一种检测乳或乳制品中致香成分的方法
文献发布时间:2023-06-19 11:57:35
技术领域
本发明涉及检测技术领域,具体为一种检测乳或乳制品中致香成分的方法。
背景技术
食品中风味成分分析的手段主要有气相色谱法(简写为GC)、超高效液相色谱法(简写为UPLC)、合相色谱法(简写为CC)、气相色谱-质谱联用法(简写为GC-MS)、超高效液相色谱-质谱联用法(简写为UPLC-MS)、合相色谱-质谱联用法(简写为CC-MS)、电子鼻法(简写为EN)等。而物质的捕集是研究基质中挥发性成分的第一步,对研究结果中成分的比例以及定量限有关键性影响。
食品中挥发性成分提取与富集的前处理方式主要有液液萃取法(简写为LLE)、超临界流体萃取法(简写为SFE)、溶剂辅助致香成分蒸馏法(简写为SAFE)、顶空法(简写为HS)、动态顶空法(简写为DHS)、顶空固相微萃取法(简写为HS-SPME)等。针对不同的食品基质其挥发性成分提取与富集方法都有局限性。对于乳及乳制品,不仅脂肪、蛋白质与糖类等非挥发性成分组成复杂,其酸类、酯类、醇类、酮类与醛类等挥发性成分的组成亦十分复杂,且部分易挥发性成分具有热敏性与易氧化性。例如脂肪族羟基酸同时含有羟基与羧基,且其位点具有活性,α-羟基酸加热时两分子羟基酸脱去两分子水形成交酯结构,β-羟基酸受热时可脱去一分子水形成不饱和烯酸,γ-羟基酸加热时羟基与羧基脱去一分子水形成内酯。
为了能真实全面的测定乳及乳制品中致香成分的组成,需要选择密闭且低温的前处理方式。此外,由于乳及乳制品中部分风味性成分含量较低,却有较低的嗅阈值浓度,对风味组成有较大贡献,需要对测定方法的条件参数进行优化,提高致香成分检测的准确性与精确性。按照官能团类别对乳及乳制品中致香成分进行分类,包括酮类(即含有官能团-CO-)、烷烃类(即含有官能团-CH
发明内容
针对现有技术存在的不足,本发明提供一种检测乳或乳制品中致香成分的方法,前处理简单、快速高效、试剂成本低、定性定量结果准确、灵敏度高,为乳与乳制品质量安全监控提供技术参考。
本发明是通过以下技术方案来实现:
一种检测乳或乳制品中致香成分的方法,当所述的致香成分为2-壬酮、十五烷、2,4-二叔丁基苯酚、月桂酸和3-乙基-5-(2-乙基丁基)-十八烷时,包括如下步骤:
步骤1,将待测的乳或乳制品用环己烷提取后溶解在体积比为1:1的环己烷和乙酸乙酯的混合液中,得到混合液;
步骤2,将混合液注入装配有紫外-可见光光度检测器的凝胶渗透色谱柱中,该色谱柱的填料为颗粒大小等于200-400μm的S-X3聚苯乙烯微球凝胶,用环己烷和乙酸乙酯的混合液进行洗脱,在16.6-18.8min内收集洗脱液,得到净化液,将净化液旋蒸至近干后用正己烷复溶,得到待测液;
步骤3,将待测液用气相色谱-质谱系统进行检测,通过气相色谱得到总离子流图,通过质谱获得一级质谱图和二级质谱图;
步骤4,先分析一级质谱图和二级质谱图,分别得到若干个一一对应的分子离子峰和碎片离子峰,再通过这些分子离子峰和碎片离子峰获得待测液中各个化合物的归属类别,最后利用二级质谱图中各个化合物的质荷比、片段丰度比与同位素丰度比,分别得到每个化合物的结构式和分子式;
步骤5,先将每个致香成分对应的标准物质的质荷比、片段丰度比与同位素丰度比与步骤4各个化合物对应的质荷比、片段丰度比与同位素丰度比进行比对,得到待测致香成分对应的质荷比,再利用每个质荷比在总离子流图中提取每一个致香成分的提取离子流图,结合每一个致香成分的提取离子流图的峰面积、标准物质的提取离子流图的峰面积、标准物质的浓度,得到各个致香成分的浓度,最后再结合各个致香成分的浓度、待测液的体积、待测的乳或乳制品的质量,得到待测的乳或乳制品中致香成分的浓度,完成乳或乳制品中致香成分的检测。
优选的,步骤1先将混匀的待测乳或乳制品试样溶解在环己烷中,在10000-12000r/min下涡旋混合1-2min,再加入饱和氯化钠水溶液,振荡15-20min,在8000-10000r/min下离心3-5min,最后取环己烷层后用体积为1:1的环己烷和乙酸乙酯混合液溶解,得到混合液。
进一步,所述试样与环己烷的比例为(1-2)g:(10-15)mL。
再进一步,所述饱和氯化钠水溶液与环己烷的体积比为(3-5):(10-15)。
优选的,步骤2中所述凝胶渗透色谱柱的净化条件如下:
凝胶渗透色谱柱为GPC净化柱,规格为20mm×210mm,流动相为体积比为1:1的环己烷和乙酸乙酯的混合液,流速为3-4mLmin
优选的,步骤2在20-25℃下将净化液旋蒸至近干,之后用3-5mL的正己烷复溶,得到待测液。
优选的,步骤3中的气相色谱条件如下:
气相色谱柱为弹性石英毛细管色谱柱,规格为30m×0.25mm,0.25μm,采用不分流恒流模式,流速为1-1.5mLmin
优选的,步骤3中的质谱条件如下:
离子源为电子轰击离子源,电子能量为70-75eV;进样口温度为230-250℃;传输线温度为230-250℃,扫描时间为3-60min,采用全扫描模式与离子扫描模式,离子化方式如下;
离子源温度为220-230℃,溶剂延迟时间为3-5min。
进一步,步骤3通过质谱的全扫描模式获得一级质谱图,通过质谱的离子扫描模式得到二级质谱图。
优选的,步骤5中待测的乳或乳制品中致香成分的浓度由下式获得;
相对现有技术,本发明具有以下有益的技术效果:
本发明一种检测乳或乳制品中致香成分的方法,由于2-壬酮、十五烷、2,4-二叔丁基苯酚、月桂酸和3-乙基-5-(2-乙基丁基)-十八烷具有强极性,依据相似相溶原理,所以先将待测的乳或乳制品用环己烷提取,这样能够从中提取到这些成分,同时环己烷可争夺乳或乳制品中蛋白质表面的水化水,破坏其胶体分子表面的水化层而使蛋白质聚集沉淀,从而达到富集这些目标致香成分的目的,通过液液萃取的方式将提取液溶解在环己烷和乙酸乙酯的混合液中,可以去除脂肪和聚糖等大分子物质,得到初步净化的混合液,再将混合液注入装配有紫外-可见光光度检测器的凝胶渗透色谱进行净化,中性、200-400μm排阻范围的聚苯乙烯微球凝胶、以及其含有的多孔的聚苯乙烯二乙烯基苯介质,可对这些分子量小且极性高的目标致香成分进行净化和分离,在对应的保留时间范围内收集洗脱液便可得到含有目标致香成分的净化液,然后以气相色谱-质谱联用的方式进行检测,之后利用质谱得到的一级质谱图和二级质谱图便可得到每个化合物的结构式和分子式,结合每个致香成分对应的标准物质的质荷比、片段丰度比与同位素丰度比与之前各个化合物的相应信息进行比对,利用每个质荷比在气相色谱得到的总离子流图中提取每一个致香成分的提取离子流图,最后用提取离子流图的峰面积、标准物质的峰面积、标准物质的浓度,可得到各个致香成分的浓度,用各个致香成分的浓度、待测液的体积、测的乳或乳制品的质量,可得到待测的乳或乳制品中致香成分的浓度。该方法定性定量结果准确、灵敏度高,为乳及乳制品中常见致香成分的筛查提供了系统的检测方法,结果显示五类致香成分在线性范围内具有良好的线性关系,相关系数均大于0.99,高(4×CCβ)、中(2×CCβ)、低(1×CCβ)三个添加水平下,基质的加标回收率达81%-90%,重复实验6次,其相对标准偏差为1.8%-5.9%。确定限(CCα)和检测容量(CCβ)分别为0.05μg kg
附图说明
图1a为本发明所述巴氏杀菌乳样品的紫外凝胶渗透色谱流出曲线;
图1b为本发明所述超高温瞬时灭菌乳的紫外凝胶渗透色谱流出曲线;
图1c为本发明所述发酵乳的紫外凝胶渗透色谱流出曲线;
图1d为本发明所述婴幼儿配方乳粉的紫外凝胶渗透色谱流出曲线。
图2a为本发明所述GPC流出液在16-16.8min的收集时间范围内对应的响应强度图;
图2b为本发明所述GPC流出液在18.2-19min的收集时间范围内对应的响应强度图;
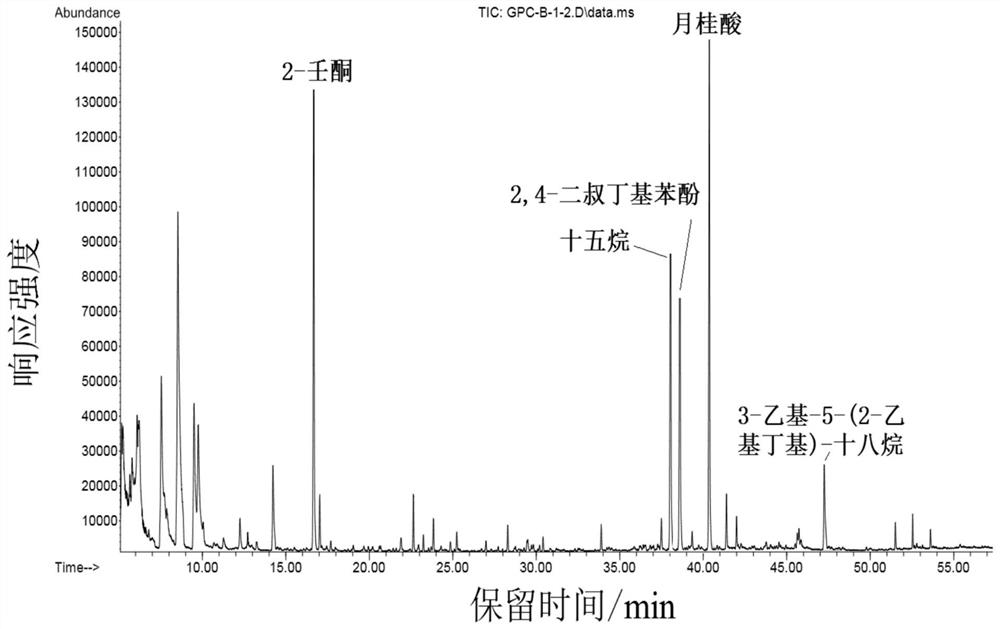
图3为巴氏杀菌乳中目标分析物的GPC净化色谱图。
具体实施方式
下面通过具体实施例对本发明原理及优势进行解释和说明,以便本领域技术人员更好的理解本发明。下述说明仅是示例性的,并不对其内容进行限定。
本发明一种利用凝胶渗透色谱检测乳或乳制品中致香成分的方法,致香成分属于香料,具体包括以下步骤:
1)利用液液萃取处理手段对被测乳及乳制品中目标待测物2-壬酮、十五烷、2,4-二叔丁基苯酚、月桂酸以及3-乙基-5-(2-乙基丁基)-十八烷进行提取处理;
称取混匀试样1-2g(精确到0.01g)于50mL具塞离心管中,加入10-15mL环己烷溶解样品,以10000r/min-12000r/min的高速涡旋混合1-2min,加入3-5mL饱和氯化钠水溶液,振荡15-20min,以8000-10000rmin
2)基于阀切换技术,运用凝胶渗透色谱对经提取浓缩后的待测目标物净化处理;
乳及乳制品样品经环己烷提取后,经自动取样装置将溶液注入凝胶渗透色谱柱中,采用环己烷-乙酸乙酯混合液(1:1,v/v)通过高压泵对样品进行洗脱,对目标化合物分析有干扰的蛋白质、脂肪相对分子质量较大,沿聚苯乙烯微球凝胶的间隙流出。凝胶渗透色谱系统配有紫外-可见光光度检测器,用于上述五种致香成分响应强度的测定,检测波长设为210nm,出峰时间在4.0min-10.0min,目标分析物2-壬酮、十五烷、2,4-二叔丁基苯酚、月桂酸与3-乙基-5-(2-乙基丁基)-十八烷的相对分子质量较小,可以进入聚苯乙烯微球凝胶内部,保留时间靠后,在16.6min-18.8min出峰;
凝胶渗透色谱净化条件:GPC净化柱(规格为20mm×210mm),填料为S-X3聚苯乙烯微球凝胶(颗粒大小为200μm-400μm);流动相:体积比为1:1的环己烷-乙酸乙酯;流速为3-4mLmin
3)运用气相色谱-质谱系统,即7890A-5975C气相色谱-质谱仪对处理得到的样品进行检测;
气相色谱条件为:毛细管色谱柱:DB-5MS(规格为30m×0.25mm,0.25μm)具体是弹性石英毛细管色谱柱,采用不分流恒流模式,流速为1-1.5mLmin
质谱条件为:电子轰击(英文简称为EI)离子源;电子能量为:70-75eV;进样口温度:230-250℃;传输线温度:230-250℃;
离子化方式为:离子源温度:220-230℃;溶剂延迟时间:3-5min;
扫描时间:3min-60min;采用全扫描模式(英文简称为SCAN)与选择离子扫描模式(英文简称为SIM),通过全扫描模式获得一级质谱图,通过选择离子扫描模式得到二级质谱图。
4)基于获得的被测样品一级质谱图与二级质谱图,通过Aglient MSDchemstation工作站提取出目标的致香成分的结构和元素组成;
通过分析一级质谱图,得到不同的分子离子峰,分析二级质谱图得到不同的碎片离子峰,再根据质量亏损机理,利用分子离子峰和碎片离子峰得到精确的相对分子质量,判断该物质的归属类别,继而利用二级质谱图中该物质的质荷比、片段丰度比与同位素丰度比,从而推断出该物质结构和元素组成,也就是该物质的结构式和分子式;
5)结合标准致香成分对应的物质建立标准谱库中的致香成分信息,包括五种标准致香成分的质荷比、片段丰度比与同位素丰度比。将这五种致香成分信息与被测物样品所得所有化合物的信息进行比对,在0.05Da的误差范围内进行乳及乳制品中致香成分结构判定。随后使用致香成分的质荷比,在步骤3气相色谱得到的总离子流图中获得每一个物质的提取离子流图,使用峰面积法进行致香成分浓度的定量,结合提取离子流图的峰面积、标准物质的峰面积、标准物质的浓度,得到各个致香成分的浓度,最后再结合各个致香成分的浓度、待测液的体积、测的乳或乳制品的质量,得到待测的乳或乳制品中致香成分的浓度,完成乳或乳制品中致香成分的检测。
由下式获得;
标准谱库的建立具体为:
1a)配制5类致香成分(2-壬酮、十五烷、2,4-二叔丁基苯酚、月桂酸以及3-乙基-5-(2-乙基丁基)-十八烷)标准品的标准溶液;
1b)运用上述步骤3所描述的气相色谱-质谱系统对致香成分的标准溶液进行筛查,采用电子轰击离子源、全扫描模式与选择离子扫描模式得到标准溶液完整的一级质谱图与二级质谱图,5种致香成分化合物的气相色谱-质谱参数见表1;
表1 5种乳及乳制品中致香成分气相色谱-质谱参数
1c)按照上述步骤4所描述的方法,通过Aglient MSD chemstation工作站对获得的一级质谱图与二级质谱图进行处理,得到标准品中各个物质的质荷比、片段丰度比与同位素丰度比,建立致香成分的标准谱库。
上述所用到的仪器、试剂、溶液的配制及具体的分析列举如下:
1.仪器
7890A-5975C气相色谱-质谱仪(美国Agilent Technologies公司)、Vortex.Genie2T型旋涡混合器(美国Scientific Industries公司)、SA31型振荡器(日本Yamata公司)、超声波清洗器(江苏昆山超声仪器有限公司)、AL204-IC型分析天平(瑞士Mettler Toledo公司)、旋转蒸发浓缩仪(瑞士Buchi公司)、ED-115型恒温干燥箱(德国Binder公司)、SevenEasy S20K型pH计(瑞士Mettler Toledo公司)、凝胶渗透色谱仪(德国J2 Scientific公司)、20mm×210mm GPC净化柱(德国J2 Scientific公司)、200-400μm S-X3聚苯乙烯微球凝胶填料(美国Bio-Beads公司)、Milli-Q Integral型纯水仪(美国Millipore公司)。
2.试剂
乳及乳制品(沃尔玛超市、家乐福等超市)、氯化钠(GR)(荷兰Akzo Nobel公司)、0.20μm微孔滤膜(美国Pall公司)、环己烷(HPLC)(美国Fisher科技公司)、乙酸乙酯(HPLC)(美国Fisher科技公司)、正己烷(HPLC)(美国Fisher科技公司)、5种致香成分及相关化学品标准物质(德国Dr.Ehrenstorfer GmbH公司、德国Sigma-Aldrich公司、德国Witega公司)。
3.溶液的配制
2-壬酮、十五烷、2,4-二叔丁基苯酚、月桂酸、3-乙基-5-(2-乙基丁基)-十八烷混合标准贮备溶液:1000mgL
4.凝胶渗透色谱净化条件的选择
根据目标分析物与杂质的保留时间,可以将相对分子质量较大的脂肪与蛋白质通过转换凝胶渗透色谱中的六通阀连通位置切入废液流路中,将待分析溶液切换至捕集环流路中,旋转蒸发浓缩,再用正己烷定容至5mL后,运用气相色谱-质谱检测。因此,六通阀连通位置切换程序的设计为凝胶渗透色谱净化的关键,图1a、图1b、图1c、图1d分别为巴氏杀菌乳、超高温瞬时灭菌乳、发酵乳、婴幼儿配方乳粉样品经环己烷提取后上样的紫外凝胶渗透色谱流出曲线。
巴氏杀菌乳样品通过凝胶渗透色谱柱紫外210nm检测,图1a中相对分子质量较大的脂肪与蛋白质在4.00-9.10min流出,待分析溶液在16.00-19.00min流出;图1b的超高温瞬时灭菌乳的紫外凝胶渗透色谱流出曲线显示,脂肪与蛋白质在4.00-9.00min流出,待分析溶液在16.00-18.80min流出;图1c的发酵乳的紫外凝胶渗透色谱流出曲线显示脂肪与蛋白质在4.70-8.90min流出,待分析溶液在16.40-18.50min流出;图1d的婴幼儿配方乳粉的紫外凝胶渗透色谱流出曲线显示脂肪与蛋白质在4.70-8.80min流出,待分析溶液在16.00-18.10min流出,因此确定凝胶渗透色谱流出液的收集时间优化的范围为16.00min-19.00min。
根据5种目标物质的质谱响应(即色谱峰峰面积)作为评价六通阀连通位置切换程序的指标。
将六通阀连通位置由废液流路切入捕集环流路的起始时间初设为16.00min,优化的步长为0.20min,缩短洗脱液流入捕集环流路的时间;固定六通阀连通位置重新切至废液的时间为19.0min,0.20min为间隔,将六通阀连通位置切换的时间逐步提前。
图2为5种化合物流出程序的优化结果。图2a显示在16.6min收集的GPC流出液响应强度最高,图2b显示18.8min收集的GPC流出液响应强度最高,这分别对应于在相应的保留时间下,5种化合物的浓度最高。若运用六通阀将目标分析物切入捕集环流路中过迟,或在目标分析化合物还未完全洗脱就将六通阀连通流路切入废液流路,待分析目标物质就会损失,造成化合物回收率下降;若运用六通阀将脂肪与蛋白质等杂质切入捕集环中过早,相对分子质量较大的杂质会进入捕集环,若目标化合物经洗脱完后,未将连通阀连接流路切入废液,分子量较小的杂质也会进入捕集环。因此控制凝胶渗透色谱的收集时间为16.60min-18.80min,实现了凝胶渗透色谱对于样品的净化。
图3表明在收集时间范围内2-壬酮、十五烷、2,4-二叔丁基苯酚、月桂酸和3-乙基-5-(2-乙基丁基)-十八烷的响应强度较高,且基质干扰基本去除,说明优化后条件可以达到良好的净化效果。
此外,实验中研究了洗脱速度对目标化合物的分离效果的影响,设置了1.0mLmin
5.分析结果
(1)方法学验证
a方法的标准曲线、线性范围及相关系数
以纵坐标y表示标准致香成分物质定量离子色谱峰峰面积,x表示标准物质浓度(mg L
b方法的定量下限、加标回收率与精密度
检测限与定量下限以确定限(CCα)与检测容量(CCβ)作为考察标准。CCα采用校准曲线法测定,其值等于能观察到响应时纵坐标上的浓度值加对应的重现性标准偏差的2.33倍。CCβ的计算方法为CCα的值与CCα取值时重现性标准偏差的1.64倍之和。据此确定限和检测容量分别为0.05μg kg
实验的准确性与精密度通过回收率与其6次平行实验结果的标准偏差评价。在基质中添加三个浓度水平下五类致香成分的系列混合标准物质溶液(CCβ,2倍CCβ和4倍CCβ),运用7890A-5975C气相色谱-质谱仪检测,计算各种致香风味物质的平均回收率与相对标准偏差。回收率与相对标准偏差结果分别为81%-90%和1.8%-5.9%,所建立的方法准确性和精密度良好。
实际样品检测
本发明在具体实施时,相对于之前的描述,采用以下具体的参数进行,
称取混匀试样2g(精确到0.01g)于50mL具塞离心管中,加入10mL环己烷溶解样品,以12000r/min的高速涡旋混合1min,加入5mL饱和氯化钠水溶液,振荡20min,以10000rmin
将收集的净化液于25℃下旋转蒸发至近干,用5mL正己烷复溶近干物,过0.20μm滤膜供上机测定;
气相色谱条件为:流速为1mLmin
质谱条件为:电子能量为:70eV;进样口温度:250℃;传输线温度:250℃;
离子化方式为:离子源温度:230℃;溶剂延迟时间:3min;
应用已建立的液液萃取-凝胶渗透色谱-气相色谱-质谱方法对市售的乳及乳制品:巴氏杀菌乳(十八个品牌的各十二个批次产品)、超高温瞬时灭菌乳(二十个品牌的各十二个批次产品)、发酵乳(十三个品牌的各十六个批次产品)与婴幼儿配方乳粉(十六个品牌的各十二个批次产品)进行分析检测,结果见表2。实验结果显示在市售的乳及乳制品中不存在2,4-二叔丁基苯酚和3-乙基-5-(2-乙基丁基)-十八烷。三类致香成分的方法学验证显示在线性范围内具有良好的线性关系,相关系数均大于0.99,高(4×CCβ)、中(2×CCβ)、低(1×CCβ)三个添加水平下,基质的加标回收率达81%-90%,重复实验6次,其相对标准偏差为1.8%-5.9%。确定限(CCα)和检测容量(CCβ)分别为0.05μg kg
表2典型乳及乳制品成分测定结果
本发明的上述实施方式为实施例,具有与本发明的权利要求书的技术思想使之相同的方法并发挥相同作用效果的技术方案,均包含在本发明内。
- 一种检测乳或乳制品中致香成分的方法
- 一种用于检测原料乳及乳制品中β-内酰胺酶的检测装置及其检测方法