一种检测多环芳烃的方法
文献发布时间:2023-06-19 18:34:06
技术领域
本发明涉及化学检测技术领域,具体涉及一种检测多环芳烃的方法。
背景技术
多环芳烃(polycyclic aromatic hydrocarbons,PAHs)是指分子中含有两个或两个以上苯环的碳氢化合物,通常存在于石化产品、木炭、木馏油、药物、染料、橡胶、塑料、润滑油、脱模剂、电容电解液、矿物油、防锈油、杀虫剂、不完全燃烧的有机化合物等材料中。石油、煤等燃料以及木材、可燃气体在不完全燃烧或高温处理条件下均可产生多环芳烃。
多环芳烃具有生物难降解性和累积性,目前已经确定部分PAHs对人体具有致癌性、致畸变性和致突变性,易导致皮肤癌、肺癌、上消化道肿瘤和动脉硬化,还能损伤生殖系统,导致不育症
目前已经证实,多环芳烃中有16种物质是高致癌的物质(具体见表1),而苯并[a]芘是PAHs中毒性最强的一种
表1 16种高关注PAHs
人造地板的材质包括塑料、橡胶、EVA等多类,由于原料或生产过程存在污染等原因,人造地板中常常检出PAHs。目前虽然国际上没有对人造地板中PAHs提出具体的限量要求,但是因为人造地板与人体直接接触,人造地板中的PAHs等有害物质会通过水系等介质慢慢渗出,并迁移至人体内,对人体健康造成危害,因此,测定PAHs的迁移量有非常重要的意义。
PAHs的挥发性不强,属于半挥性有机物,PAHs的分析通常包括样品的提取、净化、富集和测定等步骤。Winker等人
发明内容
根据第一方面,在一实施例中,提供一种检测多环芳烃的方法,包括:
萃取步骤,包括使用溶胶凝胶端羟基硅油萃取涂层,对待测溶液进行顶空固相微萃取;
解吸检测步骤,包括对萃取结束后的涂层进行热解吸,对热解吸的目标化合物进行检测。
根据第二方面,在一实施例中,提供一种溶胶凝胶端羟基硅油萃取涂层。
根据第三方面,在一实施例中,提供第二方面的溶胶凝胶端羟基硅油萃取涂层在检测多环芳烃中的用途。
依据上述实施例的一种检测多环芳烃的方法,本发明利用溶胶凝胶端羟基硅油单壁碳纳米管萃取纤维,采用气相色谱-质谱联用法,进行顶空固相微萃取,对人造地板中高关注的16种多环芳烃,在模拟汗液中的迁移量进行了分析和测定,对色谱条件和顶空固相微萃取条件进行了优化。自制萃取纤维涂层萃取效果优于进口相似类型的商用萃取纤维涂层。该方法对16种多环芳烃的检测限为0.001~0.01ng/mL,回收率为86.40~101.70%,相对标准偏差为5.74~12.03%。测得不同实际人造地板样品中多环芳烃迁移量为0.37~84.3ng/mL。
附图说明
图1.1、图1.2为盐效应的影响结果图;
图2.1、图2.2为萃取温度的影响结果图;
图3.1、图3.2为萃取时间的影响结果图;
图4.1、图4.2为解吸时间的影响结果图;
图5为16种PAHs的HS-SPME-GC/MS-SIM图;
图6为SWNTs-TSO-OH涂层与商用PDMS涂层萃取量的比较结果图;
图7~9为三个不同的实际样品的GC/MS-SIM图。
具体实施方式
下面通过具体实施方式结合附图对本发明作进一步详细说明。在以下的实施方式中,很多细节描述是为了使得本申请能被更好的理解。然而,本领域技术人员可以毫不费力的认识到,其中部分特征在不同情况下是可以省略的,或者可以由其他材料、方法所替代。在某些情况下,本申请相关的一些操作并没有在说明书中显示或者描述,这是为了避免本申请的核心部分被过多的描述所淹没,而对于本领域技术人员而言,详细描述这些相关操作并不是必要的,他们根据说明书中的描述以及本领域的一般技术知识即可完整了解相关操作。
另外,说明书中所描述的特点、操作或者特征可以以任意适当的方式结合形成各种实施方式。同时,方法描述中的各步骤或者动作也可以按照本领域技术人员所能显而易见的方式进行顺序调换或调整。因此,说明书和附图中的各种顺序只是为了清楚描述某一个实施例,并不意味着是必须的顺序,除非另有说明其中某个顺序是必须遵循的。
本文中为部件所编序号本身,例如“第一”、“第二”等,仅用于区分所描述的对象,不具有任何顺序或技术含义。
鉴于现有技术存在的缺陷,特别需要建立一种简单、快速、准确的检测方法,用于PAHs的日常监测。
SPME方法经常被用做测定水系中的PAHs
表2分析液体基质中PAHs的SPME涂层
碳纳米管(CNTs)自1991年被S.Iijima
根据第一方面,在一实施例中,提供一种检测多环芳烃的方法,包括:
萃取步骤,包括使用溶胶凝胶端羟基硅油萃取涂层,对待测溶液进行顶空固相微萃取;
解吸检测步骤,包括对萃取结束后的涂层进行热解吸,对热解吸的目标化合物进行检测。
在一实施例中,萃取步骤中,所述待测溶液包括但不限于由模拟汗液浸泡待测物后得到的溶液。
在一实施例中,所述待测物包括人造地板。
在一实施例中,所述模拟汗液包含NaHCO
在一实施例中,所述模拟汗液包含如下组分:4.2g/L NaHCO
在一实施例中,所述模拟汗液还包含溶剂,所述溶剂包含水。
在一实施例中,解吸检测步骤中,所述检测包括气相色谱-质谱检测。
在一实施例中,解吸检测步骤中,气相色谱-质谱检测使用的色谱柱为HP-5MS毛细管色谱柱,或相当者。
在一实施例中,解吸检测步骤中,气相色谱-质谱检测中,进样口温度:320℃,传输线温度:280℃;电离方式:EI;电离能量:70eV;离子源温度:150℃。
在一实施例中,解吸检测步骤中,气相色谱-质谱检测中,氦气作载气,流速1.8mL/min,不分流进样。
在一实施例中,解吸检测步骤中,气相色谱-质谱检测中,柱温:初温50℃,保留1min,以25℃/min升至200℃,然后以8℃/min升至230℃,保留5min,再以5℃/min升至316℃,保留1.05min。
在一实施例中,解吸检测步骤中,气相色谱-质谱检测中,质量扫描范围:50~450amu;选择离子监测方式;溶剂延迟:5min;5~7min,选择监测离子为m/z128;7~9min,选择监测离子为m/z152、m/z154和m/z166;9~10.5min,选择监测离子为m/z178;10.5~14min,选择监测离子为m/z202;14~20min,选择监测离子为m/z228;20~26min,选择监测离子为m/z252;26~31min,选择监测离子为m/z276和m/z278。
在一实施例中,萃取步骤中,萃取温度为25~90℃。
在一实施例中,萃取步骤中,顶空固相微萃取时间为5~120min,优选为60min。
在一实施例中,解吸检测步骤中,萃取结束后的涂层置于检测设备中进行热解吸。
在一实施例中,解吸检测步骤中,热解吸温度为300℃。
在一实施例中,解吸检测步骤中,热解吸时间≥1min,优选为1~5min。
在一实施例中,解吸检测步骤中,所述目标化合物包括但不限于萘、苊烯、苊、芴、菲、蒽、荧蒽、芘、苯并[a]蒽、
在一实施例中,萃取步骤中,溶胶凝胶端羟基硅油萃取涂层是由端羟基硅油修饰的单壁碳纳米管、四乙氧基硅烷、端羟基硅油以及聚甲基氢硅氧烷反应得到。
在一实施例中,萃取步骤中,溶胶凝胶端羟基硅油萃取涂层的制备方法如下:将端羟基硅油修饰的单壁碳纳米管、四乙氧基硅烷、端羟基硅油、聚甲基氢硅氧烷混合反应,将所得的化合物涂覆于基材表面,获得所述涂层。
在一实施例中,所述基材包括石英纤维。
根据第二方面,在一实施例中,提供一种溶胶凝胶端羟基硅油萃取涂层。
根据第三方面,在一实施例中,提供第二方面的溶胶凝胶端羟基硅油萃取涂层在检测多环芳烃中的用途。
在一实施例中,本发明利用SWNTs-TSO-OH萃取涂层,将固相微萃取技术与气相色谱质谱法相结合,选择列入监控范围的16种多环芳烃作为研究对象,对固相微萃取的盐效应、萃取时间、萃取温度、解吸时间等多种因素进行了优化,与商用的100μm的PDMS萃取纤维进行了对比,测定了人造地板在水系中迁移16种多环芳烃的检测限量、精密度及回收率。从实验结果中发现,该方法灵敏度高、检测限低、精密度及回收率良好。
实施例1
1、本实施例的原理如下:将人造地板浸泡于模拟汗液中,在常温下超声,然后取适量浸泡液,用溶胶凝胶端羟基硅油萃取涂层,进行顶空固相微萃取,再用气相色谱-质谱联用仪上解吸进样,测定人造地板在模拟汗液中多环芳烃的迁移量。
溶胶凝胶SWNTs-TSO-OH涂层的制备方法可参考文末的文献25,具体制备方法如下:
采用溶胶凝胶涂制技术,制得溶胶凝胶端羟基硅油高分子功能化单壁碳纳米管萃取涂层(sol-gel SWNTs-TSO-OH)。溶胶凝胶法制备纤维涂层包括下列四个主要步骤:(1)石英纤维表面的预处理;(2)溶胶的制备;(3)涂制;(4)涂层的老化。具体如下:
(1)石英纤维表面的预处理
第一步是除去聚酰亚胺保护层。将石英纤维浸入丙酮3h,将聚酰亚胺外涂层擦去,分别用自来水和蒸馏水冲洗15min,接着用甲醇淋洗纤维表面,吹干待用。
第二步是将脱去保护层的纤维用碱处理。其目的是为了尽可能的打开石英纤维表面的硅羟基,以提高纤维表面与溶胶凝胶体系的键合能力。将第一步处理好的纤维置于1M的NaOH溶液中1h,然后用水冲洗15min,再置于0.1M的盐酸中浸泡30min,以中和剩余的NaOH,接着依次用自来水、蒸馏水、甲醇冲洗,吹干备用。纤维除去聚酰亚胺保护层后会变得很脆,极易断裂,因此,处理过程中必须十分小心。
(2)溶胶的制备
在聚乙烯离心管中,将90mg的SWNTs-TSO-OH(端羟基硅油修饰的单壁碳纳米管,又名端羟基硅油高分子功能化单壁碳纳米管)溶解在300μL二氯甲烷中,然后加入200μL的TEOS(Tetraethoxysilane,四乙氧基硅烷,又名硅酸乙酯、正硅酸乙酯)、55μL的TSO-OH(hydroxyl-terminated silicone oil,羟基硅油,又名端羟基硅油)和25μL的PMHS[Poly(methyl hydrosiloxane),聚甲基氢硅氧烷]超声20min充分混匀后,加入180μL的TFA(三氟乙酸,95%水溶液),再超声5min,在8000rpm转速下离心3min,去掉底部的沉淀,上清液用于涂层的涂制。
(3)涂制
采用浸渍法涂制。将处理过的纤维垂直插入溶胶中放置5分钟,在纤维的表面上慢慢形成凝胶涂层,以均匀速度把纤维从溶液中提拉出来,重复此过程,每次2分钟,直到达到了需要的厚度,将纤维置于室温下的干燥器中干燥24h,然后进行老化。
(4)涂层的老化
取1cm长萃取纤维,用环氧树脂AB胶粘在订制的萃取头组件上。在氮气保护下,用程序升温的方式对萃取纤维进行老化。50℃开始以1℃/min的速度升至120℃保留1h,再以1℃/min的速度升至260℃保留1h,最后以1℃/min的速度升至340℃保留2h。在显微镜下分别测量萃取纤维的直径和空白纤维的直径,两者的差再除以2,即得到萃取纤维的厚度,单位通常为μm。
2、主要化学试剂和样品
16种多环芳烃混合标准溶液(PAHs):每种PAH浓度均为2.0g/L,购自AccuStandard公司(USA);正己烷(分析纯),氯化钠(分析纯),碳酸氢钠(分析纯),碳酸钾(分析纯),广州化学试剂厂;二次蒸馏水;人造地板样品:深圳建材市场购买。
3、主要仪器
Agilent GC6890-MS5972气相色谱-质谱联用仪(美国);
Millipore超纯水机:美国Millipore公司;
DF-2集热式恒温磁力搅拌器:浙江省乐清市乐成电器厂;
商用100μm PDMS萃取头:加拿大Supelco公司;
自制68μm溶胶凝胶SWNTs-TSO-OH涂层;
20mL顶空瓶,聚四氟乙烯搅拌磁子;
固相微萃取手柄:加拿大Supelco公司;
PAHs的分析条件:
HP-5MS毛细管柱(30m×0.25mm×0.25μm),进样口温度:320℃,传输线温度:280℃;电离方式:EI;电离能量:70eV;离子源温度:150℃;氦气(99.999%)作载气,流速1.8mL/min,不分流进样。
柱温:初温50℃,保留1min,以25℃/min升至200℃,然后以8℃/min升至230℃,保留5min,再以5℃/min升至316℃,保留1.05min。
质量扫描范围:50~450amu;选择离子监测方式;溶剂延迟:5min;5-7min,选择监测离子为m/z128;7-9min,选择监测离子为m/z152,m/z154和m/z166;9-10.5min,选择监测离子为m/z178;10.5-14min,选择监测离子为m/z 202;14-20min,选择监测离子为m/z228;20-26min,选择监测离子为m/z252;26-31min,选择监测离子为m/z276和m/z278。
4、标准工作溶液的配制
模拟汗液的配制:4.2g NaHCO
标准工作溶液的配制:用正己烷稀释2.0g/L的16种PAHs混合标准溶液,配制得到一系列如下浓度的混合标准溶液:0.001mg/L、0.01mg/L、0.1mg/L、1mg/L和5mg/L。分别取10mL的一系列浓度的混合标准溶液于10mL模拟汗液中,得到PAHs浓度分别为0.001ng/mL、0.01ng/mL、0.1ng/mL、1ng/mL和5ng/mL的工作溶液。所有的PAHs溶液于-20℃存放。
5、实验方法
条件优化:在20mL顶空瓶中加入10mL蒸馏水,加入10mL浓度合适的混合标准溶液,放入磁子,压上顶空瓶专用瓶盖。在一定的搅拌速度、温度下,纤维置于溶液的上部气体中萃取。萃取后,将纤维插入300℃的GC进样口中热解析。每次取样前,做空白测试。
样品测试:取剪碎后的人造地板样品1.5g浸泡于30mL模拟汗液中,在常温下超声30min,然后取10mL浸泡液,加入1.5gNaCl,用SWNTs-TSO-OH萃取纤维,在优化好的萃取条件下进行顶空固相微萃取。
5、结果
5.1萃取方式的选择
顶空萃取模式适用于干净基质中PAHs的测定,考虑到人造地板浸泡液成分复杂,若采用直接萃取的方式,不仅对涂层的寿命有影响,而且萃取组分多,干扰大,不利于目标化合物的分析,因此,本实施例采用顶空萃取模式。
5.2气相色谱-质谱测定及确证的分析条件选择
在选定的色谱条件下,首先通过全扫描方式(GC-MSD/SCAN)做出总离子流图(TIC),然后根据其质谱图中的碎片离子选择了丰度相对较高、分子质量较大、干扰较小的碎片离子作为测定和确证的特征目标监测离子,并针对16种多环芳烃进行GC-MSD/SIM测定。最终确定的定性和定量的特征目标监测离子见表3,每种多环芳烃的保留时间也见表3。测定时,根据其标准物和待测样品的SIM离子流图中的峰面积,采用外标法定量。确证时,可根据待测阳性检出物中碎片离子的种类作为其阳性判别的依据。
表3 16种PAHs的分子式、分子量、定性离子和定量选择离子
5.3萃取条件的优化
5.3.1盐效应
分取6份平行样品溶液,在该溶液中,每种PAHs的浓度均为0.1ng/mL,按第4节的实验方法,分别加入0、0.5、1、1.5、2、2.5g NaCl,进行HS-SPME实验,观察待测物质的萃取峰面积,结果如图1所示。
图1.1、图1.2为盐效应的影响结果图。每种PAHs的浓度均为0.1ng/mL,萃取时间:60min;萃取温度:90℃;解吸时间:4min;峰号见表3。
图1.1、图1.2的结果表明,对于1-10号分子量稍低的PAHs来说,加入2g NaCl时,获得最好的萃取效率,加入1.5g和2.5g NaCl时,萃取效率略有减少;对于11-16号分子量稍高的PAHs来说,加入1.5g NaCl时,获得最好的萃取效率,加入2.0g和2.5g NaCl时,萃取效率略有减少,特别是对于较难萃取的14-16号PAHs来说,加入1.5g NaCl时,获得的萃取效率较有优势。因此,后续实验均选择加入1.5gNaCl。
5.3.2萃取温度
为了评估萃取温度的影响,测试了萃取温度为25、30、40、60、70、80、90℃下,SWNTs-TSO-OH涂层对待测物质的萃取量(图2.1、图2.2)。
图2.1、图2.2为萃取温度的影响结果图,每种PAHs的浓度均为0.1ng/mL,每10mL样品溶液中添加1.5g NaCl;萃取时间:60min;解吸时间:4min;峰号见表3。
可见,1号PAHs(萘)的沸点较低,随着萃取温度的增加,它的萃取量反而下降,主要是因为吸附是放热过程,升温会使待测物分配系数减小,在固相中的富集量减少,2-6号PAHs随着萃取温度的增加,萃取量开始上升,后来反而下降;7-12号PAHs随着萃取温度的增加,萃取量逐步上升,到80-90℃左右,曲线趋于平缓,固相涂层、顶空相和液相三相之间达到平衡,萃取量趋于稳定。13-16号PAHs随着萃取温度的增加,萃取量一直上升,到90℃时,依旧未达到平衡,考虑可操作性和灵敏度的因素,在下述实验中,选择90℃为萃取温度。
5.3.3萃取时间
样品在HS-SPME中的分配涉及到三相之间的平衡,即:固相涂层、顶空相和液相。由于在整个过程中,样品的总量是一定的。因此,样品在三相间达到平衡时,被固相涂层所萃取的量是一定的,即不随时间而变化。测试了萃取时间分别为5、10、15、30、60、120min时,涂层对16种PAHs的吸附量(图3.1、图3.2)。
图3.1、图3.2为萃取时间的影响结果图,每种PAHs的浓度均为0.1ng/mL,每10mL样品溶液中添加1.5g NaCl;萃取温度:90℃;解吸时间:4min;峰号见表3。
由图可以看出,1-8号PAHs随着萃取时间的延长,萃取量逐步上升,30min后,三相之间达到平衡,萃取量趋于稳定。9-16号PAHs随着萃取时间的延长,萃取量一直上升,即使萃取时间为120min时,还未达到平衡,因此有必要采用非平衡态下操作与定量。Ai提出了非平衡态下的动力学模型,在搅拌充分的萃取体系中,在非平衡状态下纤维上待测物的含量不但与初始浓度有关,还与采样时间和质量迁移速率等有关。如果固定采样时间和保持搅拌条件不变,纤维上待测物的量正比于其在水相中的初始浓度。考虑到实验的方便,将60min作为萃取时间。
5.3.4解吸时间
解吸时间、涂层厚度或孔径深度、待测物的挥发性及涂层表面结构对热解吸时间均有影响。解吸温度越高,解吸速度越快,溶胶-凝胶技术制备的涂层可以耐高温,因此解吸温度可以高一些,此处为300℃;涂层表面的多孔结构有利于待测物在涂层上扩散,加快了它脱离涂层的速度。测试了不同解析时间下的PAHs的峰面积(图4.1、图4.2)。
图4.1、图4.2为解吸时间的影响结果图,每种PAHs的浓度均为0.1ng/mL,每10mL样品溶液中添加1.5g NaCl;
萃取温度:90℃;萃取时间:60min;峰号见表3。
1-6号PAHs在1min即可解析完全,7-12号PAHs在3min即可解析完全,高沸点的13-16号PAHs本来不易从涂层上解吸下来,但由于涂层有了以上的优点,它们需要的解吸时间大大缩短了,仅需约4min就能够完全解吸下来,因此,选择解吸时间为4min。
最后,优化好的萃取条件为:在20mL顶空瓶中加入10mL样品溶液,加入1.5g NaCl,放入磁子,压上顶空瓶专用瓶盖。用SWNTs-TSO-OH萃取纤维,在萃取温度90℃下,顶空固相微萃取60min。萃取后,将纤维插入300℃的GC进样口中热解析4min。
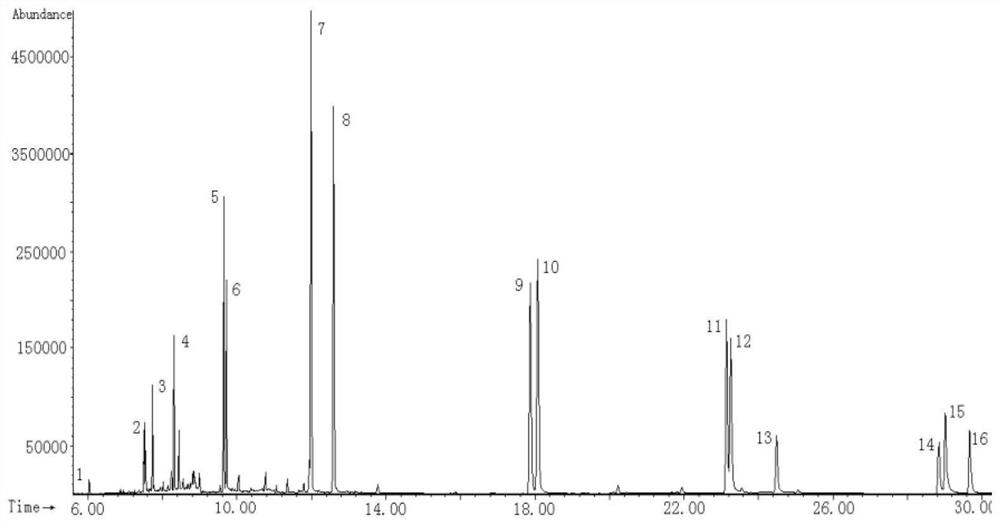
在上述优化后的萃取条件下,得到标准工作溶液的HS-SPME-GC/MS-SIM图,具体见图5所示的16种PAHs的HS-SPME-GC/MS-SIM图。图5中横坐标时间的单位为min。每种PAHs的浓度均为0.1ng/mL,每10mL样品溶液中添加1.5g NaCl;萃取温度:90℃;萃取时间:60min;解吸时间:4min;峰号见表3。
5.4与商用涂层的比较
目前,大多数文献采用100μm的PDMS萃取纤维(表2)分析水性溶液中的PAHs,因此在本节中将SWNTs-TSO-OH涂层与PDMS涂层进行了对比。
图6为SWNTs-TSO-OH涂层与商用PDMS涂层萃取量的比较结果图,条件同图5。
结果表明,除了萘之外,SWNTs-TSO-OH涂层对其余15种PAHs的萃取效率均优于PDMS涂层,特别是对于难萃取的14-16号PAHs,SWNTs-TSO-OH涂层的萃取能力优于PDMS涂层6-8倍。
6、方法的三大指标
6.1线性关系和检测限
在优化后的固相微萃取条件下,确定了各组分的线性范围和检测限(S/N=3),结果见表4。表中数据可知,方法灵敏度高,对16种多环芳烃的检测限量低至0.001~0.01ng/mL。
表4线性范围和检测限(ng/mL)
6.2精密度和回收率
采用添加法在待测样品中添加不同浓度水平的PAHs混标,进行回收率和精密度测定。结果见表5,平均回收率为86.40~101.70%,相对标准偏差为5.74~12.03%。
表5回收率和精密度实验结果
7、实际样品的测定
在上述优化后的萃取条件下,对建材市场购得的人造地板样品浸泡液中PAHs的迁移量进行了测定。表6给出了6种阳性样品的测试结果,测得不同实际人造地板中多环芳烃迁移量为0.37~84.3ng/mL。表6给出了部分样品的测试结果,图7、8、9给出了用溶胶凝胶SWNTs-TSO-OH涂层测试的实际人造地板样品浸泡液的选择离子监测图。
图7~9为三个不同的实际样品的GC/MS-SIM图。三个图中横坐标时间的单位均为min。
表6实际样品浸泡液测试结果(ng/mL)
在一实施例中,本发明利用溶胶凝胶端羟基硅油单壁碳纳米管萃取纤维,采用气相色谱-质谱联用法,进行顶空固相微萃取,对人造地板中高关注的16种多环芳烃,在模拟汗液中的迁移量进行了分析和测定,对色谱条件和顶空固相微萃取条件进行了优化。自制萃取纤维涂层萃取效果优于进口相似类型的商用萃取纤维涂层。该方法对16种多环芳烃的检测限为0.001~0.01ng/mL,回收率为86.40~101.70%,相对标准偏差为5.74~12.03%。测得不同实际人造地板样品中多环芳烃迁移量为0.37~84.3ng/mL。
参考文献:
[1]Searle E C.Chemical carcinogens[R].Second edition.Washington DC:American Chemical Society,1984.
[2]Nisbet I C T,Lagoy P K.Toxic equivalency factors(TEFs)forpolycyclic aromatic hydrocarbons(PAHs)[J].Reg.Toxicol Phamacol.,1992,16:290.
[3]Winkler E.,Buchele A.,Mueller O.,Verfahren zur bestimmung desgehaltes an polycyclischen aromatischen kohlenwasserstoffen in mais mit hilfeder kapillargaschromatographie.J.Chromatogr.,1977,138:151-164
[4]Coates J.T.,Elzerman A.W.,Extraction and determination of selectedpolycyclic a romatic hydrocarbons in plant tissues.J.Assoc.Off.Anal.Chem.,1986,69:110
[5]Llompart M.;Li K.;Fingas,M.,Headspace solid-phase microextractionfor the dete rmination of volatile and semi-volatile pollutants in water andair.J.Chromatogr.A 1998,8 24,53–61.
[6]Saraullo A.;Martos P.A.;Pawliszyn J.,Water Analysis by Solid PhaseMicroextraction Based on Physical Chemical Properties of theCoating.Anal.Chem.1997,69,1992–1998.
[7]Asakawa F.,Jitsunari F.,Choi J.,Suna S.,Takeda N.,Kitamado T.,Method for An alyzing Urinary Toluene and Xylene by Solid-PhaseMicroextraction(SPME),and Its Applic ation to Workers Using Organic Solvents.Bull.EnViron.Contam.Toxicol.1999,62,109–116.
[8]Thomas S.,Ranjan R.,Webster G.,Sarna L.,Protocol for the Analysisof High Concentrations of Benzene,Toluene,Ethylbenzene,and Xylene Isomers inWater Using Autom ated Solid-Phase Microextraction-GC-FID.Environ.Sci.Technol.1996,30,1521–1526.
[9]Meurer E.,Tomazela D.,Silva R.,Augusto F.,Eberlin M.,FiberIntroduction Mass Spectrometry:Fully Direct Coupling of Solid-PhaseMicroextraction with Mass Spectrometry.Anal.Chem.2002,74,5688–5692.
[10]Langenfeld J.,Hawthorne S.,Miller D.,Quantitative Analysis ofFuel-Related Hyd rocarbons in Surface Water and Wastewater Samples by Solid-Phase Microextraction.Anal.Chem.1996,68,144–155.
[11]FlórezMenéndez J.C.,Fernández Sánchez M.L.,Sánchez Ur’ya J.E.,Fernández Martinez E.,Sanz-Medela A.,Static headspace,solid-phasemicroextraction and headspace solid-phase microextraction for BTEXdetermination in aqueous samples by gas chromatography.Anal.Chim.Acta.2000,415,9–20.
[12]李英,李成发,白爽等,自动固相微萃取-气相色谱-质谱法测定食品接触材料中多环芳烃(PAHs)的迁移量,质谱学报.2014,35,324-329
[13]潘永红,叶元坚,蔡锦安等,固相微萃取/气相色谱-质谱联用法测定沥青基防水涂料中18种多环芳烃的迁移量,分析测试学报,2018,7,772-777
[14]Alkalde T.,Peralba M.,Zini C.,
[15]Arambarri I.,Lasa M.,Garcia R.,Millán E.,Determination of fueldialkyl ethers and BTEX in water using headspace solid-phase microextractionand gas chromatography–flame ionization detection.J.Chromatogr.A,2004,1033,193–203.
[16]Ji J.,Deng C.,Shen W.,Zhang X.,Field analysis of benzene,toluene,ethylbenzene and xylene in water by portable gas chromatography–microflameionization detector com bined with headspace solid-phasemicroextraction.Talanta 2006,69,894–899.
[17]Gaines R.B.,Ledford E.B.,Stuart J.D.,Analysis of water samplesfor trace levels of oxygenate and aromatic compounds using headspace solid-phase microextraction and comprehensive two-dimensional gaschromatography.J.Microcolumn Sep.1998,10,597–604.
[18]Popp P.,Paschke A.,Solid phase microextraction of volatileorganic compounds using carboxen-polydimethylsiloxane fibers.Chromatographia1997,46,419-424.
[19]Bocchini P.,Andalo C.,Bonfiglioli D.,Galletti G.C.,Solid-phasemicroextractiongas chromatography/mass spectrometric analysis of volatileorganic compounds in water.Rapid Commun.Mass Spectrom.1999,13,2133–2139.
[20]Cho,H.J.,Baek K.,Lee H.H.,Lee S.H.,Yang J.W.J.,Competitiveextraction of multi-component contaminants in water by Carboxen–polydimethylsiloxane fiber during solid-phase microextraction.Chromatogr.A,2003,988,177-184.
[21]Almeida C.M.M.,Boas L.V.,Analysis of BTEX and other substitutedbenzenesin water using headspace SPME-GC-FID:method validation.J.EnViron.Monitor.2004,6,80-88.
[22]Djozan Dj.,Assadi Y.,A new porous-layer activated-charcoal-coatedfused silica fiber:Application for determination of BTEX compounds in watersamples using headspace solid-phase microextraction and capillary gaschromatography.Chromatographia.,1997,45,183-189.
[23]Iijima S.,Helica microtubules of graphitic carbon.Nature,Nature,1991,354:56-58.
[24]Iijima S.,Ichihashi T,Ichihashi T.Single-shell carbon nanotubesof 1-nm diameter,Nature,1993,363:603-605.
[25]Zhang W.Y.,Sun Y.,Wu C.Y.,et al.Polymer-Functionalized Single-Walled Carbon Nanotubes as a novel sol-gel Solid-Phase Micro-extractionCoated Fiber for Determination of Poly-brominated Diphenyl Ethers in WaterSamples with Gas Chromatography-Electron Capture Detection,Anal.Chem.,2009,81(8):2912-2920
以上应用了具体个例对本发明进行阐述,只是用于帮助理解本发明,并不用以限制本发明。对于本发明所属技术领域的技术人员,依据本发明的思想,还可以做出若干简单推演、变形或替换。