硝基-4-乙酰氨基苯甲酸为原料,有机溶剂作为反应体系的溶剂,将原料溶解完全后,冷却条件下缓慢加入硼烷的有机溶液,并保持良好搅拌;
(2)硼烷的有机溶液加入完毕,撤去冷却装置,使反应体系缓慢升温,继续搅拌一段时间,反应结束后,在冷却条件下缓慢加入甲醇至无气体放出;
(3)有机溶剂减压蒸出,剩余物用碱性水溶液分散,其中的粗品用有机溶剂萃取后,洗涤,干燥,浓缩,结晶得到3-硝基-4-乙基氨基苯甲醇。
步骤(1)中反应体系的有机溶剂为四氢呋喃、二氯甲烷。
步骤(1)中硼烷的有机溶液为硼烷/四氢呋喃、硼烷/二甲硫醚。
步骤(1)中加入硼烷的有机溶剂时的体系温度控制在0-10℃,所述3-硝基-4-乙酰氨基苯甲酸与硼烷的摩尔比为1 : 2.0-3.0。
步骤(2)中反应温度为20-40℃,反应时间为2-5小时,冷却温度为5-10℃。
步骤(3)中碱性水溶液为氢氧化钠、氢氧化钾、碳酸钠、碳酸钾水溶液。
步骤(3)中萃取时所用萃取剂为乙酸乙酯、二氯甲烷。
2、二异丁基氢化铝为还原剂的具体制备方法
(1)以3-硝基-4-乙酰氨基苯甲酸为原料,有机溶剂作为反应体系的溶剂,将原料溶解完全后,冷却条件下缓慢加入二异丁基氢化铝的有机溶液,并保持良好搅拌;
(2)滴加完毕,撤去冷却装置,使反应体系缓慢升温,继续搅拌一段时间,反应结束后,在冷却条件下缓慢加入酒石酸钾钠的饱和溶液至无气体放出;
(3)加入有机溶剂萃取后,合并有机相,洗涤,干燥,浓缩,结晶得到3-硝基-4-乙基氨基苯甲醇。
步骤(1)中有机溶剂为甲苯、四氢呋喃、二氯甲烷。
步骤(1)中二异丁基氢化铝的有机溶液为二异丁基氢化铝/甲苯、二异丁基氢化铝/正己烷。
步骤(1)中反应温度为0-10℃,所述3-硝基-4-乙酰氨基苯甲酸与二异丁基氢化铝的摩尔比为1 : 5.0-10.0。
步骤(2)中反应温度为20-40℃,反应时间为2-6小时,冷却温度为5-10℃。
步骤(3)中萃取时所用萃取剂为乙酸乙酯、二氯甲烷。
3、红铝为还原剂的具体制备方法
(1)以3-硝基-4-乙酰氨基苯甲酸为原料,有机溶剂作为反应体系的溶剂,将原料溶解完全后,冷却条件下缓慢加入红铝的有机溶液,并保持良好搅拌;
(2)滴加完毕,撤去冷却装置,使反应体系缓慢升温,继续搅拌一段时间,反应结束后,在冷却条件下缓慢加入碱性水溶液淬灭;
(3)加入有机溶剂萃取后,合并有机相,洗涤,干燥,浓缩,结晶得到3-硝基-4-乙基氨基苯甲醇。
步骤(1)中有机溶剂为甲苯、四氢呋喃。
步骤(1)中红铝的有机溶液为红铝/甲苯。
步骤(1)中反应温度为0-10℃,所述3-硝基-4-乙酰氨基苯甲酸与红铝的摩尔比为1 : 5.0-7.0。
步骤(2)中反应温度为60-80℃,反应时间为1-3小时,冷却温度为5-10℃。
步骤(2)中碱性水溶液为氢氧化钠、氢氧化钾、碳酸钠、碳酸钾水溶液。
步骤(3)中萃取时所用萃取剂为乙酸乙酯、二氯甲烷。
4、还原性含锂、含钠或含钾的化合物与三氟化硼/乙醚为还原剂的具体制备方法
(1)以3-硝基-4-乙酰氨基苯甲酸为原料,有机溶剂作为反应体系的溶剂,将原料溶解完全后,冷却条件下缓慢加入含锂、含钠或含钾的还原性化合物与三氟化硼/乙醚,并保持良好搅拌;
(2)加料完毕,撤去冷却装置,使反应体系缓慢升温至回流,继续搅拌一段时间,反应结束后,在冷却条件下缓慢依次加入1/9-1/11溶剂体积的甲醇和浓度8%-12%的稀盐酸,直到无气体释放出为止,停止加入稀盐酸,加毕,继续搅拌回流0.5小时;加入稀盐酸的量由实验现象决定,需缓慢加入稀盐酸,直到无气体释放出为止,停止加入稀盐酸,可认为此时淬灭完全。
(3)有机溶剂减压蒸出,剩余物用碱性溶液氨水和有机溶剂分散,控制pH为6-8,过滤,母液分液后,水相用有机溶剂萃取,合并有机相,洗涤,干燥,浓缩,结晶得到3-硝基-4-乙基氨基苯甲醇。
步骤(1)中反应体系的有机溶剂为四氢呋喃、二氯甲烷。
由于步骤(2)使用了稀盐酸,这里有少量残留,使用碱性溶液方便控制pH值,但步骤(3)中碱性溶液优选氨水;氨水的浓度优选是25%-28%,其它浓度的氨水也可以,但是浓度较低的氨水,使用量会比较大。
步骤(3)和碱性溶液一起使用的有机溶剂是乙酸乙酯或二氯甲烷;步骤(3)中萃取时所用有机溶剂为乙酸乙酯、二氯甲烷。
步骤(1)中含锂、含钠、含钾的还原性化合物为四氢铝锂、硼氢化锂、硼氢化钠、硼氢化钾。
步骤(1)中反应温度为0-10℃,所述3-硝基-4-乙酰氨基苯甲酸、含锂、含钠、含钾的还原性化合物、三氟化硼/乙醚的摩尔比为1 : 2.0-5.0 : 3.0-4.0。
步骤(2)中反应时间为4-8小时,冷却温度为5-10℃。反应溶剂不同,回流时温度也不一样,四氢呋喃作溶剂时反应温度一般在66℃,二氯甲烷作溶剂时反应温度一般在39℃。
本发明合成方法中主要的反应式:
本发明采用以上技术方案,与现有技术相比,具有以下优点:
本发明以3-硝基-4-乙酰氨基苯甲酸为原料,在有机溶剂中与还原剂发生还原反应,合成出3-硝基-4-乙基氨基苯甲醇。该制备方法只需一步合成反应,实现两种官能团的化学转化,整个反应条件温和,不需要特殊反应设备,降低了原料成本,提高了产物收率,并且在整个反应过程中无废水排放,减少了环境污染。
附图说明
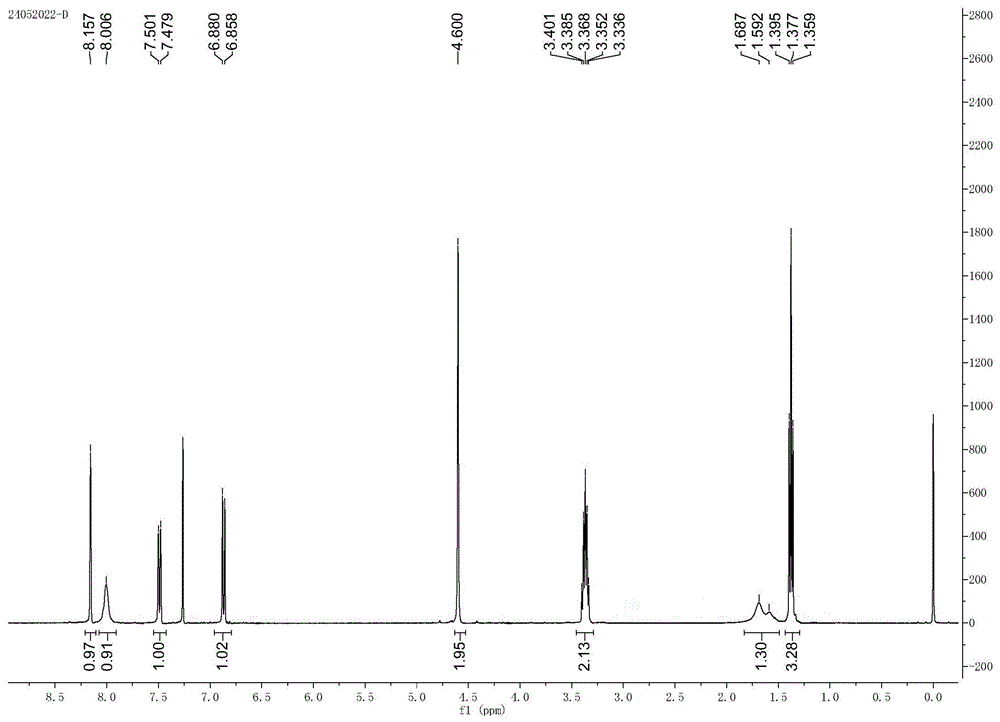
图1为发明制备的产物3-硝基-4-乙基氨基苯甲醇的核磁共振氢谱。
图2为发明制备的产物3-硝基-4-乙基氨基苯甲醇的阳离子液相质谱图,其中,179.0为3-硝基-4-乙基氨基甲苯阳离子:
具体实施方式
下面结合具体实施例对本发明进行具体描述,有必要在此指出的是本实施例只用于对本发明进行进一步说明,不能理解为对本发明保护范围的限制,该领域的技术熟练人员可以根据上述本发明的内容做出一些非本质的改进和调整。
实施例1
向无水四氢呋喃(100.0 mL)中加入3-硝基-4-乙酰氨基苯甲酸(10.1 g,45.0mmol, 1.0 equiv.),常温搅拌溶解后,冷却至0℃,搅拌下缓慢加入1 M硼烷四氢呋喃溶液(90.0 mL,90.0 mmol,2.0 equiv.),即3-硝基-4-乙酰氨基苯甲酸:硼烷四氢呋喃络合物的摩尔比为1:2,保持反应温度在0-5℃。加毕,将反应体系缓慢升温至20℃,继续反应5小时后,TLC监测反应进程,反应结束。将反应体系降温至5℃,缓慢加入无水甲醇,保持体系温度在5-10℃,至无气泡产生。减压蒸出有机溶剂,剩余物用2 N 氢氧化钠水溶液(20.0 mL)分散,其中的粗品用乙酸乙酯(20.0 mL × 3)萃取,有机相用饱和食盐水洗涤(30.0 mL ×1),无水硫酸钠干燥,过滤,母液浓缩结晶得到3-硝基-4-乙基氨基苯甲醇7.9 g,收率89%,棕色固体。
本申请的实施例中以底物3-硝基-4-乙酰氨基苯甲酸反应完即为反应结束。TLC监测某种物质是否反应完全为现有技术中的常规方法,TLC(薄层层析色谱),如果某种物质没反应完,在254nm紫外光照射下,会在TLC硅胶板上面显示出该物质的点,如果反应完了,就不会显示该物质的点。
实施例2
向无水四氢呋喃(100.0 mL)中加入3-硝基-4-乙酰氨基苯甲酸(10.1 g,45.0mmol, 1.0 equiv.),常温搅拌溶解后,冷却至0℃,搅拌下缓慢加入1 M硼烷四氢呋喃溶液(112.5 mL,112.5 mmol,2.5 equiv.),保持反应温度在5-10℃。加毕,将反应体系缓慢升温至20℃,继续反应4.5小时后,TLC监测反应结束。将反应体系降温至5℃,缓慢加入无水甲醇,保持体系温度在5-10℃,至无气泡产生。减压蒸出有机溶剂,剩余物用2 N 氢氧化钠水溶液(20.0 mL)分散,其中的粗品用乙酸乙酯(20.0 mL × 3)萃取,有机相用饱和食盐水洗涤(30.0 mL × 1),无水硫酸钠干燥,过滤,母液浓缩结晶得到3-硝基-4-乙基氨基苯甲醇8.2 g,收率93%,棕色固体。
实施例3
向无水四氢呋喃(100.0 mL)中加入3-硝基-4-乙酰氨基苯甲酸(10.1 g,45.0mmol, 1.0 equiv.),常温搅拌溶解后,冷却至0℃,搅拌下缓慢加入1 M硼烷四氢呋喃溶液(135.0 mL,135.0 mmol,3.0 equiv.),保持反应温度在0-10℃。加毕,将反应体系缓慢升温至20℃,继续反应3.5小时后,TLC监测反应结束。将反应体系降温至5℃,缓慢加入无水甲醇,保持体系温度在5-10℃,至无气泡产生。减压蒸出有机溶剂,剩余物用2 N 氢氧化钾水溶液(20.0 mL)分散,其中的粗品用二氯甲烷(50.0 mL × 3)萃取,有机相用饱和食盐水洗涤(30.0 mL × 1),无水硫酸镁干燥,过滤,母液浓缩结晶得到3-硝基-4-乙基氨基苯甲醇8.4 g,收率95%,棕色固体。
实施例4
向无水四氢呋喃(100.0 mL)中加入3-硝基-4-乙酰氨基苯甲酸(10.1 g, 45.0mmol, 1.0 equiv.),常温搅拌溶解后,冷却至0℃,搅拌下缓慢加入1 M硼烷四氢呋喃溶液(135.0 mL,135.0 mmol,3.0 equiv.),保持反应温度在0-10℃。加毕,将反应体系缓慢升温至30℃,继续反应3小时后,TLC监测反应结束。将反应体系降温至5℃,缓慢加入无水甲醇,保持体系温度在5-10℃,至无气泡产生。减压蒸出有机溶剂,剩余物用饱和碳酸钠水溶液(20.0 mL)分散,其中的粗品用乙酸乙酯(20.0 mL × 3)萃取,有机相用饱和食盐水洗涤(30.0 mL × 1),无水硫酸钠干燥,过滤,母液浓缩结晶得到3-硝基-4-乙基氨基苯甲醇8.5g,收率96%,棕色固体。
实施例5
向无水四氢呋喃(100.0 mL)中加入3-硝基-4-乙酰氨基苯甲酸(10.1 g, 45.0mmol, 1.0 equiv.),常温搅拌溶解后,冷却至0℃,搅拌下缓慢加入1 M硼烷四氢呋喃溶液(135.0 mL,135.0 mmol,3.0 equiv.),保持反应温度在0-10℃。加毕,将反应体系缓慢升温至40℃,继续反应2小时后,TLC监测反应结束。将反应体系降温至5℃,缓慢加入无水甲醇,保持体系温度在5-10℃,至无气泡产生。减压蒸出有机溶剂,剩余物用饱和碳酸钾水溶液(20.0 mL)分散,其中的粗品用乙酸乙酯(20.0 mL × 3)萃取,有机相用饱和食盐水洗涤(30.0 mL × 1),无水硫酸钠干燥,过滤,母液浓缩结晶得到3-硝基-4-乙基氨基苯甲醇8.3g,收率94%,棕色固体。
实施例6
向无水二氯甲烷(200.0 mL)中加入3-硝基-4-乙酰氨基苯甲酸(10.1 g, 45.0mmol, 1.0 equiv.),常温搅拌溶解后,冷却至0℃,搅拌下缓慢加入1 M硼烷四氢呋喃溶液(135.0 mL,135.0 mmol,3.0 equiv.),保持反应温度在0-10℃。加毕,将反应体系缓慢升温至40℃,继续反应2.5小时后,TLC监测反应结束。将反应体系降温至5℃,缓慢加入无水甲醇,保持体系温度在5-10℃,至无气泡产生。减压蒸出有机溶剂,剩余物用饱和碳酸钠水溶液(20.0 mL)分散,其中的粗品用乙酸乙酯(20.0 mL × 3)萃取,有机相用饱和食盐水洗涤(30.0 mL × 1),无水硫酸钠干燥,过滤,母液浓缩结晶得到3-硝基-4-乙基氨基苯甲醇8.0g,收率91%,棕色固体。
实施例7
向无水四氢呋喃(100.0 mL)中加入3-硝基-4-乙酰氨基苯甲酸(10.1 g,45.0mmol, 1.0 equiv.),常温搅拌溶解后,冷却至0℃,搅拌下缓慢加入2 M硼烷二甲硫醚溶液(67.5 mL,135.0 mmol,3.0 equiv.),保持反应温度在0-10℃。加毕,将反应体系缓慢升温至20℃,继续反应4.5小时后,TLC监测反应结束。将反应体系降温至5℃,缓慢加入无水甲醇,保持体系温度在5-10℃,至无气泡产生。减压蒸出有机溶剂,剩余物用2 N 氢氧化钠水溶液(20.0 mL)分散,其中的粗品用二氯甲烷(50.0 mL × 3)萃取,有机相用饱和食盐水洗涤(30.0 mL × 1),无水硫酸镁干燥,过滤,母液浓缩结晶得到3-硝基-4-乙基氨基苯甲醇8.0 g,收率90%,棕色固体。
实施例8
向无水四氢呋喃(100.0 mL)中加入3-硝基-4-乙酰氨基苯甲酸(6.7 g, 29.9mmol, 1.0 equiv.),常温搅拌溶解后,冷却至0℃,搅拌下缓慢加入硼氢化钠(5.7 g,150.0mmol,5.0 equiv.),48%三氟化硼/乙醚(17.0 g,120.0 mmol,4.0 equiv.),保持反应温度在0-5℃。加毕,将反应体系缓慢升温至回流,继续反应5小时后,TLC监测反应结束。将反应体系降温至5℃,缓慢加入无水甲醇和2 N 盐酸,保持体系温度在5-10℃,至无气泡产生,加热回流0.5小时。减压蒸出有机溶剂,剩余物用质量浓度为25%的浓氨水(100.0 mL),乙酸乙酯(80.0 mL)分散,过滤,母液分液,水相用乙酸乙酯(20.0 mL × 3)萃取,合并有机相,用饱和食盐水洗涤(30.0 mL × 1),无水硫酸钠干燥,过滤,母液浓缩结晶得到3-硝基-4-乙基氨基苯甲醇5.1 g,收率86%,棕色固体。
实施例9
向无水四氢呋喃(100.0 mL)中加入3-硝基-4-乙酰氨基苯甲酸(6.7 g, 29.9mmol, 1.0 equiv.),常温搅拌溶解后,冷却至0℃,搅拌下缓慢加入硼氢化钠(2.3 g,59.8mmol,2.0 equiv.),48%三氟化硼/乙醚(12.7 g,89.7mmol,3.0 equiv.),保持反应温度在5-10℃。加毕,将反应体系缓慢升温至回流,继续反应8小时后,TLC监测反应结束。将反应体系降温至5℃,缓慢加入无水甲醇和2 N 盐酸,保持体系温度在5-10℃,至无气泡产生,加热回流0.5小时。减压蒸出有机溶剂,剩余物用浓氨水(100.0 mL),乙酸乙酯(80.0 mL)分散,过滤,母液分液,水相用乙酸乙酯(20.0 mL × 3)萃取,合并有机相,用饱和食盐水洗涤(30.0 mL × 1),无水硫酸钠干燥,过滤,母液浓缩结晶得到3-硝基-4-乙基氨基苯甲醇4.2g,收率71%,棕色固体。
实施例10
向无水四氢呋喃(100.0 mL)中加入3-硝基-4-乙酰氨基苯甲酸(6.7 g, 29.9mmol, 1.0 equiv.),常温搅拌溶解后,冷却至0℃,搅拌下缓慢加入硼氢化锂(2.6 g,119.6mmol,4.0 equiv.),48%三氟化硼/乙醚(12.7 g,89.7 mmol,3.0 equiv.),保持反应温度在0-10℃。加毕,将反应体系缓慢升温至回流,继续反应6小时后,TLC监测反应结束。将反应体系降温至5℃,缓慢加入无水甲醇和2 N 盐酸,保持体系温度在5-10℃,至无气泡产生,加热回流0.5小时。减压蒸出有机溶剂,剩余物用浓氨水(100.0 mL),乙酸乙酯(80.0 mL)分散,过滤,母液分液,水相用乙酸乙酯(20.0 mL × 3)萃取,合并有机相,用饱和食盐水洗涤(30.0 mL × 1),无水硫酸钠干燥,过滤,母液浓缩结晶得到3-硝基-4-乙基氨基苯甲醇4.9g,收率83%,棕色固体。
实施例11
向二氯甲烷(150.0 mL)中加入3-硝基-4-乙酰氨基苯甲酸(6.7 g, 29.9 mmol,1.0 equiv.),常温搅拌溶解后,冷却至0℃,搅拌下缓慢加入硼氢化钾(5.6 g,104.6 mmol,3.5 equiv.),48%三氟化硼/乙醚(14.8 g,104.6 mmol,3.5 equiv.),保持反应温度在0-10℃。加毕,将反应体系缓慢升温至回流,继续反应7小时后,TLC监测反应结束。将反应体系降温至5℃,缓慢加入无水甲醇和2 N 盐酸,保持体系温度在5-10℃,至无气泡产生,加热回流0.5小时。减压蒸出有机溶剂,剩余物用浓氨水(100.0 mL),二氯甲烷(80.0 mL)分散,过滤,母液分液,水相用二氯甲烷(20.0 mL × 3)萃取,合并有机相,用饱和食盐水洗涤(30.0 mL× 1),无水硫酸镁干燥,过滤,母液浓缩结晶得到3-硝基-4-乙基氨基苯甲醇3.9 g,收率67%,棕色固体。
实施例12
向无水四氢呋喃(100.0 mL)中加入3-硝基-4-乙酰氨基苯甲酸(6.7 g, 29.9mmol, 1.0 equiv.),常温搅拌溶解后,冷却至0℃,搅拌下缓慢加入四氢铝锂(4.5 g,119.6mmol,4.0 equiv.),48%三氟化硼/乙醚(12.7 g,89.7 mmol,3.0 equiv.),保持反应温度在0-10℃。加毕,将反应体系缓慢升温至回流,继续反应4小时后,TLC监测反应结束。将反应体系降温至5℃,缓慢加入无水甲醇和2 N 盐酸,保持体系温度在5-10℃,至无气泡产生,加热回流0.5小时。减压蒸出有机溶剂,剩余物用浓氨水(100.0 mL),乙酸乙酯(80.0 mL)分散,过滤,母液分液,水相用乙酸乙酯(20.0 mL × 3)萃取,合并有机相,用饱和食盐水洗涤(30.0 mL × 1),无水硫酸钠干燥,过滤,母液浓缩结晶得到3-硝基-4-乙基氨基苯甲醇4.6g,收率79%,棕色固体。
实施例13
向无水甲苯(100 mL)中加入3-硝基-4-乙酰氨基苯甲酸(6.7 g, 29.9 mmol, 1.0equiv.),常温搅拌溶解后,冷却至0℃,搅拌下缓慢加入1.5 M 二异丁基氢化铝/甲苯(99.7mL,149.5 mmol,5.0 equiv.),保持反应温度在0-5℃。加毕,将反应体系缓慢升温至20℃,继续反应6小时后,TLC监测反应结束。将反应体系降温至5℃,缓慢加入酒石酸钾钠的饱和溶液,保持体系温度在5-10℃,至无气泡产生。分液,水相用乙酸乙酯(20.0 mL × 3)萃取,合并有机相,用饱和食盐水洗涤(30.0 mL × 1),无水硫酸钠干燥,过滤,母液浓缩结晶得到3-硝基-4-乙基氨基苯甲醇3.8 g,收率65%,棕色固体。
实施例14
向无水甲苯(100 mL)中加入3-硝基-4-乙酰氨基苯甲酸(6.7 g, 29.9 mmol, 1.0equiv.),常温搅拌溶解后,冷却至0℃,搅拌下缓慢加入1.5 M 二异丁基氢化铝/甲苯(149.5 mL,224.3 mmol,7.5 equiv.),保持反应温度在5-10℃。加毕,将反应体系缓慢升温至30℃,继续反应4小时后,TLC监测反应结束。将反应体系降温至5℃,缓慢加入酒石酸钾钠的饱和溶液,保持体系温度在5-10℃,至无气泡产生。分液,水相用乙酸乙酯(20.0 mL ×3)萃取,合并有机相,用饱和食盐水洗涤(30.0 mL × 1),无水硫酸钠干燥,过滤,母液浓缩结晶得到3-硝基-4-乙基氨基苯甲醇4.7 g,收率80%,棕色固体。
实施例15
向无水甲苯(100.0 mL)中加入3-硝基-4-乙酰氨基苯甲酸(6.7 g, 29.9 mmol,1.0 equiv.),常温搅拌溶解后,冷却至0℃,搅拌下缓慢加入1.5 M 二异丁基氢化铝/甲苯(199.3 mL,299.0 mmol,10.0 equiv.),保持反应温度在5-10℃。加毕,将反应体系缓慢升温至40℃,继续反应2小时后,TLC监测反应结束。将反应体系降温至5℃,缓慢加入酒石酸钾钠的饱和溶液,保持体系温度在5-10℃,至无气泡产生。分液,水相用乙酸乙酯(20.0 mL ×3)萃取,合并有机相,用饱和食盐水洗涤(30.0 mL × 1),无水硫酸钠干燥,过滤,母液浓缩结晶得到3-硝基-4-乙基氨基苯甲醇4.9 g,收率83%,棕色固体。
实施例16
向无水四氢呋喃(100.0 mL)中加入3-硝基-4-乙酰氨基苯甲酸(6.7 g, 29.9mmol, 1.0 equiv.),常温搅拌溶解后,冷却至0℃,搅拌下缓慢加入1 M 二异丁基氢化铝/正己烷(224.3 mL,224.3 mmol,7.5 equiv.),保持反应温度在0-10℃。加毕,将反应体系缓慢升温至40℃,继续反应4小时后,TLC监测反应结束。将反应体系降温至5℃,缓慢加入酒石酸钾钠的饱和溶液至无气体放出,保持体系温度在5-10℃,至无气泡产生。分液,水相用乙酸乙酯(20.0 mL × 3)萃取,合并有机相,用饱和食盐水洗涤(30.0 mL × 1),无水硫酸钠干燥,过滤,母液浓缩结晶得到3-硝基-4-乙基氨基苯甲醇4.5 g,收率77%,棕色固体。
实施例17
向二氯甲烷(150.0 mL)中加入3-硝基-4-乙酰氨基苯甲酸(6.7 g, 29.9 mmol,1.0 equiv.),常温搅拌溶解后,冷却至0℃,搅拌下缓慢加入1 M 二异丁基氢化铝/正己烷(224.3 mL,224.3 mmol,7.5 equiv.),保持反应温度在0-10℃。加毕,将反应体系缓慢升温至40℃,继续反应5小时后,TLC监测反应结束。将反应体系降温至5℃,缓慢加入酒石酸钾钠的饱和溶液至无气体放出,保持体系温度在5-10℃,至无气泡产生。分液,水相用二氯甲烷(20.0 mL × 3)萃取,合并有机相,用饱和食盐水洗涤(30.0 mL × 1),无水硫酸镁干燥,过滤,母液浓缩结晶得到3-硝基-4-乙基氨基苯甲醇4.0 g,收率68%,棕色固体。
实施例18
向无水甲苯(100 mL)中加入3-硝基-4-乙酰氨基苯甲酸(6.7 g, 29.9 mmol, 1.0equiv.),常温搅拌溶解后,冷却至0℃,搅拌下缓慢加入3.6 M 红铝/甲苯(49.8 mL,179.4mmol,6.0 equiv.),保持反应温度在0-5℃。加毕,将反应体系缓慢升温至70℃,继续反应2小时后,TLC监测反应结束。将反应体系降温至5℃,搅拌下缓慢加入2 N 氢氧化钠溶液(200mL),保持体系温度在5-10℃。分液,水相用乙酸乙酯(20 mL × 3)萃取,合并有机相,用饱和食盐水洗涤(30 mL × 1),无水硫酸钠干燥,过滤,母液浓缩结晶得到3-硝基-4-乙基氨基苯甲醇5.0 g,收率85%,棕色固体。
实施例19
向无水甲苯(100 mL)中加入3-硝基-4-乙酰氨基苯甲酸(6.7 g, 29.9 mmol, 1.0equiv.),常温搅拌溶解后,冷却至0℃,搅拌下缓慢加入3.6 M 红铝/甲苯(41.5 mL,149.5mmol,5.0 equiv.),保持反应温度在5-10℃。加毕,将反应体系缓慢升温至70℃,继续反应3小时后,TLC监测反应结束。将反应体系降温至5℃,搅拌下缓慢加入2 N 氢氧化钾溶液(200mL),保持体系温度在5-10℃。分液,水相用乙酸乙酯(20 mL × 3)萃取,合并有机相,用饱和食盐水洗涤(30 mL × 1),无水硫酸钠干燥,过滤,母液浓缩结晶得到3-硝基-4-乙基氨基苯甲醇4.6 g,收率78%,棕色固体。
实施例20
向无水甲苯(100 mL)中加入3-硝基-4-乙酰氨基苯甲酸(6.7 g, 29.9 mmol, 1.0equiv.),常温搅拌溶解后,冷却至0℃,搅拌下缓慢加入3.6 M 红铝/甲苯(41.5 mL,149.5mmol,5.0 equiv.),保持反应温度在5-10℃。加毕,将反应体系缓慢升温至80℃,继续反应3小时后,TLC监测反应结束。将反应体系降温至5℃,搅拌下缓慢加入饱和碳酸钾溶液(200mL),保持体系温度在5-10℃。分液,水相用乙酸乙酯(20 mL × 3)萃取,合并有机相,用饱和食盐水洗涤(30 mL × 1),无水硫酸钠干燥,过滤,母液浓缩结晶得到3-硝基-4-乙基氨基苯甲醇4.7 g,收率80%,棕色固体。
实施例21
向无水四氢呋喃(100 mL)中加入3-硝基-4-乙酰氨基苯甲酸(6.7 g, 29.9 mmol,1.0 equiv.),常温搅拌溶解后,冷却至0℃,搅拌下缓慢加入3.6 M 红铝/甲苯(58.1 mL,209.3 mmol,7.0 equiv.),保持反应温度在0-10℃。加毕,将反应体系缓慢升温至60℃,继续反应1小时后,TLC监测反应结束。将反应体系降温至5℃,搅拌下缓慢加入饱和碳酸钠溶液(200 mL),保持体系温度在5-10℃。分液,水相用二氯甲烷(20 mL × 3)萃取,合并有机相,用饱和食盐水洗涤(30 mL × 1),无水硫酸钠干燥,过滤,母液浓缩结晶得到3-硝基-4-乙基氨基苯甲醇5.0 g,收率86%,棕色固体。
另外本发明制备的产物3-硝基-4-乙基氨基苯甲醇的核磁共振氢谱和阳离子液相质谱图分别如图1和图2所示。
以上所述的仅是本发明的优选实施方式,但本发明的保护范围并不局限于此,应当指出,对于本领域的及任何熟悉本技术领域的技术人员来说,在不脱离本发明整体构思前提下,根据本发明的技术方案及其发明构思加以等同替换或改变,及作出的若干改变和改进,这些也应该视为本发明的保护范围。