一种4-溴-6-氯烟酸的制备方法
文献发布时间:2023-06-19 19:30:30
技术领域
本发明涉及医药、农药中间体的合成领域,尤其是涉及一种4-溴-6-氯烟酸的制备方法。
背景技术
4-溴-6-氯烟酸是一种重要的医药、农药中间体。原有合成的方法是以4-溴-2-氯-3-(三甲基硅烷基)-吡啶为原料,用二氧化碳在深冷条件下上羧基,然后,脱三甲基硅烷得到4-溴-6-氯烟酸(WO2014074675A1)。此方法缺点是:原料来源不易得到,价格高;且须无水无氧和超低温等比较苛刻条件;由于原料价格高,两步总收率也不高仅为60%,操作条件苛刻等因素该路线并不实用,很难实现规模化生产。其合成路线如下所示:
上述工艺存在起始原料价格昂贵,工艺过程操作复杂等原因,而导致工业化成本高,不适合规模化生产的问题。
发明内容
本发明所要解决的技术问题是提供一种工艺线路简单,反应条件温和、安全,生产成本较低且总收率高的4-溴-6-氯烟酸的制备方法。
本发明解决上述技术问题所采用的技术方案为:一种4-溴-6-氯烟酸的制备方法,包括以下步骤:
(1)将2-氯-4-氨基-5-甲基吡啶溶于稀酸中,然后加入催化剂和溴化物,滴加亚硝酸钠水溶液进行重氮化上溴,得到2-氯-4-溴-5-甲基吡啶;
(2)将2-氯-4-溴-5-甲基吡啶和氧化试剂反应得到4-溴-6-氯烟酸。
进一步,步骤(1)具体为:将2-氯-4-氨基-5-甲基吡啶溶于稀酸中,搅拌溶清后,降温到T1-30-100℃,加入催化剂和溴化物,搅拌半小时,降温到T2-30-40℃,然后缓慢滴加亚硝酸钠水溶液,控制温度不高于T2,滴完后,保持温度在T2下再反应一小时;反应完全后,反应液用甲基叔丁基醚萃取三次,合并有机相,用饱和碳酸氢钠水溶液洗,饱和食盐水再清洗,活性炭脱色,无水硫酸钠干燥,过滤,滤饼用甲基叔丁基醚淋洗两次合并有机相,浓缩至干,得到淡黄色油状物2-氯-4-溴-5-甲基吡啶。
进一步,所述的稀酸为稀硫酸、稀氢溴酸、稀硝酸或稀磷酸;所述的催化剂为硫酸铜,所述的溴化物为溴化钠或溴化钾。
进一步,所述的2-氯-4-氨基-5-甲基吡啶、所述的催化剂、所述的溴化物与所述的亚硝酸钠的物质的量比为1:(1-3):(2-5):(2-5)。
进一步,所述的2-氯-4-氨基-5-甲基吡啶、所述的催化剂、所述的溴化物与所述的亚硝酸钠的物质的量比为1:2:2.5:3。
进一步,所述的T1的温度为20℃;T2的温度为-5℃-0℃。
进一步,步骤(2)具体为:将2-氯-4-溴-5-甲基吡啶加入含有溶剂的反应瓶中,控制温度10-180℃下加入或通入氧化剂后,控制温度为20-100℃直至反应完全,得到4-溴-6-氯烟酸,其中所述的2-氯-4-溴-5-甲基吡啶与所述的氧化剂的物质的量比为1:(1-6)。
进一步,所述的氧化剂为氧气、氯气、重铬酸钠、重铬酸钾、高锰酸钾或硝酸。
进一步,所述的溶剂根据不同的氧化剂选用不同的溶剂:所述的氧化剂为高锰酸钾,选用水为溶剂;所述的氧化剂为氧气,选用甲醇、乙醇或异丙醇为溶剂;所述的氧化剂为重重铬酸钠或重铬酸钾,选用硫酸为其溶剂。
当氧化剂为重重铬酸钠或重铬酸钾时,选用硫酸为其溶剂;上述步骤(2)反应完全后,将得到的反应液缓慢倒入到冰块中,搅拌半小时,有固体析出,过滤,滤饼用水淋洗至中性,得到类白色固体4-溴-6-氯烟酸(重铬酸钠或重铬酸钾氧化法);
当氧化剂为高锰酸钾时,选用水为其溶剂;上述步骤(2)反应完全后,将反应液冷却至30℃,过滤,滤饼用冷水洗淋,得到滤液和淋洗液,用6N的盐酸调pH至3,形成沉淀,过滤,得到的固体水洗,烘干,得到类白色固体4-溴-6-氯烟酸(高锰酸钾氧化法);
当氧化剂为氧气时,选用甲醇、乙醇或异丙醇为溶剂;上述步骤(2)反应完全后,过滤,将得到的反应混合液减压蒸去溶剂,残余物加入冷水,搅拌,用10wt%的盐酸调pH值至3,析出大量白色固体,过滤,得到的固体水洗,烘干,得到类白色固体4-溴-6-氯烟酸(空气/氧气氧化法)。
与现有技术相比,本发明的优点在于:本发明一种4-溴-6-氯烟酸的制备方法,以价格低廉的2-氯-4-氨基-5-甲基吡啶(它是合成奥美拉唑的关键中间体(见申请公开号CN103193704A),已规模化生产,其来源广泛成本低)为原料,经过重氮上溴和甲基氧化两步反应得到目标产物。本合成路线原料来源广泛且价格低廉、工艺简单、反应条件温和,总收率高达82%,适合规模化大生产,具有广阔的应用前景。
附图说明
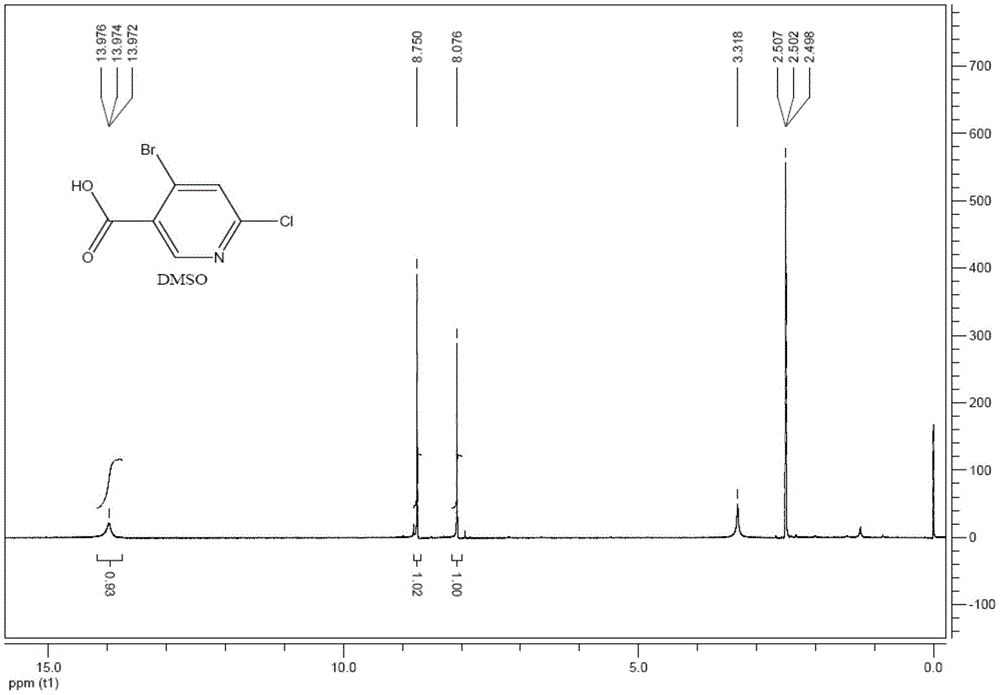
图1为本发明制备的4-溴-6-氯烟酸的核磁共振图谱。
具体实施方式
以下结合附图实施例对本发明作进一步详细描述。
具体实施例
一种4-溴-6-氯烟酸的制备方法,包括以下步骤:
1、将2-氯-4-氨基-5-甲基吡啶溶于稀酸中,然后加入催化剂和溴化物,滴加亚硝烟钠水溶液进行重氮化上溴,得到2-氯-4-溴-5-甲基吡啶;
2、将2-氯-4-溴-5-甲基吡啶和氧化试剂反应得到4-溴-6-氯烟酸,其合成路线如下:
上述步骤(1)具体为:将2-氯-4-氨基-5-甲基吡啶溶于稀酸中,搅拌溶清后,降温到T1-30-100℃,加入催化剂和溴化物,搅拌半小时,降温到T2-30-40℃,然后缓慢滴加亚硝酸钠水溶液,控制温度不高于T2,滴完后,保持温度在T2下再反应一小时;反应完全后,反应液用甲基叔丁基醚萃取三次,合并有机相,用饱和碳酸氢钠水溶液洗,饱和食盐水再清洗,活性炭脱色,无水硫酸钠干燥,过滤,滤饼用甲基叔丁基醚淋洗两次合并有机相,浓缩至干,得到淡黄色油状物2-氯-4-溴-5-甲基吡啶。其中稀酸为稀硫酸、稀氢溴酸、稀硝酸或稀磷酸;催化剂为硫酸铜,溴化物为溴化钠或溴化钾;2-氯-4-氨基-5-甲基吡啶、;催化剂、;溴化物与;亚硝酸钠的物质的量比为1:(1-3):(2-5):(2-5)。
上述步骤(2)具体为:将2-氯-4-溴-5-甲基吡啶加入含有溶剂的反应瓶中,控制温度10-180℃下加入或通入氧化剂后,控制温度为20-100℃直至反应完全,得到4-溴-6-氯烟酸,其中2-氯-4-溴-5-甲基吡啶与氧化剂的物质的量比为1:(1-3)。氧化剂为氧气、氯气、重铬酸钠、重铬酸钾、高锰酸钾或硝酸。
当氧化剂为重重铬酸钠或重铬酸钾时,选用硫酸为其溶剂;上述步骤(2)反应完全后,将得到的反应液缓慢倒入到冰块中,搅拌半小时,有固体析出,过滤,滤饼用水淋洗至中性,得到类白色固体4-溴-6-氯烟酸(重铬酸钠或重铬酸钾氧化法);
当氧化剂为高锰酸钾时,选用水为其溶剂;上述步骤(2)反应完全后,将反应液冷却至30℃,过滤,滤饼用冷水洗淋,得到滤液和淋洗液,用6N的盐酸调pH至3,形成沉淀,过滤,得到的固体水洗,烘干,得到类白色固体4-溴-6-氯烟酸(高锰酸钾氧化法);
当氧化剂为氧气时,选用甲醇、乙醇或异丙醇为溶剂;上述步骤(2)反应完全后,过滤,将得到的反应混合液减压蒸去溶剂,残余物加入冷水,搅拌,用10wt%的盐酸调pH值至3,析出大量白色固体,过滤,得到的固体水洗,烘干,得到类白色固体4-溴-6-氯烟酸(空气/氧气氧化法)。
实施例1
1、2-氯-4-溴-5-甲基吡啶的合成
准备10L三口瓶,机械搅拌,温度计,滴液漏斗。先配好硫酸水溶液(548.8g,5.6mol,水4L),再将其加入到10L反应瓶中,开启搅拌,冰盐浴,加入原料2-氯-4-氨基-5-甲基吡啶(290g,2.0mol)搅拌溶清,然后在T1<20℃时加入五水硫酸铜(1004g,4mol),溴化钠(515g,5mol),搅拌半小时,在T2<0℃,滴加亚硝酸钠水溶液(441.6g,6.4mol,水1L),控制T2<0℃,体系放热明显,缓慢滴加,约两小时滴完;滴完,保温反应一小时。取样TLC(EA:PE=1:1),反应完全后,将反应液用甲基叔丁基醚萃取三次(1.5L×3),合并有机相,用饱和碳酸氢钠水溶液洗有机相一次(1L×1),饱和食盐水洗一次(1L×1),活性炭脱色,无水硫酸钠干燥,过滤,滤饼用甲基叔丁基醚淋洗两次(500ml×2),合并有机相,浓缩至干,得到380g淡黄色油状物,y=92%,纯度为97%。
实施例2-6与实施例1的区别在于,2-氯-4-氨基-5-甲基吡啶,硫酸铜,溴化钠,亚硝酸钠的物质的量不改变,仅改变反应温度,制备的2-氯-4-溴-5-甲基吡啶的具体反应条件,收率及纯度如下表1所示。
表1反应温度对产物(2-氯-4-溴-5-甲基吡啶)收率及产物纯度的影响
由上述表1可知,实施例3-6与实施例1不同之处在于,在其它条件都不变,反应温度增加,收率和纯度都在下降,温度的增加会产生越来越多的2-氯-4-羟基-5-甲基吡啶、2,4-二溴-5-甲基吡啶和2-溴-4-氯-5-甲基吡啶等副产物,所以温度越高产物收率和纯度会下降得更快。
虽然实施2与实施例1的收率和纯度相差不多,但从经济效益考虑,选择施例1的反应温度较为经济,它为较佳的反应温度。因此,T1的温度最佳为20℃;T2的温度最佳为-5℃-0℃。
表2反应物的摩尔比例不同对产物(2-氯-4-溴-5-甲基吡啶)收率及产物纯度的影响
由上述表2可知,实施例7-15与实施例1不同之处在于,硫酸铜和溴化钠的当量比不同,当硫酸铜和溴化钠的反应当量由1eq增加至3eq,收率由40%上升至90%以上,纯度也由55.7%上升至95.4%以上;当硫酸铜反应当量增加至5eq,对收率和纯度影响不大,即适量的催化剂(硫酸铜)参与反应有助于反应正向进行;适量的催化剂参与下,溴化钠的反应当量由1eq增加至5eq,收率由40%上升至94%,即适当过量的溴化试剂是有助于反应正向进行,但当反应当量超过2.5eq后,再增大溴化钠的量时,产物纯度是在不断下降,可见过量的溴化试剂在有助于反应正向进行同时,副产物增长也明显;2.5eq是收率和纯度最佳的一个平衡点。
2、4-溴-6-氯烟酸的合成
重铬酸钠氧化
准备5L三口瓶,机械搅拌,温度计,冰水浴,先向反应瓶中加入2L浓硫酸,开启搅拌,冰水浴,T<5℃以下,加入上步得到的中间体2-氯-4-溴-5-甲基吡啶(206.5g,1mol),放热至10℃,体系呈淡黄色澄清液;分批加入重铬酸钠(447g,1.5mol),控制T<40℃,约3小时加完,加毕,搅拌一小时;取样TLC(EA:PE=1:3,UV),产物极性变大很多,取样后,样品加入到冰水中淬灭再检测。反应完毕,将反应液缓慢倒入到8kg冰块中,搅拌半小时,有固体析出,过滤,滤饼用水淋洗至中性,得到类白色固体4-溴-6-氯烟酸210g,y=89%。,其核磁共振图谱如图1所示。
实施例16
同上述实施例1,其区别在于4-溴-6-氯烟酸的合成采用高锰酸钾氧化,具体步骤如下:准备5L三口瓶,机械搅拌,温度计,油浴锅,先向反应瓶中加入3L冷水,开启搅拌,加入上步得到的中间体2-氯-4-溴-5-甲基吡啶(150g,0.73mol),升温至80℃,温度稳定后,分批加入高锰酸钾(500g,3.2mol),控制温度不高95℃,约3小时加完,加毕,95℃保温过夜反应;次日,冰水浴冷却至30℃后过滤,取滤液,滤饼二氧化锰用500mL冷水洗淋,将得到的滤液和淋洗液混合后用6N的盐酸调pH至3,形成沉淀,过滤,得到的固体水洗,烘干,得到类白色固体4-溴-6-氯烟酸70g,y=40.6%。
实施例17
同上述实施例1,其区别在于4-溴-6-氯烟酸的合成采用氧气氧化,具体步骤如下:2L压力釜中加入1.2L甲醇和上步得到的中间体2-氯-4-溴-5-甲基吡啶(150g,0.73mol),乙酸钴0.02g,乙酰丙酮钴0.02g,碳酸铯15g,氮气置换2次釜内的空气,搅拌,通入氧气反应,压力0.5MPa,温度80℃下,保温反应20小时。反应完毕,过滤,得到的反应混合液减压蒸去溶剂,残余物加入800mL水,搅拌,用10wt%的盐酸调pH值至3,析出大量白色固体,过滤,得到的固体水洗,烘干,得到类白色固体4-溴-6-氯烟酸138g,y=80%。
上述说明并非对本发明的限制,本发明也并不限于上述举例。本技术领域的普通技术人员在本发明的实质范围内,做出的变化、改型、添加或替换,也应属于本发明的保护范围。