一种测定肺炎球菌多糖蛋白结合物中游离蛋白及DMAP残留量的方法
文献发布时间:2024-04-18 20:01:23
技术领域
本发明属于生物技术领域,具体涉及一种可以同时检测游离蛋白及结合反应产生的4-二甲氨基吡啶的方法。
背景技术
结合疫苗将该荚膜多糖与蛋白载体通过化学反应制备成多糖蛋白结合物,从而间接使其分子量增大、空间结构复杂,由T细胞非依赖抗原成为T细胞依赖抗原,预防接种人群将会扩大,同时将会大大提高多糖的抗原性,提高免疫保护水平。
但结合物制备过程中,部分载体蛋白未能与多糖进行结合,以蛋白单体或聚体形式存在,这部分载体蛋白称为游离蛋白。而多糖蛋白结合疫苗中游离蛋白含量超过规定限度,会影响疫苗的免疫原性,并增加副反应发生率。WHO推荐在细菌多糖蛋白结合疫苗中游离蛋白含量应小于总蛋白含量的5%。因此,结合疫苗中游离蛋白的含量是疫苗质量的主要参数之一,也是疫苗质量控制的重要指标。
同时,结合物制备过程中,连接多糖与载体蛋白的常用方式之一是异脲键连接反应。1-氰基,4-二甲氨基吡啶四氟硼酸盐(1-cyano-4-dimethylamino-pyridinium,CDAP)是一种可用于异脲键连接反应中多糖活化的试剂,其通过氰根活化多糖链上的邻羟基,生成活化多糖和4-二甲氨基吡啶(DMAP)。
活化机理:
CDAP活化法多应用于脑膜炎球菌结合疫苗、流行性嗜血杆菌结合疫苗、肺炎球菌结合疫苗及A族乙型溶血性链球菌疫苗等多糖-蛋白结合疫苗的工艺。与溴化氰活化法(多糖活化率为12%)比较,使用CDAP多糖的活化率可高达50%,且其毒性较低,更易储存;CDAP虽能在水溶液的环境中活化多糖,但在水溶液中并不稳定,在中性水溶液中数小时即降解,而在碱性水溶液中则会迅速降解,降解产物为DMAP。因此,无论是参与活化反应的CDAP还是过量的CDAP,在结合反应完成后,CDAP均是以其降解产物DMAP的形式存在于结合物中。因此DMAP应纳入相关疫苗的杂质检测中。
游离蛋白最常使用的检测方法为2015版《中国药典》记载的HPLC法;DMAP法多采用以C18色谱柱,乙腈等有机溶剂作为流动相的反相色谱柱;但结合物为水溶性物质,作为样品与有机流动相混合后易出现沉淀,造成色谱柱的不可逆性损坏。因此样品必须超滤离心等预处理才能进样。而且结合物在以乙腈作为流动相的C18反相色谱柱上的峰型不佳,且出现溶剂倒峰,使DMAP峰型不易观察和积分而影响定量结果的准确性。
本发明优化了色谱条件,采用了与样品兼容性更好的空间排阻凝胶色谱柱,可在同一色谱柱上实现了游离蛋白、DMAP的保留和分离,实现同一试验中对游离蛋白与DMAP的双重定量,避免不同类型色谱柱的更换,简化了实验步骤,节省实验耗材、缩短检测周期,可对结合工艺中游离蛋白及DMAP进行有效监测,为结合疫苗的质量控制提供了重要依据。因可实现CDAP与DMAP在色谱柱的保留与分离,结合光谱扫描中的特征性吸收峰,也可判断样品中CDAP的降解程度。
发明内容
针对现有技术上存在的问题,本发明的目的在于提供一种可以同时测定肺炎球菌多糖蛋白结合疫苗中游离蛋白及4-二甲氨基吡啶残留量的方法。
肺炎球菌多糖蛋白结合疫苗的型别组成为但不限于1、2、3、4、5、6A、6B、7F、8、9N、9V、10A、11A、12F、14、15B、17F、18C、19A、19F、20、22F、23F、33F、24F、35B型肺炎球菌荚膜多糖,蛋白为载体蛋白,选自但不限于白喉类毒素、破伤风类毒素。具体技术方案如下:
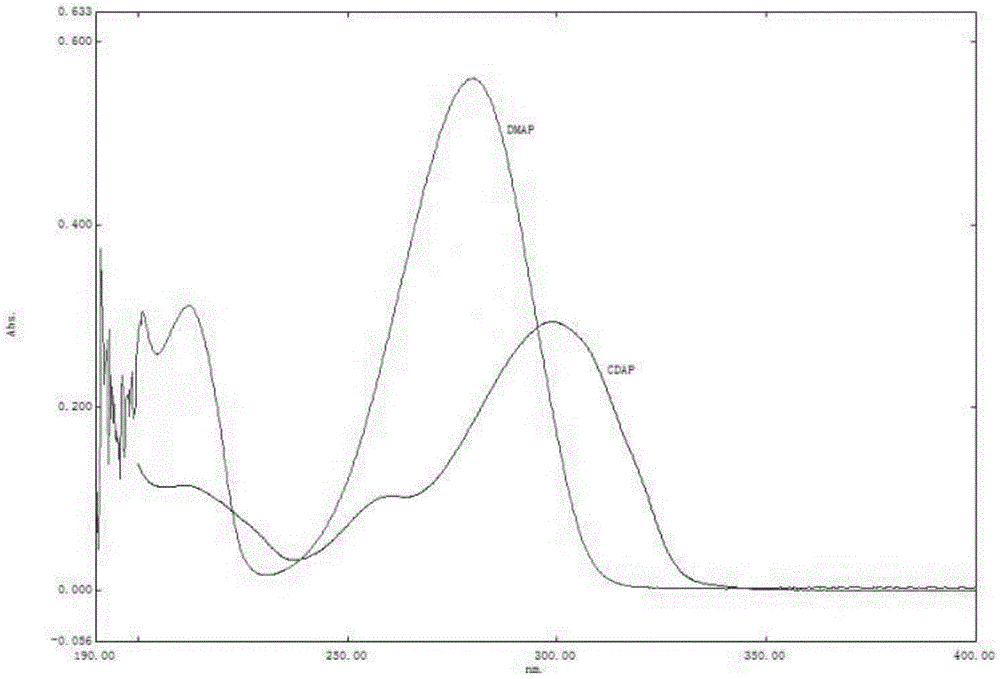
通过查阅文献“中国生物制品学杂志,2021(2):152-160”和对DMAP对照品溶液进行200-400nm的紫外光谱扫描(如图1所示),发现CDAP的特征性吸收峰在300nm,DMAP的特征性吸收峰在280nm,DMAP和蛋白的紫外特征性吸收峰一致,若能在色谱柱上实现DMAP和游离蛋白、DMAP和结合物的保留和分离,即可实现DMAP的定量。参考2015版《中国药典》记载的HPLC法测定结合物中游离蛋白的方法,本发明建立的方法,可实现在结合物、游离蛋白及DMAP在同一色谱柱的保留和分离。
为此,本发明提供一种测定肺炎球菌多糖蛋白结合物中游离蛋白及4-二甲氨基吡啶残留量的方法,所述的检测方法为高效液相色谱法,色谱条件为:保护柱为TSKgelguardcolumn PWXL(6.0mm ID×40mm),分析柱为TSKgel G5000PWXL(7.8mm ID×300mm);流动相为0.85%氯化钠溶液,检测波长280nm,柱温30-40℃,流速0.5ml/min,上样体积为100μl。
本发明所述的方法,包括以下步骤:
1)游离蛋白对照品的配制
游离蛋白对照品为肺炎多糖蛋白结合疫苗的载体蛋白,用0.85%的氯化钠溶液稀释至终浓度为24μg/ml;
2)4-二甲氨基吡啶对照品的配制
准确称取4-二甲氨基吡啶,容量瓶定容配制终浓度为10μg/ml的4-二甲氨基吡啶溶液;
3)供试品溶液的配制
取肺炎球菌多糖蛋白结合物适量,用0.85%的氯化钠溶液稀释至蛋白浓度为200μg/ml;
4)测定
将各游离蛋白对照品工作液,4-二甲氨基吡啶对照品工作液,供试品溶液注入高效液相色谱仪,得到色谱图,根据色谱图的峰面积绘制各标准曲线,标准曲线代入法计算供试品溶液中游离蛋白和4-二甲氨基吡啶的含量。
其中的色谱条件如上所述。
本发明所述的方法,其中,
1)游离蛋白对照品的配制中,游离蛋白对照品为肺炎多糖蛋白结合疫苗的载体蛋白,经发酵、提纯、脱毒而成;
2)供试品溶液的配制,其中,所述肺炎球菌多糖蛋白结合物选自1、2、3、4、5、6A、6B、7F、8、9N、9V、10A、11A、12F、14、15B、17F、18C、19A、19F、20、22F、23F、24F、33F、35B,共26个型的肺炎多糖DT及TT结合物原液。
本发明所述的方法,其中,游离蛋白对照品的配制中,游离蛋白选自:破伤风类毒素TT、白喉类毒素DT。
本发明所述的方法,其中,所述的制备蛋白标准曲线的对照品工作液浓度为24ug/ml,其制备为:根据载体蛋白(白喉类毒素或破伤风类毒素)的蛋白含量测定值,用流动相0.85%的氯化钠溶液系列稀释至终浓度为24μg/ml。
本发明所述的方法,其中,所述的制备4-二甲氨基吡啶标准曲线的对照品工作液浓度为10ug/ml,其制备为:准确称取4-二甲氨基吡啶标准品0.1g,用0.85%的氯化钠溶液溶解并定容至100ml;混匀后取出1ml至100ml容量瓶中用流动相0.85%的氯化钠溶液定容至刻度线,终浓度为10μg/ml。
本发明所述的方法,其中,所述蛋白对照品分别用流动相0.85%的氯化钠溶液稀释至浓度为6μg/ml、9μg/ml、12μg/ml、18μg/ml、24μg/ml以绘制标准曲线。
本发明所述的方法,其中,所述4-二甲氨基吡啶对照品分别用流动相0.85%的氯化钠溶液稀释至浓度为2μg/ml、4μg/ml、6μg/ml、8μg/ml、10μg/ml以绘制标准曲线。
本发明所述的方法,其中,所述计算供试品溶液中游离蛋白和4-二甲氨基吡啶的含量,方法为,以不同浓度的游离蛋白工作液为横坐标,其对应的峰面积为纵坐标绘制标准曲线,将样品的游离蛋白的特征性吸收峰面积代入曲线计算样品的游离蛋白含量;以不同浓度的4-二甲氨基吡啶工作液为横坐标,其对应的峰面积为纵坐标绘制标准曲线,将样品的4-二甲氨基吡啶的特征性吸收峰面积代入曲线计算样品的4-二甲氨基吡啶含量。
本发明所述的方法,其中,本发明可通过光谱扫描图和液相色谱图综合判断CDAP的降解状态。多糖活化过程中,一部分CDAP参与活化反应,转化为4-二甲氨基吡啶;剩余过量的CDAP在碱性条件下2h后,绝大部分也转换成4-二甲氨基吡啶;和文献分析一致;
本发明所述的方法,其中,流动相为0.85%的氯化钠溶液,以配制1000ml总液量为例,配方为:称取8.5g氯化钠,补加纯化水至1000ml。pH 5.5-7.0;
本发明所述的方法,其中,为使蛋白和多糖蛋白结合物这种生物大分子保持原有的空间构像,且能在色谱柱上有效分离,通过实例证明所述pH为6.2±0.2;
本发明所述的方法,其中,柱温30℃、35℃、40℃时,蛋白和多糖蛋白结合物均能得到很好地保留和分离,且结果一致,因柱温较高时会降低色谱柱对结合物的吸附作用,从而有利于维持柱效,延长色谱柱的使用寿命,综合考虑色谱柱的长期稳定性和检测成本,柱温设定为35℃;
本发明和现有技术相比,优点如下:本发明能实现在同一试验中游离蛋白与DMAP的双重定量,简化了实验步骤,节省实验耗材、缩短检测周期,保证了肺炎球菌结合疫苗的有效性和安全性;同时对于DMAP含量的测定方面,与C18反相色谱柱相比,避免了样品遇有机溶剂产生沉淀,影响柱效,从而减少色谱柱使用寿命;由于DMAP是强极性的碱性化合物,在反相色谱模式下保留时间短,且峰型不佳,如图2,本发明方法无溶剂倒峰的出现,目的峰无拖尾,更易观察和积分,还具有灵敏度高、准确性高,重复性好等特点,如图3。以下通过实验数据说明本发明的优点:
C18反相色谱柱法和本发明方法测定3型肺炎多糖蛋白结合物结果比较
与C18反相色谱柱法比较,本方法定量限低,能测定较低浓度的DMAP样品;样品加标回收率更接近100%,说明准确性高;批间精密度的RSD可达到0.06%,说明精密度高;分析原因为C18反相色谱柱法测定样品中的DMAP含量时,由于前后倒锋出现,会干扰DMAP目的峰基线,从而导致积分时面积误差,从而影响结果的准确度及精密度;且单位浓度的DMAP在C18反相色谱柱上的保留峰面积远低于本发明的方法,造成灵敏度低、定量限偏高,对于一些低浓度样品无法检出的结果。
附图说明
图1为DMAP对照品与CDAP对照品的扫描光谱图
图2A,C18反相色谱柱测定2型肺炎球菌多糖蛋白结合物原液色谱图
图2B,C18反相色谱柱测定3型肺炎球菌多糖蛋白结合物原液色谱图
图3A,本发明方法测定2型肺炎球菌多糖蛋白结合物原液色谱图
图3B,本发明方法测定3型肺炎球菌多糖蛋白结合物原液色谱图
图4A,2型肺炎多糖TT结合物原液TT2-20220607中各成分的出峰位置的比较色谱图
图4B,3型肺炎多糖TT结合物原液TT3-20220605中各成分的出峰位置的比较色谱图
图4C,19A型肺炎多糖DT结合物原液DT19A-20200601中各成分的出峰位置的比较色谱图
图4D,19F型肺炎多糖DT结合物原液DT19F-20200701中各成分的出峰位置的比较色谱图
图5A,室温溶解的CDAP(溶解后立即进样检测)的液相色谱峰图
图5B,室温溶解2小时后的CDAP(溶解后2h后进样检测)的液相色谱峰图
具体实施方式
下面结合实施例对本发明提供的用高效液相色谱法同时测定肺炎球菌多糖蛋白结合物中游离蛋白及4-二甲氨基吡啶残留量的方法作进一步详细、完整地说明。下面描述的试验例和实施例是示例性的,仅用于解释本发明,但不作为本发明的限制。
1、仪器和试剂
Waters 2489UV/可见光检测器、Alliance e2695型高效液相色谱仪、岛津UV-1800紫外分光光度计、液相色谱柱TSKgel G5000PWXL、保护柱TSKgel guardcolumnPWXL、Millex针头式过滤器、过滤装置、滤膜、十万级电子天平(Sartorius)。
DMAP(Sigma,纯度≥99%),CDAP(Sigma,纯度≥98%)、破伤风类毒素(TT)、白喉类毒素(DT)、氯化钠(国药,分析纯)、乙腈(Sigma,纯度≥99.9%)、三乙胺(Sigma,纯度≥99.5%)、甘氨酸(Sigma,纯度≥99%)
2、样品
2.1蛋白对照品的配制
本发明所采用的蛋白对照品为肺炎多糖蛋白结合疫苗的载体蛋白(破伤风类毒素TT、白喉类毒素DT),均经发酵、提纯、脱毒而成,为企业自制;根据其蛋白浓度,用流动相0.85%的氯化钠溶液系列稀释至终浓度为24μg/ml。
2.2DMAP对照品的配制
准确称取DMAP 0.1g,用0.85%的氯化钠溶液溶解并定容至100ml;混匀后取出1ml至100ml容量瓶中用流动相0.85%的氯化钠溶液定容至刻度线,终浓度为10μg/ml。
2.3其他试剂的配制
2.3.1 0.375%乙腈溶液:量取0.375ml乙腈溶液,加纯化水至100ml。
2.3.2 0.03%三乙胺溶液:量取0.03ml三乙胺溶液,加纯化水至100ml。
2.3.3 7.5%甘氨酸溶液:准确称取甘氨酸0.75g,溶解于10ml纯化水中。
2.3.4 40μg/ml CDAP对照品溶液:准确称取CDAP 400μg,溶解于10ml纯化水中。
2.4待检样品
本发明以2型、3型、6A型、14型、33F型、35B型肺炎多糖TT结合物原液、19A型、19F型肺炎多糖DT结合物原液为例,但不限于这8个型别,也不限于多糖蛋白结合物载体蛋白的类型;据结合物的蛋白浓度用流动相0.85%的氯化钠溶液稀释至蛋白浓度为200μg/ml;
3、光谱扫描条件:以0.85%的氯化钠溶液为空白,进行基线扫描;紫外光谱扫描波长范围200-400nm,扫描速度:中速;采样间隔1nm;
4、色谱条件:色谱柱为TSKgel G5000PWXL分析柱,保护柱TSKgelguardcolumnPWXL,流动相为0.85%氯化钠溶液,pH6.2±0.2,检测波长280nm,流速0.5ml/min,柱温35℃;
5、专属性
分别取2.4中所述的4种预稀释的结合物原液和载体蛋白破伤风类毒素(TT)、白喉类毒素(DT)、DMAP对照品及样品中可能含有的残留溶剂乙腈、三乙胺、甘氨酸,分别作为进样样品,使用100μl上样环上样,色谱条件参考上述4所述。根据出峰位置及分离度,如表1及图4,残留溶剂及结合物原液在目的峰游离蛋白(DT或TT)及DMAP相应位置处均无色谱峰,不干扰游离蛋白(DT或TT)及DMAP的测定;游离蛋白(DT或TT)峰及DMAP峰与相邻峰的分离度均大于1.5,且理论塔板数均大于3000,可判断该方法能特征性检出游离蛋白(DT或TT)及DMAP;
表1结合物原液中各成分的出峰范围及与邻近峰的分离度
注:试剂峰为乙腈、三乙胺、甘氨酸所出的非特征性吸收峰。
6、定量限
取已知浓度的载体蛋白TT与DT,及DMAP对照品,分别用0.85%氯化钠溶液逐级稀释,均进样检测,当信噪比接近10时对应的浓度,即为该方法的定量限。结果表明,浓度分别为0.5μg/ml的TT的信噪比为10.5,1.2μg/ml的DT的信噪比为15,0.02μg/ml的DMAP的信噪比为20,则该方法TT的定量限为0.5μg/ml、DT的定量限为1.2μg/ml、DMAP的定量限为0.02μg/ml。
7、准确性
7.1载体蛋白TT与DT的准确性检测
取载体蛋白为TT的2型、3型肺炎多糖蛋白(TT)结合物原液及载体蛋白为DT的19A型、19F型肺炎多糖蛋白(DT)结合物原液为供试品,分别向其中加入与其载体蛋白相对应的、不同浓度的已知量的TT或DT对照品,每个加标浓度水平平行测定3次。通过测定加标前后供试品中载体蛋白的含有量、比较加标前后的差值与对照品加入量,计算加标回收率及RSD。从表2中数据可知,TT的加标回收率在98.11%~104.60%,从表3中数据可知,DT的加标回收率在95.43%~103.26%,均符合《中国药典》(2020年版)中9101药品质量标准分析方法验证指导原则的要求,表明该方法对于检测游离蛋白(TT和DT)残量有很好的准确性。
表2供试品的TT加标回收率结果
表3供试品的DT加标回收率结果
7.2DMAP的准确性检测
取载体蛋白为TT的3型肺炎多糖蛋白(TT)结合物原液及载体蛋白为DT的19A型肺炎多糖蛋白(DT)结合物原液为供试品,分别向其中加入3种不同浓度的已知量的DMAP对照品,每个加标浓度水平平行测定3次。通过测定加标前后供试品中DMAP的含有量、比较加标前后的差值与对照品加入量,计算加标回收率及RSD。从表4中数据可知,DMAP的加标回收率在97.11%~98.42%,符合《中国药典》(2020年版)中9101药品质量标准分析方法验证指导原则的要求,表明该方法对于检测DMAP残量有很好的准确性。
表4供试品的DMAP加标回收率结果
8、精密度
8.1载体蛋白TT与DT的精密度检测
分别取载体蛋白为TT的6A型肺炎多糖蛋白(TT)结合物原液、2型肺炎多糖蛋白(TT)结合物原液及载体蛋白为DT的19A型肺炎多糖蛋白(DT)结合物原液、19F型肺炎多糖蛋白(DT)结合物原液为供试品,由3名试验员于不同时间内各测定一次,每次试验各样品平行测定6次;以6次测定结果的相对标准偏差参考方法的重复性;以不同天、不同实验员平行测定18次,计算18次测定结果的相对标准偏差,考察方法的中间精密度。结果(表5、表6)表明,同一实验员平行6次测定载体蛋白TT的RSD值为1.02%~1.74%,不同实验员平行测定18次结果的RSD值为2.69%、1.33%;同一实验员平行6次测定载体蛋白DT的RSD值为1.40%~1.87%,不同实验员平行测定18次结果的RSD值为1.87%、2.49%;
表5供试品的TT重复性和中间精密度结果
/>
表6供试品的DT重复性和中间精密度结果
/>
8.2DMAP的精密度检测
取载体蛋白为TT的3型肺炎多糖蛋白(TT)结合物原液及载体蛋白为DT的19A型肺炎多糖蛋白(DT)结合物原液为供试品,由3名试验员于不同时间内各测定一次,每次试验各样品平行测定6次;以6次测定结果的相对标准偏差参考方法的重复性;以不同天、不同实验员平行测定18次,计算18次测定结果的相对标准偏差,考察方法的中间精密度。表7结果表明,同一实验员平行6次测定的RSD值为0.05%~0.50%,不同实验员平行测定18次结果的RSD值为0.83%、3.17%。
表7供试品的DMAP重复性和中间精密度结果
9、线性和范围
9.1载体蛋白DT与TT的线性和范围
将浓度分别为6μg/ml、9μg/ml、12μg/ml、18μg/ml、24μg/ml的DT与TT对照品工作液分别进样,以浓度为横坐标,峰面积为纵坐标绘制标准曲线,得出DT的标准曲线Y=3857.4X-174.8,相关系数R
9.2DMAP的线性和范围
将浓度分别为2μg/ml、4μg/ml、6μg/ml、8μg/ml、10μg/ml的DMAP对照品工作液分别进样,以浓度为横坐标,峰面积为纵坐标绘制标准曲线,得出DMAP的标准曲线Y=363108X-31426,相关系数R
10、耐用性
10.1载体蛋白DT与TT的耐用性
柱温为30、35、40℃条件下,分别以19A、19F型肺炎多糖蛋白(DT)结合物原液、35B、33F型肺炎多糖蛋白(TT)结合物原液为供试品,分别进样,代入相应载体蛋白的标准曲线,计算不同温度下各原液载体蛋白含量的RSD。表8结果表明,YTT35B-20211104、YTT33F-20211104批原液在进样温度为30、35、40℃的检测结果的RSD分别为2.33%、2.42%;表明该方法在30~40℃的温度变化对TT含量检测无显著影响。DT19A-20220601、DT19F-20200701批原液在进样温度为30、35、40℃的检测结果的RSD分别为1.48%、2.55%,表明该方法在30~40℃的温度变化对DT含量检测无显著影响。
表8.载体蛋白测定的耐用性结果
10.2DMAP的耐用性
柱温为30、35、40℃条件下,分别以19A、19F型肺炎多糖蛋白(DT)结合物原液、35B、33F型肺炎多糖蛋白(TT)结合物原液为供试品,分别进样,代入DMAP的标准曲线,计算不同温度下各原液DMAP含量的RSD。表9结果表明,各型原液在进样温度为30、35、40℃的检测结果的RSD为0.49%~2.94%;表明该方法在30~40℃的温度变化对DMAP含量检测无显著影响。
表9.DMAP测定的耐用性结果
实施例1、色谱柱的选择
(1)供试品及对照品工作液的制备:供试品19A型肺炎多糖蛋白(TT)结合物原液按其蛋白浓度用流动相0.85%的氯化钠溶液稀释至蛋白浓度为200μg/ml;取上述同一供试品添加TT蛋白对照品溶液和DMAP对照品溶液,用流动相稀释至结合物的蛋白浓度为200μg/ml,添加的TT和DMAP终浓度分别为10μg/ml、1μg/ml;10μg/ml TT蛋白对照品溶液;1μg/mlDMAP对照品溶液;上述供试品和对照品溶液分别进行进样,每个样品进样2次,上样体积100ul;
(2)色谱条件:色谱柱分别为TSKgel G5000PW
(3)结果分析:比较不同色谱柱对同一样品TT、DMAP及19A型肺炎多糖蛋白(TT)结合物原液的分离效果(分离度)、峰型(对称性)、信噪比S/N及加标回收率,筛选最适合色谱柱;结果可见,TSKgel G3000SW
Ultrahydrogel 2000分析柱可进行结合物与游离蛋白TT的分离,TT和DMAP分离度分别为2.0和3.3,游离蛋白TT和DMAP的峰型均佳,对称性为1.25和1.20;TT与DMAP峰的S/N分别为27.7和2050.3;但TT的回收率仅为46.0%,远低于回收率标准范围90~108%;
TSKgel G5000PW
实施例2、流动相的选择
(1)供试品及对照品工作液的制备:供试品19A型肺炎多糖蛋白(TT)结合物原液按其蛋白浓度分别用三种流动相0.85%的氯化钠溶液、0.1MPBS溶液、50mmoL Tris-HCl溶液稀释至蛋白浓度为200μg/ml;取上述同一供试品添加TT蛋白对照品溶液和DMAP对照品溶液,用三种流动相稀释至结合物的蛋白浓度为200μg/ml,添加的TT和DMAP终浓度分别为10μg/ml、1μg/ml;用三种流动相分别稀释至终浓度为10μg/ml TT蛋白对照品溶液和1μg/mlDMAP对照品溶液;上述供试品和对照品溶液分别进行进样,每个样品进样2次,上样体积100ul;
(2)色谱条件:色谱柱为TSKgel G5000PWXL分析柱,流动相为0.85%氯化钠溶液、0.1moLPBS溶液、50mmoL Tris-HCl溶液,检测波长280nm,流速0.5ml/min,柱温35℃;
(3)结果分析:比较不同流动相对同一样品TT、DMAP及19A型肺炎多糖蛋白(TT)结合物原液的分离效果(分离度)、峰型(对称性)、信噪比S/N及加标回收率,筛选最适合流动相;结果可见Tris-HCl溶液可进行结合物与游离蛋白TT的分离,分离度为2.9,游离蛋白TT峰对称性1.31,S/N为463.6,回收率为104.6%;但该色谱柱未能实现DMAP的保留,未出现DMAP的特征性吸收峰;
0.1moL PBS溶液可进行结合物与游离蛋白TT的分离,TT和DMAP分离度分别为2.3和3.7,游离蛋白TT和DMAP的峰对称性为0.98和1.42;TT与DMAP峰的S/N分别为48.0和1371.2;但TT的回收率为85.4%,DMAP的回收率为106.2%,TT的回收率不符合回收率标准范围90~108%;。
0.85%氯化钠溶液可进行结合物与游离蛋白TT的分离,分离度为2.1,游离蛋白TT和DMAP的峰对称性为0.99和1.43;TT峰面积为71645,S/N为67.6,TT回收率为100.6%,可实现TT的完全回收;DMAP分离度为3.2,峰面积为1592713,S/N为1732.5,DMAP回收率为98.0%,可实现DMAP的良好回收;为了实现游离蛋白及DMAP的双重定量,综合游离蛋白及DMAP的分离效果,因此选择0.85%溶液作为流动相。
实施例3、2型肺炎多糖蛋白(TT)结合物原液、3型肺炎多糖蛋白(TT)结合物原液的游离蛋白与DMAP含量的测定
(1)供试品及标准曲线的制备:供试品2型肺炎多糖蛋白(TT)结合物原液、3型肺炎多糖蛋白(TT)结合物原液按其蛋白浓度分别用流动相0.85%的氯化钠溶液稀释至蛋白浓度为200μg/ml;TT蛋白对照品溶液分别用流动相稀释至浓度为6μg/ml、9μg/ml、12μg/ml、18μg/ml、24μg/ml五个浓度梯度;DMAP对照品溶液分别用流动相稀释至2μg/ml、4μg/ml、6μg/ml、8μg/ml、10μg/ml五个浓度梯度;上述供试品和对照品溶液分别进行进样,每个样品平行进样2次,上样体积100ul;
(2)色谱条件:色谱柱为TSKgel G5000PWXL分析柱,流动相为0.85%氯化钠溶液,pH6.2±0.2,检测波长280nm,流速0.5ml/min,柱温35℃;
(3)供试品的载体蛋白TT及DMAP含量的计算:记录各标准曲线点及供试品的色谱图,对特征性峰面积进行积分,计算供试品的游离蛋白及DMAP的含量。以不同浓度(6μg/ml、9μg/ml、12μg/ml、18μg/ml、24μg/ml)的TT为横坐标,其对应的峰面积为纵坐标绘制TT标准曲线,将样品的游离蛋白的特征性吸收峰面积代入曲线计算样品的游离蛋白含量。以不同浓度(2μg/ml、4μg/ml、6μg/ml、8μg/ml、10μg/ml)的DMAP为横坐标,其对应的峰面积为纵坐标绘制标准曲线,将样品的DMAP的特征性吸收峰面积代入曲线计算样品的DMAP含量。本次试验的TT的标准曲线Y=10268X-27598,2型肺炎多糖蛋白(TT)结合物原液、3型肺炎多糖蛋白(TT)结合物原液的载体蛋白TT的积分面积分别为29615μV·s、19881μV·s,代入曲线方程,求得游离蛋白TT的含量分别为7.572μg/ml、4.624μg/ml;DMAP的标准曲线Y=363108X-31426,2型肺炎多糖蛋白(TT)结合物原液、3型肺炎多糖蛋白(TT)结合物原液的DMAP的积分面积分别为637418μV·s、1942065μV·s,代入曲线方程,求得供试品的DMAP含量分别为1.942μg/ml、5.435μg/ml;
实施例4、19A型肺炎多糖蛋白(DT)结合物原液、19F型肺炎多糖蛋白(DT)结合物原液的游离蛋白与DMAP含量的测定
(1)供试品及标准曲线的制备:供试品19A型肺炎多糖蛋白(DT)结合物原液、19F型肺炎多糖蛋白(DT)结合物原液按其蛋白浓度分别用流动相0.85%的氯化钠溶液稀释至蛋白浓度为200μg/ml;DT蛋白对照品溶液分别用流动相稀释至浓度为6μg/ml、9μg/ml、12μg/ml、18μg/ml、24μg/ml五个浓度梯度;DMAP对照品溶液分别用流动相稀释至2μg/ml、4μg/ml、6μg/ml、8μg/ml、10μg/ml五个浓度梯度;上述供试品和对照品溶液分别进行进样,每个样品平行进样2次,上样体积100ul;
(2)色谱条件:色谱柱为TSKgel G5000PWXL分析柱,流动相为0.85%氯化钠溶液,pH6.2±0.2,检测波长280nm,流速0.5ml/min,柱温35℃;
(3)供试品的载体蛋白DT及DMAP含量的计算:记录各标准曲线点及供试品的色谱图,对特征性峰面积进行积分,计算供试品的游离蛋白及DMAP的含量。以不同浓度(6μg/ml、9μg/ml、12μg/ml、18μg/ml、24μg/ml)的DT为横坐标,其对应的峰面积为纵坐标绘制DT标准曲线,将样品的游离蛋白的特征性吸收峰面积代入曲线计算样品的游离蛋白含量。以不同浓度(2μg/ml、4μg/ml、6μg/ml、8μg/ml、10μg/ml)的DMAP为横坐标,其对应的峰面积为纵坐标绘制标准曲线,将样品的DMAP的特征性吸收峰面积代入曲线计算样品的DMAP含量。本次试验的DT的标准曲线Y=3857.4X-174.8,19A型肺炎多糖蛋白(DT)结合物原液、19F型肺炎多糖蛋白(DT)结合物原液的载体蛋白DT的积分面积分别为6328μV·s、15008μV·s,代入曲线方程,求得游离蛋白DT的含量分别为5.686μg/ml、3.936μg/ml;DMAP的标准曲线Y=359934x-36114R
实施例3、CDAP在乙腈溶液和PH9.6氯化钠溶液中降解的研究
(1)供试品的制备:分别称取CDAP 0.0407g、0.0398g,分别用0.375%乙腈与PH9.6的0.85%氯化钠溶液溶解并稀释至终浓度为40μg/ml;称取DMAP0.0405g,用0.85%氯化钠溶液溶解并稀释至终浓度为40μg/ml;上述供试品溶解后立即进样,室温静置2小时后再次进样,上样体积均为100ul;
(2)光谱扫描条件:以0.85%的氯化钠溶液和0.375%乙腈分别作为空白,进行基线扫描;紫外光谱扫描波长范围200-400nm,扫描速度:中速;采样间隔1nm;
(3)色谱条件:色谱柱为TSKgel G5000PWXL分析柱,流动相为0.85%氯化钠溶液,pH6.2±0.2,检测波长280nm,流速0.5ml/min,柱温35℃;
(4)CDAP在乙腈溶液和PH9.6氯化钠溶液中降解分析:乙腈溶解的CDAP溶液与0.85%氯化钠溶液溶解CDAP溶液,溶解后立刻进行光谱扫描与液相色谱分析,光谱扫描结果均显示在300nm有最大吸收峰;液相色谱分析显示乙腈溶解的CDAP在25.4min出现特征性吸收峰;0.85%氯化钠溶液溶解CDAP显示有两个未完全分离的吸收峰如图5A,因在液相色谱中运行一次单一样品的时间约40min,提示出峰时0.85%氯化钠溶液溶解CDAP已有部分降解。室温放置2小时后,再进行光谱扫描与液相色谱分析,光谱扫描结果显示乙腈溶解的CDAP溶液仍在300nm有最大吸收峰,0.85%氯化钠溶液溶解CDAP溶液的最大吸收峰由300nm移至280nm;液相色谱分析显示乙腈溶解的CDAP溶液仍在25.4min出现特征性吸收峰,而0.85%氯化钠溶液溶解CDAP溶液的特征性吸收峰由25.4min移至26.6min,且与DMAP的特征性吸收峰的保留时间一致,如图5B,提示CDAP在乙腈溶液中较稳定,室温放置2小时后仍未出现降解;而CDAP在PH9.6的0.85%氯化钠溶液中不稳定,室温放置2小时后可完全降解为DMAP,结合文献“中国生物制品学杂志,2021(2):152-160”中对CDAP及DMAP的核磁氢谱及碳谱的研究,与该文献所得结论一致。
最后有必要再次说明的是:以上实施例只用于对本发明的技术方案作进一步详细的说明,不能理解为对本发明保护范围的限制,本领域的技术人员根据本发明的上述内容作出一些非本质的改进和调整均属于本发明的保护范围。
- 一种物料传递机构、传递方法和传递机器人
- 动力传递机构的管理装置和动力传递机构的管理方法