新型苯二氮杂*衍生物及其用途
文献发布时间:2023-06-19 09:23:00
相关申请
本申请要求2018年5月29日提交的美国临时申请第62/677,498号的权益,其内容通过引用整体并入本文。
发明背景
抗体-药物缀合物(ADC)是新兴的一类对多种癌症都有疗效的强大的抗肿瘤药物。ADC通常包括三种不同的特征:细胞结合剂或靶向部分;接头;和细胞毒性剂。ADC的接头组分是开发靶向抗癌剂的重要特征,所述抗癌剂具有所需的靶特异性,即在肿瘤细胞中具有高活性,但在健康细胞中具有低活性。靶向部分与细胞毒性剂(如果未靶向,可能损害健康组织)的联合使用也改变了此类细胞毒性剂所需特征的演算。因此,需要用于制备ADC的改进的接头和细胞毒性剂。
发明内容
在某些方面,本文提供了具有式(I)结构的化合物:
或其药学上可接受的盐,
其中:
A
R
Z
n
在一些实施方案中,式(I)的化合物选自:
或其药学上可接受的盐。
在某些方面,本文提供包含式(I)的化合物、可切割的接头和靶向剂的缀合物,其中可切割的接头将化合物可切割地连接至活性剂靶向剂。在优选实施方案中,靶向剂是细胞结合剂。
在某些方面,本文提供了具有式(II)、(IIa)和(IIb)的结构的化合物:
或其药学上可接受的盐,
其中:
A
R
Z
Z
L
X
Ar代表环,诸如芳基、杂芳基、环烷基或杂环烷基,优选为芳基或杂芳基;
Y
TG为触发基团,当被激活时,生成能够与SO
每个R
每个R
两个R
n
w和x各自独立地为值为0或1的整数;并且
y为值为0至5的整数。
在某些方面,本文提供了具有式(III)的结构的缀合物:
(D-L)
(III)
或其药学上可接受的盐,
其中:
LG为连接基团;
CB为细胞结合剂;
cb和dl各自独立地为值为1至约20,优选1至约10,诸如为1的整数;并且
每个D-L独立地为具有式(I)或(II)的化合物的结构的基团。
本公开还涉及包含式(I)、(II)、(IIa)、(IIb)的化合物或式(III)的缀合物和载体(例如,药学上可接受的载体)的组合物(例如,药物组合物)。
在某些方面,本公开提供了将活性剂递送至细胞的方法,包括施用式(III)的缀合物或其药物组合物,其中选择靶向部分以结合与靶细胞相关联的分子。在某些方面,本发明提供了式(III)的缀合物及其药物组合物,用于将活性剂递送至细胞的方法中,其中选择靶向部分以结合与靶细胞相关联的分子。特别地,本发明的化合物、缀合物和组合物可用在于哺乳动物(例如,人)中抑制异常细胞生长或治疗增殖性病症,诸如其中靶细胞是癌细胞且靶向部分经选择以结合与癌细胞相关联(且不与健康细胞相关联,或至少优先与肿瘤细胞而非健康细胞相关联)的分子。
式(III)的缀合物及其药物组合物可用于治疗哺乳动物(例如,人)的病症诸如癌症、类风湿性关节炎、多发性硬化、移植物抗宿主病(GVHD)、移植排斥、狼疮、肌炎、感染、免疫缺陷诸如AIDS和炎性疾病。
附图简述
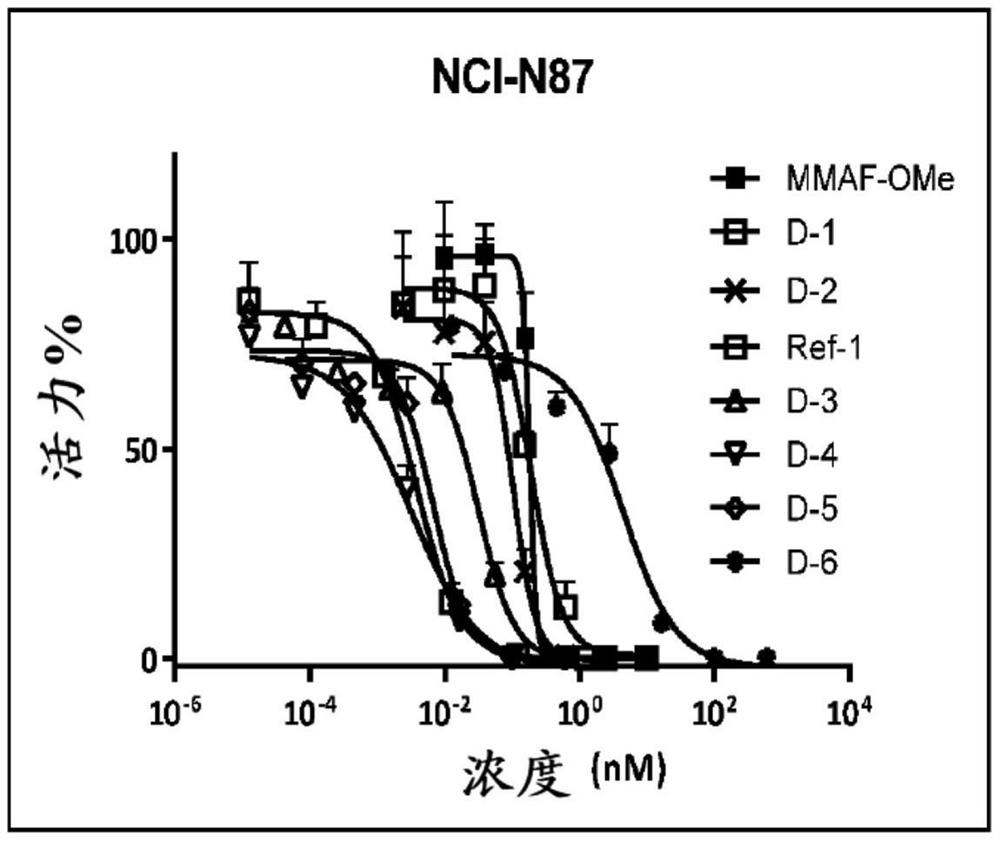
图1显示化合物D-1、D-2、D-3、D-4、D-5和D-6对NCI-N87的体外细胞毒性活性。
图2显示了化合物D-1、D-2、D-3、D-4、D-5和D-6对JIMT-1的体外细胞毒性活性。
图3显示化合物D-1、D-2、D-3和D-5对Calu-6的体外细胞毒性活性。
图4显示了化合物D-1、D-2、D-3和D-5对A-498的体外细胞毒性活性
图5显示了化合物D-1、D-2、D-3和D-5对HCT-116的体外细胞毒性活性。
图6显示了化合物D-1、D-2、D-3和D-5对MIA-PaCa-2的体外细胞毒活性。
图7显示了化合物D-1、D-2、D-3和D-5对PC-3的体外细胞毒性活性。
图8显示了化合物D-1、D-2、D-3和D-5对HEK-293E的体外细胞毒性活性。
图9显示了化合物D-1、D-2、D-3和D-5对SK-OV-3的体外细胞毒性活性。
图10显示了针对NCI-N87的缀合物T-2-AB、T-3-AB和T-7-AB的体外分析。
图11显示了针对JIMT-1的缀合物T-2-AB、T-3-AB和T-7-AB的体外分析。
图12显示了针对SK-BR-3的缀合物T-2-AB、T-3-AB和T-7-AB的体外分析。
图13和14显示了缀合物T-2-AB的体内测试。
具体实施方式
一些吡咯并苯二氮
PBD的一般结构为:
它们的芳香族A环和吡咯并C环两者中的取代基的数目、类型和位置不同,C环的饱和程度也不同。在B-环中,在N10-C11位(其为负责烷化DNA的亲电中心)存在亚胺(N=C)、甲醇胺(NH-CH(OH))或甲醇胺甲基醚(NH-CH(OMe))。所有已知的天然产物在手性CI la位置都具有(S)-构型,当从C环向A环看时,所述构型为它们提供了右手扭转(right-handedtwist)。这赋予它们适当的三维形状以实现与B型DNA的小沟的等螺旋度(isohelicity),导致在结合位点处的适贴配合(snug fit)(Kohn,In Antibiotics III.Springer-Verlag,New York,第3-11页(1975);Hurley和Needham-VanDevanter,Acc.Chem.Res.,19:230-237(1986))。它们在小沟中形成加合物的能力使其能够干扰DNA加工,因此它们可用作抗肿瘤剂。
吲哚啉苯并二氮
本文提供了包含PBD或IBD化合物和任选的可切割接头的化合物及其缀合物,及其用途。在某些实施方案中,本文公开的化合物和缀合物包含活性剂(例如,式(I)的化合物)和SO
定义
除非本文另有定义,否则本申请中使用的科学和技术术语应具有本领域普通技术人员通常理解的含义。通常,本文所述的与化学、细胞和组织培养、分子生物学、细胞和癌症生物学、神经生物学、神经化学、病毒学、免疫学、微生物学、药理学、遗传学以及蛋白质和核酸化学结合使用的术语以及所述学科的技术是本领域公知的和常用的术语及技术。
除非另有说明,本公开的方法和技术通常根据本领域公知的常规方法并且如在贯穿本说明书引用和论述的各种一般和更具体的参考文献中所述来执行。参见,例如,“Principles of Neural Science”,McGraw-Hill Medical,New York,N.Y.(2000);Motulsky,“Intuitive Biostatistics”,Oxford University Press,Inc.(1995);Lodish等人,“Molecular Cell Biology,第4版”,W.H.Freeman&Co.,New York(2000);Griffiths等人,“Introduction to Genetic Analysis,第7版”,W.H.Freeman&Co.,N.Y.(1999);和Gilbert等人,“Developmental Biology,第6版”,Sinauer Associates,Inc.,Sunderland,MA(2000).
除非本文另有定义,否则本文使用的化学术语均按照本领域常规用法使用,如由“The McGraw-Hill Dictionary of Chemical Terms”,Parker S.,编辑,McGraw-Hill,SanFrancisco,C.A.(1985)所列举的。
在本申请中提及的所有上述以及任何其它出版物、专利和已公布的专利申请明确地通过引用并入本文。如有冲突,以本说明书(包括其具体定义)为准。
术语“试剂”在本文中用于表示化学化合物(例如有机或无机化合物、化合物的混合物)、生物大分子(诸如核酸、抗体,包括其部分以及人源化抗体、嵌合抗体和人抗体及单克隆抗体、蛋白质或其部分,例如肽、脂质、碳水化合物)或由生物材料诸如细菌、植物、真菌或动物(特别是哺乳动物)细胞或组织制备的提取物。剂包括例如其结构已知的剂和其结构未知的剂。
“患者”、“受试者”或“个体”可互换使用,指人或非人动物。这些术语包括哺乳动物,诸如人、灵长类动物、家畜动物(包括牛、猪等)、伴侣动物(例如,犬科动物、猫科动物等)和啮齿动物(例如,小鼠和大鼠)。
“治疗”疾患或患者是指采取步骤以获得有益或期望的结果,包括临床结果。如本文中所用以及本领域所充分理解的,“治疗”是用于获得有益的或期望的结果,包括临床结果的方法。有益的或期望的临床结果可包括但不限于缓解或改善一种或多种症状或疾患、减轻疾病程度、稳定(即不恶化)疾病状态、防止疾病传播、延迟或减慢疾病进展、改善或减轻疾病状态以及缓解(无论是部分还是完全),无论是可检测的还是不可检测的。“治疗”还可意指与未接受治疗的预期生存期相比延长生存期。
术语“预防”是本领域公认的,并且当与疾患诸如局部复发(例如,疼痛)、疾病诸如癌症、综合征(syndrome complex)诸如心力衰竭或任何其它医学病症相关时,在本领域中是公知的,并且包括施用组合物,所述组合物相对于未接受所述组合物的受试者减少受试者中医学疾患症状的频率或延迟其发作。因此,癌症的预防包括例如根据统计学上和/或临床上显著的量,例如,相对于未治疗的对照群体,减少接受预防性治疗的患者群体中可检测的癌性生长的数量,和/或相对于未治疗的对照群体,延迟治疗群体中可检测的癌性生长的出现。
向受试者“施用”物质、化合物或剂或物质、化合物或剂“的施用”可使用本领域技术人员已知的多种方法之一来进行。例如,可通过静脉内、动脉内、皮内、肌内、腹膜内、皮下、眼内、舌下、口服(通过摄入)、鼻内(通过吸入)、脊柱内、大脑内和经皮肤(通过吸收,例如通过皮肤导管)施用化合物或剂。化合物或剂还可适当地通过可再装填或可生物降解的聚合物装置或其它装置(例如,贴片和泵或制剂)引入,所述装置提供化合物或剂的延长、缓慢或受控释放。施用还可例如进行一次、多次和/或在一个或多个延长的时期内进行。
向受试者施用物质、化合物或剂的适当方法还将取决于例如受试者的年龄和/或身体状况以及化合物或剂的化学和生物性质(例如,溶解度、消化率、生物利用度、稳定性和毒性)。在一些实施方案中,化合物或剂通过口服施用,例如通过摄入向受试者施用。在一些实施方案中,口服施用的化合物或剂是缓释制剂或缓慢释放制剂,或使用用于这种缓慢或延长释放的装置施用。
如本文中所用,短语“联合施用”是指两种或多种不同治疗剂的任何施用形式,所述施用形式使得在先前施用的治疗剂在体内仍有效的同时施用第二种剂(例如,两种剂在患者体内同时有效,这可包括两种剂的协同作用)。例如,不同的治疗性化合物可以在同一制剂中施用或以单独的制剂同时或依次施用。因此,接受这种治疗的个体可受益于不同治疗剂的联合作用。
药物或剂的“治疗有效量”或“治疗有效剂量”是指当向受试者施用时将具有预期治疗效果的药物或剂的量。一个剂量的施用不一定产生完全的治疗效果,完全的治疗效果只有在一系列剂量施用后才可能产生。因此,可以一次或多次施用治疗有效量。受试者所需的精确有效量将取决于,例如,受试者的体型大小、健康状况和年龄,以及所治疗疾患的性质和程度,诸如癌症或MDS。熟练技术人员可通过常规实验容易地确定给定状况下的有效量。
如本文中所用,术语“烯基”是指含有至少一个双键的脂肪族基团,并旨在包括“未取代的烯基”和“取代的烯基”,后者是指在烯基的一个或多个碳上具有取代氢的取代基的烯基部分。此类取代基可出现在包含在或未包含在一个或多个双键中的一个或多个碳上。此外,如下文所论述,此类取代基包括所有设想用于烷基的取代基,除非稳定性是禁止的。例如,设想了一个或多个烷基、碳环基、芳基、杂环基或杂芳基对烯基的取代。
“烷基”基团或“烷烃”是完全饱和的直链或支链非芳族烃。通常,除非另有定义,否则直链或支链烷基具有1至约20个碳原子,优选1至约10个碳原子。直链和支链烷基的实例包括甲基、乙基、正丙基、异丙基、正丁基、仲丁基、叔丁基、戊基、己基、戊基和辛基。C
此外,如说明书、实施例和权利要求通篇中所用,术语“烷基”(或“低级烷基”)旨在包括“未取代的烷基”和“取代的烷基”,其后者是指在烃主链的一个或多个碳上具有替代氢的取代基的烷基部分。如果没有另外说明,此类取代基可包括例如卤素(例如,氟)、羟基、羰基(诸如羧基、烷氧基羰基、甲酰基或酰基)、硫代羰基(诸如硫酯、硫代乙酸酯或硫代甲酸酯)、烷氧基、磷酰基、磷酸酯、膦酸酯、次膦酸酯、氨基、酰氨基、脒、亚胺、氰基、硝基、叠氮基、巯基、烷硫基、硫酸酯、磺酸酯、氨磺酰基(sulfamoyl)、磺酰氨基(sulfonamido)、磺酰基(sulfonyl)、杂环基、芳烷基或芳族或杂芳族部分。在优选实施方案中,取代的烷基上的取代基选自C
当与化学部分诸如酰基、酰氧基、烷基、烯基、炔基或烷氧基结合使用时,术语“C
如本文中所用,术语“酰胺”是指其中
每个R
术语“胺”和“氨基”是本领域公认的,并且是指未取代的和取代的胺及其盐,例如可以由下式表示的部分
其中每个R
如本文中所用,术语“芳基”包括取代或未取代的单环芳族基团,其中环的每个原子是碳。优选地,所述环是6元或10元环,更优选为6元环。术语“芳基”还包括具有两个或更多个环的多环环体系,其中两个或更多个碳对于两个邻接的环是共同的,其中至少一个环是芳族的,例如,另外的环可以是环烷基、环烯基、环炔基、芳基、杂芳基和/或杂环基。芳基包括苯、萘、菲、苯酚、苯胺等。
“环烷基”基团为完全饱和的环烃。“环烷基”包括单环和双环环。通常,除非另有定义,否则单环烷基具有3至约10个碳原子,更常见地为3至8个碳原子。双环环烷基的第二环可选自饱和环、不饱和环和芳族环。环烷基包括其中在两个环之间共有一个、两个或三个或更多个原子的双环分子。术语“稠合环烷基”是指其中每个环与另一个环共有两个相邻原子的双环环烷基。稠合双环环烷基的第二环可选自饱和环、不饱和环和芳族环。“环烯基”基团为含有一个或多个双键的环状烃。
如本文中所用,术语“卤代”和“卤素”意指卤素,包括氯、氟、溴和碘。
术语“杂芳基(heteroaryl)”和“杂芳基(hetaryl)”包括取代或未取代的芳族单环结构,优选5至7元环,更优选5至6元环,其环结构包括至少一个杂原子,优选1至4个杂原子,更优选1或2个杂原子。术语“杂芳基(heteroaryl)”和“杂芳基(hetaryl)”还包括具有两个或更多个环的多环环体系,其中两个或更多个碳对于两个邻接的环是共同的,其中至少一个环是杂芳族的,例如,另外的环可以是环烷基、环烯基、环炔基、芳基、杂芳基和/或杂环基。杂芳基包括例如吡咯、呋喃、噻吩、咪唑、噁唑、噻唑、吡唑、吡啶、吡嗪、哒嗪和嘧啶等。
如本文中所用,术语“杂原子”意指除碳或氢外的任何元素的原子。优选杂原子是氮、氧和硫。
术语“杂环基”、“杂环”和“杂环的”是指取代或未取代的非芳族环结构,优选为3元至10元环,更优选为3元至7元环,其环结构包括至少一个杂原子,优选1至4个杂原子,更优选1或2个杂原子。术语“杂环基”和“杂环的”还包括具有两个或更多个环的多环环体系,其中两个或更多个碳对于两个邻接的环是共同的,其中至少一个环是杂环的,例如,另外的环可以是环烷基、环烯基、环炔基、芳基、杂芳基和/或杂环基。杂环基包括例如哌啶、哌嗪、吡咯烷、四氢吡喃、四氢呋喃、吗啉、内酯类、内酰胺类等。
当与化学部分诸如酰基、酰氧基、烷基、烯基、炔基或烷氧基结合使用时,术语“低级”意指包括其中取代基中有十个或更少,优选六个或更少的非氢原子的基团。例如,“低级烷基”是指含有十个或更少,优选六个或更少碳原子的烷基。在某些实施方案中,本文定义的酰基、酰氧基、烷基、烯基、炔基或烷氧基取代基分别为低级酰基、低级酰氧基、低级烷基、低级烯基、低级炔基或低级烷氧基,无论它们单独出现还是与其它取代基组合出现,诸如在羟基烷基和芳烷基的叙述中(在该情况下,例如当计算烷基取代基中的碳原子数时,不计算芳基中的原子数)。
术语“取代的”是指在主链的一个或多个碳上具有替代氢的取代基的部分。应当理解,“取代”或“被取代的”包括隐含的条件,即这种取代与被取代的原子和取代基的允许价一致,并且取代产生稳定的化合物,例如,其不会自发地经历诸如通过重排、环化、消除等的转化。如本文中所用,设想了术语“取代的”包括有机化合物的所有可允许的取代基。在广泛方面,可允许的取代基包括有机化合物的无环和环状、支化和非支化、碳环和杂环、芳族和非芳族取代基。对于适当的有机化合物,可允许的取代基可以是一个或多个以及可以相同或不同。为了本发明的目的,杂原子诸如氮可具有氢取代基和/或满足杂原子的价态的本文所述有机化合物的任何可允许的取代基。取代基可包括本文所述的任何取代基,例如卤素、羟基、羰基(诸如羧基、烷氧基羰基、甲酰基或酰基)、硫代羰基(诸如硫酯、硫代乙酸酯或硫代甲酸酯)、烷氧基、磷酰基、磷酸酯、膦酸酯、次膦酸酯、氨基、酰氨基、脒、亚胺、氰基、硝基、叠氮基、巯基、烷硫基、硫酸酯、磺酸酯、氨磺酰基、磺酰氨基、磺酰基、杂环基、芳烷基或芳族或杂芳族部分。在优选实施方案中,取代的烷基上的取代基选自C
“保护基团”是指当与分子中的反应性官能团连接时,掩蔽、降低或阻止该官能团的反应性的一组原子。通常,在合成过程中可以根据需要选择性地除去保护基团。保护基团的实例可见于Greene和Wuts,Protective Groups in Organic Chemistry,第3版,1999,John Wiley&Sons,NY和Harrison等人,Compendium of Synthetic Organic Methods,第1-8卷,1971-1996,John Wiley&Sons,NY。代表性氮保护基团包括但不限于甲酰基、乙酰基、三氟乙酰基、苄基、苄氧基羰基(“CBZ”)、叔丁氧羰基(“Boc”)、三甲基甲硅烷基(“TMS”)、2-三甲基甲硅烷基-乙磺酰基(“TES”)、三苯甲基和取代的三苯甲基、烯丙氧基羰基、9-芴甲氧羰基(“FMOC”)、硝基-藜芦基氧羰基(“NVOC”)等。代表性羟基保护基团包括但不限于其中羟基被酰化(酯化)或烷基化的那些,诸如苄基醚和三苯甲基醚,以及烷基醚、四氢吡喃基醚、三烷基甲硅烷基醚(例如,TMS或TIPS基团)、乙二醇醚,诸如乙二醇和丙二醇衍生物和烯丙基醚。
如本文中所用,术语“调节”包括功能或活性(诸如细胞增殖)的抑制或压制以及功能或活性的增强。
短语“药学上可接受的”是本领域公认的。在某些实施方案中,该术语包括组合物、赋形剂、佐剂、聚合物和其它材料和/或剂型,其在合理的医学判断范围内,适合用于与人类和动物的组织接触,而没有过度的毒性、刺激、过敏反应或其它问题或并发症,与合理的效益/风险比相称。
“药学上可接受的盐”或“盐”在本文中用于指适用于患者治疗或与患者治疗相容的酸加成盐或碱加成盐。
如本文中所用,术语“药物可接受的酸加成盐”意指由式I表示的任何碱化合物的任何无毒有机或无机盐。形成合适盐的示例性无机酸包括盐酸、氢溴酸、硫酸和磷酸,以及金属盐诸如正磷酸单氢钠和硫酸氢钾。形成合适盐的示例性有机酸包括单羧酸、二羧酸和三羧酸,诸如乙醇酸、乳酸、丙酮酸、丙二酸、琥珀酸、戊二酸、富马酸、苹果酸、酒石酸、柠檬酸、抗坏血酸、马来酸、苯甲酸、苯乙酸、肉桂酸和水杨酸以及磺酸,诸如对甲苯磺酸和甲磺酸。可形成单酸盐或二酸盐,并且此类盐可以以水合、溶剂化或基本无水的形式存在。一般而言,式I的化合物的酸加成盐与其游离碱形式相比在水和各种亲水有机溶剂中更易溶解,并且通常显示出更高的熔点。合适的盐的选择是本领域技术人员已知的。其它非学上可接受的盐,例如草酸盐,可用于例如分离供实验室使用或随后转化为药学上可接受的酸加成盐的式I的化合物。
如本文中所用,术语“药物可接受的碱性加成盐”意指式I所示的任何酸化合物或其任何中间体的任何无毒有机或无机碱加成盐。形成合适盐的示例性无机碱包括氢氧化锂、氢氧化钠、氢氧化钾、氢氧化钙、氢氧化镁或氢氧化钡。形成合适盐的示例性有机碱包括脂肪族、脂环族或芳香族有机胺,诸如甲胺、三甲胺和甲基吡啶或氨。合适的盐的选择是本领域技术人员已知的
在本公开的方法和组合物中有用的许多化合物在其结构中具有至少一个立构中心(stereogenic center)。该立构中心可以以R或S构型存在,所述R和S符号按PureAppl.Chem.(1976),45,11-30中描述的规则使用。本公开设想了化合物、其盐、前药或混合物的所有立体异构形式,诸如对映异构体和非对映异构体形式(包括所有可能的立体异构体混合物)。参见,例如,WO 01/062726。
此外,某些含有烯基的化合物可以以Z(同侧(zusammen))或E(异侧(entgegen))异构体的形式存在。在每种情况下,本公开包括混合物和单独的各异构体。
所述化合物中的一些还可以以互变异构形式存在。尽管在本文所述的公式中未明确指出,但此类形式意欲包含在本发明的范围内。
“前药”或“药学上可接受的前药”是指施用后在宿主中代谢(例如水解或氧化)以形成本公开的化合物(例如,式I化合物)的化合物。前药的典型实例包括在活性化合物的功能部分上具有生物不稳定或可裂解(保护)基团的化合物。前药包括可被氧化、还原、胺化、脱氨基、羟基化、脱羟基化、水解、脱水、烷基化、脱烷基化、酰化、脱酰化、磷酸化或脱磷酸化以产生活性化合物的化合物。在美国专利6,875,751、7,585,851和7,964,580中公开了使用酯或氨基磷酸酯作为生物不稳定或可裂解(保护)基团的前药的实例,所述美国专利的公开内容通过引用并入本文。本公开的前药被代谢以产生式I的化合物。本公开在其范围内包括本文所述化合物的前药。选择和制备合适的前药的常规程序描述于例如“Design ofProdrugs”H.Bundgaard编辑,Elsevier,1985中。
如本文中所用,短语“药学上可接受的载体”意指药学上可接受的材料、组合物或媒介物,诸如液体或固体过滤器、稀释剂、赋形剂、溶剂或用于配制药用或治疗用途的药物的包封材料。
术语“异常细胞生长”和“增殖性病症”在本申请中可互换使用。除非另有说明,否则如本文中所用,“异常细胞生长”是指不依赖于正常调控机制(例如,失去接触抑制)的细胞生长。这包括,例如,以下的异常生长:(1)通过表达突变的酪氨酸激酶或过表达受体酪氨酸激酶而增殖的肿瘤细胞(多个肿瘤);(2)其中发生异常酪氨酸激酶激活的其它增殖性疾病的良性和恶性细胞;(3)通过受体酪氨酸激酶增殖的任何肿瘤;(4)通过异常丝氨酸/苏氨酸激酶激活而增殖的任何肿瘤;和(5)其中发生异常丝氨酸/苏氨酸激酶激活的其它增殖性疾病的良性和恶性细胞。
术语“癌症”和“癌性”是指或描述哺乳动物中特征通常在于细胞生长不受调控的生理状况。“肿瘤”包含一个或多个癌细胞。癌症的实例包括但不限于癌、淋巴瘤、母细胞瘤、肉瘤和白血病或淋巴样恶性肿瘤(lymphoid malignancies)。此类癌症的更特定实例包括鳞状细胞癌(例如,上皮鳞状细胞癌)、包括小细胞肺癌、非小细胞肺癌(“NSCLC”)、肺腺癌和肺鳞状细胞癌在内的肺癌、腹膜癌、肝细胞癌、包括胃肠癌在内的胃癌(gastric cancer)或胃癌(stomach cancer)、胰腺癌、胶质母细胞瘤、宫颈癌、卵巢癌、肝癌、膀胱癌、肝瘤、乳腺癌、结肠癌、直肠癌、结直肠癌、子宫内膜癌或子宫癌、唾液腺癌、肾癌(kidney cancer)或肾癌(renal cancer)、前列腺癌、外阴癌、甲状腺癌、肝癌、肛门癌、阴茎癌、急性白血病以及头/脑和颈癌。
本发明的化合物和缀合物
本发明提供具有式(I)的结构的化合物:
或其药学上可接受的盐,
其中:
A
R
Z
n
在一些实施方案中,A
其中:
R
R
R
Ar
o为值为1或2的整数;并且
p为值为0至5的整数。
在一些此类实施方案中,Ar
在一些实施方案中,A
在一些实施方案中,A
在一些实施方案中,A
R
R
R
Ar
R
每个R
q为值为1或2的整数;并且
r为值为0至5的值的整数。
在一些实施方案中,A
W
R
每个W
R
在一些此类实施方案中,A
在某些实施方案中,式(I)的化合物是具有式(Ia)、(Ib)或(Ic)的结构的化合物:
或其药学上可接受的盐,其中:
R
R
R
R
R
Ar
R
每个R
Ar
R
每个R
o和q为各自独立地具有1或2的值的整数。
在一些此类实施方案中,Ar
在一些实施例中,R
在一些实施方案中,Z
在某些实施方案中,Z
在其它实施方案中,Z
Y
R
R
Y为值为1至约10的整数。在优选实施方案中,Z
在更优选实施方案中,Z
在其它实施方案中,Z
Y
R
R
R
z为值为0至约10的整数。
在某些优选实施方案中,Z
在某些实施方案中,式(I)的化合物选自:
或其药学上可接受的盐。
在一些实施方案中,本文提供包了含式(I)的化合物、可切割的接头和靶向剂的缀合物,其中可切割的接头将化合物可切割地连接至靶向剂。在优选实施方案中,靶向剂是细胞结合剂。在一些实施方案中,接头包含在预定条件下经历化学反应(例如,物理化学反应和/或生物反应)以释放亲核杂原子的官能团,和位于亲核杂原子附近的SO
本文还提供了具有式(II)、(IIa)和(IIb)结构的化合物
或其药学上可接受的盐,
其中:
A
R
Z
n
Z
L
X
Ar表示环,诸如芳基、杂芳基、环烷基或杂环烷基,优选为芳基或杂芳基;
Y
TG为触发基团,当被激活时生成能够与SO
w和x各自独立地为值为0或1的整数;
每个R
每个R
两个R
y为值为1至约10的整数。
在一些实施方案中,X
在其它实施方案中,Ar为芳基。在优选实施方案中,苯基或萘基。
在又一其它实施方案中,Z
在其它实施方案中,x为0。
在各种实施方案中,式(II)、(IIa)和(IIb)的变量与上述式(I)中定义的相同。
在美国临时申请第62/597,226号中提供了缀合物、连接基团、触发基团等的其它实施方案,所述美国临时申请通过引用特此整体并入。
如上所述,在某些实施方案中,本文公开的化合物和缀合物能够通过激活触发基团的化学反应之后的分子内环化反应来解离由式(II)表示的一种或多种活性剂。在某些实施方案中,化学反应是物理化学反应和/或生物化学反应。
在一些实施方案中,本文公开的化合物和缀合物包含在Ar上相对于X(例如,O)的相邻原子处引入的亲核官能团(Y或Y’)。通常,亲核官能团被触发基团(TG)掩蔽,如下文进一步详述。激活后,触发基团释放亲核官能团,与附近的SO
例如,当Y为-Y'-TG且Q与SO
当Q为
在一些实施方案中,释放时的Q
在一些实施方案中,本文公开的化合物和缀合物是在化学和生理方面稳定的。在一些此类实施方案中,本文公开的化合物和缀合物在血液中几乎没有活性剂解离的状态下到达所需靶细胞,从而选择性地释放药物。
在一些实施方案中,本发明的缀合物包括触发基团(TG)。TG为能够通过化学反应诸如生物反应裂解,优选选择性裂解的基团。通常,触发基团用于掩蔽Y或Y’基团的亲核性质,从而为本文公开的化合物和缀合物提供稳定性(例如,通过阻止缀合物在到达靶位置或经历预定触发条件之前自蚀(self-immolation)或分子内环化)。激活后,触发基团释放亲核Y基团,并允许发生自蚀或分子内环化,如上所述的。
在一些实施方案中,TG包含可被TEV、胰蛋白酶、凝血酶、组织蛋白酶B、组织蛋白酶D、组织蛋白酶K、半胱天冬酶1、基质金属蛋白酶(MMP)等识别的序列(诸如肽序列)或部分,其可被酶(例如,氧化还原酶、转移酶、水解酶、裂解酶、异构酶、连接酶等)水解和/或可包括选自磷酸二酯、磷脂、酯、β-半乳糖、β-葡萄糖、岩藻糖、寡糖等的部分。
在一些实施方案中,TG包含可在亲核试剂条件下裂解的反应性化学部分或官能团(例如,甲硅烷基醚、2-N-酰基硝基苯磺酰胺、不饱和乙烯基硫化物、激活后的磺酰胺、丙二醛-吲哚衍生物、乙酰丙酸酯(levulinoyl ester)、腙或酰基腙)。
在一些实施方案中,TG可包含可在碱性试剂条件下裂解的反应性化学部分或官能团(例如,2-氰乙基酯、乙二醇基二琥珀酸酯(ethylene glycolyl disuccinate)、2-磺酰基乙基酯、烷基硫代酯或噻吩基酯)。
在一些实施方案中,TG可包含可通过光照射裂解的反应性化学部分或官能团(例如,2-硝基苄基衍生物、苯酰基酯、8-喹啉基苯磺酸酯、香豆素、磷酸三酯、双芳基腙或双满双硫代丙酸衍生物(bimane bi-thiopropionic acid derivative))。
在一些实施方案中,TG可包含可被还原剂条件裂解的反应性化学部分或官能团(例如,羟胺、二硫化物、乙酰丙酸酯、硝基或4-硝基苄基衍生物)。
在一些实施方案中,TG可包含可使用酸性条件裂解的反应性化学部分或官能团(例如,糖、叔丁基氨基甲酸酯类似物、二烷基或二芳基二烷氧基硅烷、原酸酯、缩醛、乌头基、腙、β-硫代丙酸酯、氨基磷酸酯、亚胺、三苯甲基、乙烯基醚、聚缩酮和2-(二苯基膦基)苯甲酸烷基酯衍生物;烷基酯、8-羟基喹啉酯和吡啶甲酸酯)。
在一些实施方案中,TG可包含可在氧化条件下裂解的反应性化学部分或官能团(例如,硼酸盐、邻二醇、对甲氧基苄基衍生物或硒化合物)。
在某些优选实施方案中,TG包含糖,其可在酸性或酶促条件下裂解。在某些优选实施方案中,触发基团为-NO
在一些实施方案中,本文公开的化合物和缀合物包含糖触发基团,例如选自以下的触发基团:
其中每个R
作为触发基团的保护基团
在一些实施方案中,TG为能够被化学反应、物理化学反应和/或生物反应裂解的基团。在某些实施方案中,TG为保护基团。在一些此类实施方案中,保护基为胺基保护基团、醇保护基团或硫醇保护基。
在某些实施方案中,胺保护基团是能够用于有机合成的一般保护基团,包括但不限于:氨基甲酸间硝基苯酯、氨基甲酸3,5-二甲氧基苄酯、氨基甲酸邻硝基苯酯、氨基甲酸苯基(邻硝基苯基)甲酯、氨基甲酸烷基酯、氨基甲酸9-芴基甲酯、氨基甲酸2,2,2-三氯乙酯、氨基甲酸2-三甲基甲硅烷基乙酯(Teoc)、氨基甲酸叔丁酯(Boc)、氨基甲酸乙烯酯(Voc)、氨基甲酸烯丙酯(Alloc)、氨基甲酸1-异丙基烯丙酯(Ipaoc)、氨基甲酸8-喹啉酯、氨基甲酸N-羟基哌啶酯、氨基甲酸苄酯、氨基甲酸对甲氧基苄酯、氨基甲酸对硝基苄酯、氨基甲酸二苯基甲酯、乙酰胺、氯乙酰胺、三氯乙酰胺、苯基乙酰胺、苯甲酰胺、N-邻苯二甲酰亚胺、N-2,3-二苯基马来酰亚胺、N-2,5-二甲基吡咯、N-1,1-二甲基硫代亚甲基胺、N-亚苄基胺、苯亚磺酰胺、邻硝基苯亚磺酰胺、三苯基甲基亚磺酰胺、对甲苯磺酰胺、甲磺酰胺等。但不限于此。
在某些实施方案中,醇保护基团是能够用于有机合成的一般保护基团,其包括但不限于:甲醚、甲氧基甲基醚(MOM醚)、苄基氧基甲基醚(BOM醚)、2-(三甲基硅烷基)乙氧基甲基醚(SEM醚)、苯基硫代甲基醚(PTM醚)、2,2-二氯-1,1-二氟乙基醚、对溴苯酰基醚、氯丙基甲基醚、异丙基醚、环己基醚、4-甲氧基苄基、2,6-二氯苄基醚、4-(二甲基氨基羰基)苄基醚、9-蒽基甲醚、4-甲基吡啶基醚、甲硫基甲基醚(MTM醚)、2-甲氧基乙氧基甲基醚(MEM醚)、双(2-氯乙氧基)甲基醚、四氢吡喃基醚(THP醚)、四氢硫代吡喃基醚、4-甲氧基四氢吡喃基醚、四氢呋喃基醚、1-乙氧基乙基醚、1-甲基-1-甲氧基乙基醚、2-(苯基硒基)乙基醚)、叔丁基醚、烯丙基醚、苄基醚、邻硝基苄基醚、三苯基甲基醚、α-萘基二苯基甲基醚、对-甲氧基苯基二苯基甲基醚、9-(9-苯基-10-氧代)蒽基醚、三甲基甲硅烷基醚(TMS醚)、异丙基二甲基甲硅烷基醚、叔丁基二甲基甲硅烷基醚(TBDMS醚)、叔丁基二苯基甲硅烷基醚、三苄基甲硅烷基醚、三异丙基甲硅烷基醚、甲酸酯、乙酸酯、三氯乙酸酯、苯氧基乙酸酯、异丁酸酯、新戊酸酯、金刚烷酸酯、苯甲酸酯、2,4,6-三甲基苯甲酸(Mesitoate)酯、碳酸甲酯、碳酸2,2,2-三氯乙酯、碳酸烯丙酯、碳酸对硝基苯酯、苄基碳酸酯、对硝基苄基碳酸酯、S-苄基碳酸酯、N-苯基氨基甲酸酯、硝酸酯、2,4-二硝基苯基亚磺酸酯、二甲基膦酰酯(DMP酯)、二甲基硫代膦酰酯(MPT酯)、甲磺酸芳基酯、甲苯磺酸芳基酯等,但不限于此。
在某些实施方案中,硫醇保护基团能够用于有机合成,包括但不限于:S-苄基硫醚、S-对甲氧基苄基硫醚、S-邻或对羟基或乙酰氧基苄基硫醚、S-对硝基苄基硫醚、S-4-吡啶甲基硫醚、S-2-吡啶甲基N-氧化物硫醚、S-9-蒽基甲基硫醚、S-9-芴基甲基硫醚、S-甲氧基甲基单缩硫醛、A-乙酰基衍生物、S-苯甲酰基衍生物、S-(N-乙基氨基甲酸酯)、S-(N-甲氧基甲基氨基甲酸酯)等,但不限于此。
在一些实施方案中,本文公开的化合物和缀合物包含通过共价键连接每个CB与Ar的连接基团。典型的连接基团是稳定的、不可水解的部分,诸如,例如C
(i)亚烷基部分中的至少一个-CH
(ⅱ)至少一个杂亚芳基包含在亚烷基部分中;
(ⅲ)至少一个氨基酸部分、糖键、肽键或酰胺键包含在亚烷基部分中;以及
(ⅳ)亚烷基可进一步被一个或多个选自由以下组成的组的取代基取代:C
在某些实施方案中,所述连接单元包括以下中的至少两种,更优选至少三种:
(i)至少一种选自-NH-、-C(=O)、-O-、-S-和-P-的杂原子;
(ii)至少一种杂亚芳基;
(iii)至少一种氨基酸部分、糖键、肽键或酰胺键;以及
(ⅳ)亚烷基可进一步被一个或多个选自由以下组成的组的取代基取代:C
在其它实施方案中,连接每个CB和Ar的连接基团包含通过点击化学反应产生的官能团。
在替代实施方案中,连接单元包含能够参与点击化学反应的反应性官能团。
点击化学反应是可在温和条件下进行的反应,并且对于在生物分子中不常见的官能团(例如,叠氮化物基团、乙炔基团等)具有极高的选择性。因此,该反应可在复合触发基团、靶向部分等存在的情况下进行。另外,点击化学具有高反应特异性。例如,叠氮化物基团与乙炔基团之间的点击化学反应可选择性地进行,而不受分子中存在的其它官能团的干扰。例如,叠氮化物-乙炔点击化学可以以高产率提供三唑部分。
因此,在一些实施方案中,连接每个CB与Ar的连接基团包括
在其它实施方案中,连接每个CB与Ar的连接单元是由式(A)表示的连接基团:
**-L
(A)
其中:
*为与CB的附接点;
**为与Ar的附着点;
W
W
P
L
Y
R
R
X为-NHC(=O)-(CH
W
W
W
Y
X’为-O-、-S-、-NH-或-CH
CB与如上定义的相同;
b、c、d、e、f、g、h、i和j各自独立地为值为1至约10的整数;
k和y各自独立地为值为0至约10的整数;
Y
X”为-O-、-S-、-NH-或-CH
o和q为值为1至约10的整数。
在一些实施方案中,P
其中:
R
R
R
X”为-O-、-S-、-NH-或-CH
Z和CB与上述定义相同;
p为值为1至约10的整数;
s和s”为值为0至约10的整数;
s’为值为1至约10的值的整数;并且
m为值为0或1的整数。
在式(B)或(C)的一些实施方案中:
R
p为值为1至约10的整数;
s为值为0至约10的整数;
R
R
s”为值为0至约10的整数;
s’为值为1至约10的整数;
m为值为0或1的整数;
X”’为-O-、-S-、-NH-或-CH
Z”为连接CB与R
在式(B)或(C)的一些此类实施方案中:
R
R
Z”为连接单元的反应性前体,其选自异氰酸酯、异硫氰酸酯、2-吡啶基二硫化物、卤代乙酰胺(-NHC(O)CH
在式(B)或(C)的其它此类实施方案中:
R
R
Z”为连接CB与R
在一些实施方案中,Y
其中:
W
R
R
X
W
c、d、e、f、g、h、i和j各自独立地为值为1至约10的整数;
X”为-O-、-S-、-NH-或-CH
L
在某些实施方案中,连接每个CB与Ar的连接单元为包含通过共价键彼此连接的(CH
W
W
P
L
Y
X”为O-、-S-、-NH-或-CH
Y
W
a为0至10;
b、c和d各自独立地为值为1至约10的整数;并且
o和q各自独立地为值为1至约10的整数。
在一些实施方案中,R
在一些实施方案中,连接每个CB和Ar的连接单元是由式(A)表示的连接基团:
**-L
(A)
其中:
*为CB的附接点;并且
**为Ar的附接点。
在一些此类实施方案中,P
其中:
R
p为值为1至约10的整数;并且
s和s”各自独立地为值为0至约10的整数。
在一些实施方案中,P
其中:
R
p为值为1至约10的整数;
s为值为0至约10的整数;
R
R
s”为值为0至约10的整数;
s’为值为1至约10的整数;
m为值为0或1的整数;
X”’为-O-、-S-、-NH-或-CH
Z”为连接CB与R
在P
R
R
Z”为连接单元的反应性前体,其选自异氰酸酯、异硫氰酸酯、2-吡啶基二硫化物、卤代乙酰胺(-NHC(O)CH
在P
R
R
Z”为连接CB与R
在替代实施方案中,连接CB和Ar的连接单元为由式(IIIb)、(IIIc)、(IIId)、(IIIe)、(IIIf)、(IIIh)、(IIIh)或(IIIj)表示的连接基团:
其中:
R
X
W
R
R
R
s和s”各自独立地为值为0至约10的整数;
m为值为0或1的整数;
X
b、c、d、e、g、h、o和q各自独立地为值为1至约10的整数。
在式(IIIb)、(IIIc)、(IIId)、(IIIe)、(IIIf)、(IIIh)、(IIIh)或(IIIj)的一此类实施方案中:
R
R
Z”为连接CB与R
本发明的化合物和缀合物还可包含配体或靶向部分CB。在一些实施方案中,配体或靶向部分是任何分子识别元件,其可以通过例如非共价键合(诸如氢键合、金属配位、疏水力、范德华力、π-π相互作用、卤素键合、静电和/或电磁效应)与至少一种其他分子发生特定的相互作用。在某些实施方案中,CB选自纳米颗粒、免疫球蛋白、核酸、蛋白质、寡肽、多肽、抗体、抗原性多肽的片段、重复体(repebody)等。
本发明的化合物和缀合物可包含一个或多个靶向部分。也就是说,变量cb可具有选自1、2、3、4、5、1-10或1-20的整数值。
在一些实施方案中,CB包含通过共价键(例如,肽键)缀合的两个或更多个独立选择的天然氨基酸或非天然氨基酸,并且肽可以包含通过肽键缀合的2个、3个、4个、5个、6个、7个、8个、9个、10个、11个、12个、13个、14个、15个、16个、17个、18个、19个、20个或更多个天然氨基酸或非天然氨基酸。在一些实施方案中,配体包含较短的氨基酸序列(例如,天然蛋白质的片段或合成多肽片段)以及全长蛋白质(例如,工程化前的蛋白质)。
在一些实施方案中,CB选自抗体、激素、药物、抗体类似物(例如,非-IgG)、蛋白质、寡肽、多肽等,其与受体结合。在某些实施方案中,CB选择性地将药物靶向特定的器官、组织或细胞。在其它实施方案中,与正常细胞相比,CB特异性结合在癌细胞中过表达的受体,并且可以分类为单克隆抗体(mAb)或抗体片段和低分子非抗体。优选地,CB选自肽、肿瘤细胞特异性肽、肿瘤细胞特异性适体、肿瘤细胞特异性碳水化合物、肿瘤细胞特异性单克隆抗体、多克隆抗体和在文库筛选中鉴定的抗体片段。
示例性配体或靶向部分包括但不限于肉碱、肌醇、硫辛酸、吡哆醛、抗坏血酸、烟酸、泛酸、叶酸、核黄素、硫胺素、生物素、维生素B
靶标
在一些实施方案中,分子识别元件的一种或多种靶标与一种或多种特定细胞或组织类型特异性相关联。在一些实施方案中,靶标与一种或多种特定疾病状态特异性相关联。在一些实施方案中,靶标与一个或多个特定发育阶段特异性相关联。例如,细胞类型特异性标志物在该细胞类型中的表达水平通常为参考细胞群中的表达水平的至少2倍。在一些实施方案中,细胞类型特异性标志物存在的水平为其在参考群体中的平均表达的至少3倍、至少4倍、至少5倍、至少6倍、至少7倍、至少8倍、至少9倍、至少10倍、至少50倍、至少100倍或至少1000倍。细胞类型特异性标志物的检测或测量可使得将一种或多种目标细胞类型与许多、大多数或所有其它类型的细胞区分开来成为可能。在一些实施方案中,如本文所述,靶标可包括蛋白质、碳水化合物、脂质和/或核酸。
在一些实施方案中,如果物质特异性结合靶向部分,诸如核酸靶向部分,则该物质被认为是“靶向的”。在一些实施方案中,靶向部分,诸如核酸靶向部分,在严格条件下特异性结合靶。
在某些实施方案中,本文所述的缀合物和化合物包含靶向部分,其特异性结合与器官、组织、细胞、细胞外基质组分和/或细胞内区室相关联的一种或多种靶标(例如抗原)。在一些实施方案中,本文所述的缀合物和化合物包含靶向部分,其特异性结合与特定器官或器官系统相关联的靶标。在一些实施方案中,本文所述的缀合物和化合物包含靶向部分,其特异性结合一种或多种细胞内靶标(例如,细胞器、细胞内蛋白质)。在一些实施方案中,本文所述的缀合物和化合物包含靶向部分,其特异性结合与患病器官、组织、细胞、细胞外基质组分和/或细胞内区室相关的靶标。在一些实施方案中,本文所述的缀合物和化合物包含靶向部分,其特异性结合与特定细胞类型(例如,内皮细胞、癌细胞、恶性细胞、前列腺癌细胞等)相关联的靶标。
在一些实施方案中,本文所述的缀合物和化合物包含靶向部分,其结合对一种或多种特定组织类型(例如,肝组织对比前列腺组织)特异的靶标。在一些实施方案中,本文所述的缀合物和化合物包含靶向部分,其结合对一种或多种特定细胞类型(例如,T细胞对比B细胞)特异的靶标。在一些实施方案中,本文所述的缀合物和化合物包含靶向部分,其结合对一种或多种特定疾病状态(例如,肿瘤细胞对比健康细胞)特异的靶标。在一些实施方案中,本文所述的缀合物和化合物包含靶向部分,其结合对一个或多个特定发育阶段(例如,干细胞对比已分化的细胞)特异的靶标。
在一些实施方案中,靶标可以是专门或主要与一种或几种细胞类型、一种或几种疾病和/或一个或几种发育阶段相关联的标志物。细胞类型特异性标志物在该细胞类型中的表达水平通常为参考细胞群中的至少2倍,所述参考细胞群可由例如以近似相等的量包含来自多个(例如,5至10个或更多)不同组织或器官的细胞的混合物组成。在一些实施方案中,细胞类型特异性标志物存在的水平为其在参考群体中的平均表达的至少3倍、至少4倍、至少5倍、至少6倍、至少7倍、至少8倍、至少9倍、至少10倍、至少50倍、至少100倍或至少1000倍。细胞类型特异性标志物的检测或测量可使得将一种或多种目标细胞类型与许多、大多数或所有其它类型的细胞区分开来成为可能。
在一些实施方案中,靶标包括蛋白质、碳水化合物、脂质和/或核酸。在一些实施方案中,靶标包含蛋白质和/或其特征部分,诸如肿瘤标志物、整联蛋白、细胞表面受体、跨膜蛋白、细胞间蛋白、离子通道、膜转运蛋白、酶、抗体、嵌合蛋白、糖蛋白等。在一些实施方案中,靶标包括碳水化合物和/或其特征部分,诸如糖蛋白、糖(例如,单糖、二糖、多糖)、糖萼(即,大多数真核细胞外表面上富含碳水化合物的外围区)等。在一些实施方案中,靶标包括脂质和/或其特征部分,诸如油、脂肪酸、甘油酯、激素、类固醇(例如,胆固醇、胆汁酸)、维生素(例如,维生素E)、磷脂、鞘脂、脂蛋白等。在一些实施方案中,靶标包括核酸和/或其特征部分,诸如DNA核酸、RNA核酸、经修饰的DNA核酸、经修饰的RNA核酸、包括DNA、RNA、经修饰的DNA和经修饰的RNA的任何组合的核酸。
本领域中已知许多标志物。常见标志物包括细胞表面蛋白,例如受体。示例性受体包括但不限于转铁蛋白受体;LDL受体;生长因子受体,诸如表皮生长因子受体家族成员(例如,EGFR、Her2、Her3、Her4)或血管内皮生长因子受体、细胞因子受体、细胞粘附分子、整联蛋白、选择素和CD分子。所述标志物可以是仅存在于恶性细胞上或以更高的量存在于恶性细胞上的分子,例如肿瘤抗原。
抗体-药物结合物(ADC)
在一些实施方案中,CB为抗体,Q为药物。因此,本文公开的化合物和缀合物可用于将抗体与药物部分缀合,以形成抗体-药物缀合物(ADC)。由于ADC能够选择性地将一种或多种药物部分递送至靶组织,诸如肿瘤相关抗原,因此抗体-药物缀合物(ADC)可提高治疗疾病(例如癌症)的治疗效果。因此,在某些实施方案中,本发明提供了用于治疗用途(例如,癌症治疗)的ADC。
本发明的ADC包含与一个或多个药物部分连接的抗体。ADC的特异性由抗体的特异性来定义。在一个实施方案中,抗体与一种或多种细胞毒性药物连接,所述细胞毒性药物被内部递送至癌细胞。
下面提供了可用于本发明的ADC的药物实例。术语“药物”、“剂”和“药物部分”在本文中可互换使用。术语“连接的”和“缀合的”在本文中也可互换使用,表示抗体与部分共价连接。
在某些方面,本公开涉及ADC、包含ADC的组合物、治疗方法和配制ADC组合物的方法。ADC包含与细胞毒性化合物缀合的抗体或抗体片段。在一些实施方案中,通过接头将细胞毒性化合物与抗体缀合。在其它实施方案中,将细胞毒性化合物与抗体直接连接。下面描述本公开所包含的抗体、接头和细胞毒性化合物的类型。
在一些实施方案中,ADC具有下式(式(III)):
(D-L)
(III)
或其药学上可接的受盐,
其中:
LG为连接基团;
CB为细胞结合剂;
cb和dl各自独立地为值为1至约20,优选1至约10的整数;并且
每个D-L独立地为具有式(I)或(II)的化合物的结构的基团。
ADC的抗体可以是通常特异性地但不一定特异性地结合目标靶细胞表面上表达的抗原的任何抗体。抗原不必能够,但是在一些实施方案中,能够将与其结合的ADC内化到细胞中。目标靶细胞可包括其中需要诱导细胞凋亡的细胞。靶抗原可以是在目标靶细胞上表达的任何蛋白质、糖蛋白、多糖、脂蛋白等,但通常是这样的蛋白质,其仅在靶细胞上表达而在正常或健康细胞上不表达,或者与正常或健康细胞相比在靶细胞上过表达,使得ADC选择性地靶向特定的目标细胞,例如肿瘤细胞。本领域技术人员将会理解,所选择的特定的抗原以及由此特定的抗体将取决于所需目标靶细胞的身份。在具体的实施方案中,ADC的抗体是适于向人施用的抗体。
抗体(Ab)和免疫球蛋白(Ig)是具有相同结构特征的糖蛋白。虽然抗体表现出对特定靶标的结合特异性,但免疫球蛋白包括抗体和其它缺乏靶标特异性的抗体样分子。天然抗体和免疫球蛋白通常为约150,000道尔顿的异四聚糖蛋白,由两条相同的轻(L)链和两条相同的重(H)链组成。每条重链的一端具有可变结构域(VH),后面是许多恒定结构域。每条轻链的一端具有可变结构域(VL),并且另一端具有恒定结构域。
“VH”是指抗体的免疫球蛋白重链(包括Fv、scFv或Fab的重链)的可变区。“VL”是指免疫球蛋白轻链(包括Fv、scFv、dsFv或Fab的轻链)的可变区。
术语“抗体”在本文中以最广泛的意义使用,是指特异性结合特定抗原或与特定抗原发生免疫反应的免疫球蛋白分子,包括多克隆、单克隆、基因工程或以其它方式修饰的形式的抗体,包括但不限于鼠抗体、嵌合抗体、人源化抗体、异源缀合的抗体(heteroconjugateantibody)(例如,双特异性抗体、双抗体、三抗体和四抗体),以及抗体的抗原结合片段,包括例如Fab’、F(ab’)2、Fab、Fv、rIgG和scFv片段。术语“scFv”是指单链Fv抗体,其中来自常规抗体的重链和轻链的可变结构域已连接形成一条链。
抗体可以是鼠抗体、人抗体、人源化抗体、嵌合抗体或源自其它物种的抗体。抗体是由免疫系统产生的蛋白质,其能够识别并结合特定的抗原。(Janeway,C.,Travers,P.,Walport,M.,Shlomchik(2001)Immuno Biology,第5版,Garland Publishing,New York)。靶抗原通常具有许多结合部位,也称为表位,由多种抗体上的CDR识别。每种特异性结合不同表位的抗体都有不同的结构。因此,一种抗原可能具有不止一种相应的抗体。抗体包括全长免疫球蛋白分子或全长免疫球蛋白分子的免疫活性部分,即包含抗原结合部位的分子,所述抗原结合部位免疫特异性地结合目标靶标或其部分的抗原,此类靶标包括但不限于癌细胞或产生与自身免疫性疾病相关联的自身免疫抗体的细胞。本文公开的免疫球蛋白可以是免疫球蛋白分子的任何类型(例如,IgG、IgE、IgM、IgD和IgA)、类别(例如,IgG1、IgG2、IgG3、IgG4、IgA1和IgA2)或亚类。免疫球蛋白可源自任何物种。然而,一个方面,免疫球蛋白来源于人、鼠或兔。
术语“抗体片段”是指全长抗体的一部分,通常为靶结合区域或可变区。抗体片段的实例包括Fab、Fab′、F(ab′)2和Fv片段。“Fv”片段是包含完整靶识别和结合部位的最小抗体片段。该区域由紧密的非共价缔合的一个重链可变结构域和一个轻链可变结构域的二聚体(VH-VL二聚体)组成。正是在这种构型中,每个可变结构域的三个CDR相互作用以在VH-VL二聚体的表面上限定靶结合部位。通常,六个CDR赋予抗体靶结合特异性。然而,在一些情况下,甚至单个可变结构域(或仅包含三个对靶标特异的CDR的Fv的一半)也可具有识别和结合靶标的能力。“单链Fv”或“scFv”抗体片段在单个多肽链中包含抗体的VH和VL结构域。一般来说,Fv多肽还在VH与VL结构域之间包含多肽接头,这使得scFv能够形成用于靶标结合的所需结构。“单结构域抗体”由单个VH或VL结构域组成,所述结构域表现出足够的对靶标的亲和力。在具体的实施方案中,单结构域抗体是骆驼化抗体(参见,例如,Riechmann,1999,Journal of Immunological Methods 231:25-38)。
Fab片段包含轻链的恒定区和重链的第一个恒定区(CH1)。Fab’片段与Fab片段相异在于在重链CH1结构域的羧基末端添加了几个残基,包括来自抗体铰链区的一个或多个半胱氨酸。F(ab′)片段是由F(ab′)2胃蛋白酶消化产物的铰链半胱氨酸处的二硫键断裂产生的。抗体片段的额外化学偶联是本领域普通技术人员已知的。
轻链和重链可变区都有互补决定区(CDR),也称为高变区。可变结构域的更高度保守的部分被称为框架(FR)。如本领域中已知的,根据上下文和本领域已知的各种定义,描绘抗体高变区的氨基酸位置/边界可以变化。可变结构域内的一些位置可被视为混合高变位置(hybrid hypervariable position),因为根据一组标准,这些位置可被视为在高变区内,而根据一组不同的标准,这些位置可被视为在高变区外。这些位置中的一个或多个也可在扩展的高变区中找到。每条链中的CDR由FR区紧密地保持在一起,并且与来自另一条链的CDR一起,促成抗体的靶结合部位形成(参见Kabat等人,Sequences of Proteins ofImmunological Interest(National Institute of Health,Bethesda,Md.1987)。如本文中所用,除非另有说明,否则免疫球蛋白氨基酸残基的编号是根据Kabat等人的免疫球蛋白氨基酸残基编号系统进行的。
在某些实施方案中,本发明的ADC的抗体是单克隆抗体。术语“单克隆抗体”(mAb)是指源自单个拷贝或克隆(包括例如任何真核、原核或噬菌体克隆)而不是籍以产生其的方法的抗体。优选地,本公开的单克隆抗体存在于同质或基本上同质的群体中。单克隆抗体既包括完整分子以及抗体片段(例如,Fab和F(ab’)2片段),其能够特异性结合蛋白质。Fab和F(ab’)2片段缺少完整抗体的Fc片段,从动物循环中清除更快,并且可能比完整抗体具有更少的非特异性组织结合(Wahl等人,1983,J.Nucl.Med 24:316)。可用于本公开的单克隆抗体可使用本领域已知的多种技术制备,包括使用杂交瘤、重组和噬菌体展示技术或其组合。本公开的抗体包括嵌合抗体、灵长类化抗体、人源化抗体或人抗体。
虽然在大多数情况下,抗体仅由遗传编码的氨基酸组成,但在一些实施方案中,可以特异性地掺入非编码的氨基酸。在Tian等人,2014,Proc Nat’l Acad Sci USA 111(5):1766-1771和Axup等人,2012,Proc Nat’l Acad Sci USA 109(40):16101-16106中论述了可掺入抗体中用于控制化学计量和附接位置的非编码氨基酸以及制备此类经修饰的抗体的方法的实例,所述文献的全部内容通过引用并入本文。
在某些实施方案中,本文所述的ADC的抗体是嵌合抗体。如本文中所用,术语“嵌合”抗体是指具有源自非人免疫球蛋白(诸如大鼠或小鼠抗体)的可变序列和人免疫球蛋白恒定区(通常选自人免疫球蛋白模板)的抗体。用于产生嵌合抗体的方法是本领域已知的。参见,例如,Morrison,1985,Science 229(4719):1202-7;Oi等人,1986,BioTechniques 4:214-221;Gillies等人,1985,J.Immunol.Methods125:191-202;美国专利第5,807,715号、第4,816,567号和第4,816397号,所述参考文献和美国专利通过引用整体并入本文。
在某些实施方案中,本文所述的ADC的抗体是人源化抗体。非人(例如,鼠)抗体的“人源化”形式是嵌合免疫球蛋白、免疫球蛋白链或其片段(诸如Fv、Fab、Fab’、F(ab’)2或抗体的其它靶结合亚结构域),其包含源自非人免疫球蛋白的最小序列。一般而言,人源化抗体将包含基本上所有的至少一个(通常是两个)可变结构域,其中所有或基本上所有的CDR区对应于非人免疫球蛋白的那些可变结构域,并且所有或基本上所有的FR区是人免疫球蛋白序列的那些FR区。人源化抗体还可包含免疫球蛋白恒定区(Fc)的至少一部分,通常是人免疫球蛋白共有序列的一部分。抗体人源化的方法是本领域已知的。参见,例如,Riechmann等人,1988,Nature 332:323-7;美国专利第5,530,101号、第5,585,089号、第5,693,761号、第5,693,762和属于Queen等人的美国专利第6,180,370号;EP239400;PCT公布WO91/09967;美国专利第5,225,539号;EP592106、EP519596;Padlan,1991,Mol.Immunol.,28:489-498;Studnicka等人,1994,Prot.Eng.7:805-814;Roguska等人,1994,Proc.Natl.Acad Sci.USA91:969-973;以及美国专利第5,565,332号,将所有所述文献、专利、公布据此通过引用整体并入。
在某些实施方案中,本文所述的ADC的抗体是人抗体。完全“人”抗体对于人患者的治疗性治疗可以是合乎需要的。如本文中所用,“人抗体”包括具有人免疫球蛋白的氨基酸序列的抗体,并包括从人免疫球蛋白文库或从不表达内源免疫球蛋白的一种或多种人免疫球蛋白的转基因动物分离的抗体。人抗体可通过本领域已知的多种方法制备,所述方法包括使用源自人免疫球蛋白序列的抗体文库的噬菌体展示方法。参见美国专利第4,444,887号、第4,716,111号、第6,114,598号、第6,207,418号、第6,235,883号、第7,227,002号、第8,809,151号和美国公布申请第2013/189218号,所述专利或公布申请的内容通过引用整体并入本文。还可使用转基因小鼠来产生人抗体,所述转基因小鼠不能表达功能性内源免疫球蛋白但能表达人免疫球蛋白基因。参见,例如,美国专利第5,413,923号、第5,625,126号、第5,633,425号、第5,569,825号、第5,661,016号、第5,545,806号、第5,814,318号、第5,885,793号、第5,916,771号、第5,939,598号、第7,723,270号、第8,809,051和美国公布的申请第2013/117871号,所述专利或申请通过引用整体并入本文。另外,诸如Medarex(Princeton,N.J.)、Astellas Pharma(Deerfield,Ill.)和Regeneron(Tarrytown,N.Y.)等公司可以签约以使用与上述技术相似的技术提供针对所选择抗原的人抗体。可使用称作“导向选择”的技术产生识别所选择表位的完全人抗体。在这种方法中,选择的非人单克隆抗体(例如小鼠抗体)用于指导识别相同表位的完全人抗体的选择(Jespers等人,1988,Biotechnology12:899-903)。
在某些实施方案中,本文所述的ADC的抗体是灵长类化抗体。术语“灵长类化抗体”是指包含猴可变区和人恒定区的抗体。用于产生灵长类化抗体的方法是本领域已知的。参见,例如,美国专利第5,658,570号、第5,681,722号和第5,693,780号,所述专利通过引用整体并入本文。
在某些实施方案中,本文所述的ADC的抗体是双特异性抗体或双重可变结构域抗体(DVD)。双特异性和DVD抗体是单克隆(通常是人或人源化的)抗体,其对至少两种不同的抗原具有结合特异性。例如,在美国专利第7,612,181号(其公开内容通过引用并入本文)中描述了DVD。
在某些实施方案中,本文所述的ADC的抗体是衍生抗体。例如,但不限于,衍生抗体通常通过糖基化、乙酰化、聚乙二醇化、磷酸化、酰胺化、通过已知的保护/封闭基团的衍生化、蛋白水解切割、与细胞配体或其它蛋白质的键联等来进行修饰。许多化学修饰中的任一种都可通过已知技术来进行,所述技术包括但不限于特定的化学裂解、乙酰化、甲酰化、衣霉素的代谢合成等。另外,所述衍生物可以例如使用ambrx技术(例如,参见,Wolfson,2006,Chem.Biol.13(10):1011-2)来包含一种或多种非天然氨基酸。
在某些实施方案中,本文所述的ADC的抗体具有经修饰以相对于相应的野生型序列改变至少一种恒定区介导的生物效应子功能的序列。例如,在一些实施方案中,,抗体可被修饰来相对于未修饰的抗体降低至少一种恒定区介导的生物效应子功能,例如减少的与Fc受体(FcR)的结合。可通过在FcR相互作用所必需的特定区域中突变抗体的免疫球蛋白恒定区区段来减少FcR结合(参见,例如,Canfield和Morrison,1991,J.Exp.Med 173:1483-1491;以及Lund等人,1991,J.Immunol.147:2657-2662)。
在某些实施方案中,相对于未修饰的抗体,本文所述的ADC的抗体经修饰以获得或改善至少一种恒定区介导的生物效应子功能,例如,以增强FcγR相互作用(参见,例如,美国2006/0134709)。例如,根据本文所述的方法,可以产生具有恒定区的抗体,所述恒定区以比相应的野生型恒定区更大的亲和力结合FcγRIIA、FcγRIIB和/或FcγRIIIA。
在某些特定的实施方案中,本文所述的ADC的抗体是结合肿瘤细胞的抗体,诸如针对细胞表面受体或肿瘤相关抗原(TAA)的抗体。在试图发现用于癌症诊断和疗法的有效细胞靶标的过程中,研究人员已经寻求鉴定跨膜多肽或另外地肿瘤相关多肽,与一种或多种正常非癌细胞相比,所述多肽在一种或多种特定类型癌细胞的表面上特异性表达。通常,与非癌细胞表面相比,此类肿瘤相关多肽在癌细胞表面上更丰富地表达。这种细胞表面受体和肿瘤相关抗原是本领域已知的,并且可以使用本领域公知的方法和信息制备用于产生抗体。
示例性细胞表面受体和TAA
本文所述的ADC的抗体可靶向的细胞表面受体和TAA的实例包括但不限于下表1中列出的各种受体和TAA。为方便起见,将与这些抗原相关的信息(其均为本领域所知)列于下面,所述信息包括名称、别称、Genbank登录号和一个或多个主要参考文献,遵循国家生物技术信息中心(NCBI)的核酸和蛋白质序列鉴定惯例。对应于所列细胞表面受体和TAA的核酸和蛋白质序列可在公共数据库如GenBank中获得。
表I.
示例性抗体
与本公开的ADC一起使用的示例性抗体包括但不限于3F8(GD2)、阿巴伏单抗(Abagovomab)(CA-125(模仿))、阿德木单抗(EpCAM)、阿夫珠单抗(Afutzumab)(CD20)、阿来珠单抗聚乙二醇(Alacizumab pegol)(VEGFR2)、ALD518(IL-6)、阿仑珠单抗(Alemtuzumnab)(CD52)、阿妥莫单抗-喷替酸(Altumomab pentetate)(CEA)、阿马妥昔单抗(Amatuximab)(间皮素)、马安那莫单抗(Anatumomnab mafenatox)(TAG-72)、阿泊珠单抗(Apolizumab)(HLA-DR)、阿西莫单抗(CEA)、巴维昔单抗(磷脂酰丝氨酸)、贝妥莫单抗(CD22)、贝利木单抗(BAFF)、贝索单抗(CEA相关抗原)、贝伐珠单抗(VEGF-A)、比伐珠单抗-美登素(Bivatuzumab mertansine)(CD44 v6)、兰妥莫单抗(CD19)、本妥昔单抗(Brentuximab vedotin)((CD30(TNFRSF8))、坎妥珠单抗-美登素(Cantuzumabmertansine)(Mucin CanAg)、坎妥珠单抗-丹参素(cantuzumab ravtansine)(MUC1)、卡罗单抗喷地肽(Capromab pendetide)(前列腺癌细胞)、卡芦单抗(MCP-1)、卡妥索单抗(EpCAM,CD3)、CC49(Tag-72)、cBR96-DOX ADC(Lewis-Y抗原)、西妥昔单抗(EGFR)、西他珠单抗-bogatox(Citatuzumab bogatox)(EpCAM)、西妥木单抗(IGF-1受体)、克莱维足单抗四氢西泮(Clivatuzumab tetraxetan)(MUC1)、可那妥木单抗(Conatumumab)(TRAIL-E2)、达西珠单抗(CD40)、达洛珠单抗(胰岛素样生长因子1受体)、达拉他珠单抗((CD38(环状ADP核糖水解酶))、地昔单抗(DLL4)、地诺单抗(RANKL)、地莫单抗(B淋巴瘤细胞)、卓齐妥单抗(Drozitumab)(DR5)、杜斯基单抗(Dusigitumab)(ILGF2)、依美昔单抗(Ecromeximab)(D3神经节苷脂)、依库珠单抗(Eculizumab)(C5)、依决洛单抗(Edrecolomab)(EpCAM)、埃罗妥珠单抗(Elotuzumab)(SLAMF7)、艾西莫单抗(IL-6)、恩那珠单抗(Enavatuzumab)(TWEAK受体)、Enoticumab(DLL4)、恩昔妥昔单抗(Ensituximab)(5AC)、依匹莫单抗-西坦(Epitumomab cituxetan)(Episialin)、依帕珠单抗(CD22)、厄妥索单抗((HER2/neu,CD3))、依那西珠单抗(Etancizumab)(整联蛋白αvβ3)、法勒珠单抗(Farletuzumab)(叶酸受体1)、FBTA05(CD20)、非卡妥珠单抗(Ficlatuzumab)(HGF)、弗吉妥姆单抗(Figitumumab)(IGF-1受体)、Flanvotumab((TYRP1(糖蛋白75))、非苏木单抗(Fresolimumab)(TGF-1)、加利昔单抗(Galiximab)(CD80)、Ganitumab(IGF-I)、吉妥珠单抗奥唑米星(Gemtuzumabozogamicin)(CD33)、吉瑞昔单抗(Girentuximab)((碳酸酐酶9(CA-IX))、格莱木单抗-维多汀(Glembatumumab vedotin)(GPNMB)、替伊莫单抗-泰坦(Ibritumomab tiuxetan)(CD20)、伊库鲁单抗(VEGFR-1)、伊戈伏单抗(CA-125)、IMAB362(CLDN18.2)、伊格单抗(EGFR)、英达妥昔单抗-丹参素(Indatuximab ravtansine)(SDC1)、英妥木单抗(CD51)、伊珠单抗奥加米星(Inotuzumab ozogamicin)(CD22)、伊匹木单抗(CD152)、伊妥木单抗((CD30(TNFRSF8))、拉贝珠单抗(CEA)、兰博单抗(Lambrolizumab)(PDCD1)、来沙木单抗(Lexatumumab)(TRAIL-R2)、林妥珠单抗(CD33)、洛妥珠单抗-美登素(Lorvotuzumab mertansine)(CD56)、鲁卡木单抗(Lucatumumab)(CD40)、鲁昔单抗(Lumiliximab)((CD23(IgE受体))、马帕木单抗(TRAIL-R1)、马格妥昔单抗(Margetuximab)(ch4DS)、马妥珠单抗(EGFR)、米拉珠单抗(CD74)、米妥莫单抗(GD3神经节苷脂)、莫加单抗(Mogamulizumab)(CCR4)、莫妥单抗假毒素(Moxetumomab pasudotox)(CD22)、他那可单抗(Nacolomab tafenato)(C2-42抗原)、他那莫单抗(Naptumomab estafenatox)(5T4)、纳那妥单抗(Narnatumab)(RON)、那他珠单抗(整联蛋白α4)、奈昔木单抗(EGFR)、耐斯伐库单抗(Nesvacumab)(血管生成素2)、尼妥珠单抗(Nimotuzumab)(EGFR)、纳武单抗(IgG4)、奥卡珠单抗(Ocaratuzumab)(CD20)、奥法木单抗、奥拉图单抗(PDGF-Rα)、恩妥珠单抗(Onartuzumab)(人散射因子受体激酶)、恩妥昔单抗(Ontuxizumab)(TEM1)、奥珠单抗-莫纳托(Oportuzumab monato)(EpCAM)、奥瑞戈单抗(Oregovomab)(CA-125)、奥特托珠单抗(Otlertuzumab)(CD37)、帕尼单抗(Panitumumab)(EGFR)、Pankomab(MUC1的肿瘤特异性糖基化)、帕沙妥珠单抗(Parsatuzumab)(EGFL7)、帕曲妥单抗(Patritumab)(HER3)、培妥莫单抗(Pemtumomab)(MUC1)、培妥珠单抗(Pertuzumab)(HER2/neu)、匹地珠单抗(Pidilizumab)(PD-1)、品妥珠单抗维多汀(Pinatuzumab vedotin)(CD22)、普立木单抗(Pritumumab)(波形蛋白)、雷妥莫单抗(N-羟乙酰神经氨酸)、拉吉妥姆单抗(Radretumab)(纤连蛋白额外结构域-B)、雷莫芦单抗(VEGFR2)、利妥木单抗(Rilotumumab)(HGF)、利妥昔单抗(CD20)、罗妥木单抗(Robatumumab)(IGF-1受体)、萨马珠单抗(Samalizumab)(CD200)、沙妥莫单抗喷地肽(Satumomab pendetide)(TAG-72)、司里班妥单抗(Seribantumab)(ERBB3)、西罗珠单抗(FAP)、SGN-CD19A(CD19)、SGN-CD33A(CD33)、司妥昔单抗(IL-6)、索利托单抗(EpCAM)、索能单抗(Sonepcizumab)(鞘氨醇-1-磷酸酯)、他贝芦单抗(Tabalumb)(BAFF)、他卡妥珠单抗四氢西泮(Tacatuzumab tetraxetan)(甲胎蛋白)、塔普单抗-帕他莫(Taplitumomab paptox)(CD19)、替妥莫单抗(生腱蛋白C)、替妥木单抗(Teprotumumab)(CD221)、TGN1412(CD28)、替西莫单抗(CTLA-4)、替加珠单抗(Tigatuzumab)(TRAIL-R2)、TNX-650(IL-13)、托维图单抗(Tovetumab)(CD40a)、曲妥珠单抗(HER2/neu)、TRBS07(GD2)、曲美木单抗(CTLA-4)、曲妥珠单抗-西莫白介素(Tucotuzumab celmoleukin)(EpCAM)、乌布妥昔单抗(Ublituximab)(MS4A)、乌瑞芦单抗(Urelumab)(4-1BB)、凡他尼布(Vandetanib)(VEGF)、Vantictumab(Frizzled受体)、伏洛昔单抗(整联蛋白α5β1)、伏司妥珠单抗-马佛多汀(Vorsetuzumabmafodotin)(CD70)、伏妥莫单抗(肿瘤抗原CTAA16.88)、扎鲁木单抗(Zalutumumab)(EGFR)、扎木单抗(Zanolimumab)(CD4)和扎土希单抗(Zatuximab)(HER1)。
制造抗体的方法
ADC的抗体可以通过在宿主细胞中重组表达免疫球蛋白轻链和重链基因来制备。例如,为了重组表达抗体,用一种或多种携带编码抗体免疫球蛋白轻链和重链的DNA片段的重组表达载体转染宿主细胞,使得轻链和重链在宿主细胞中表达,并任选地分泌到培养宿主细胞的培养基中,可从该培养基中回收抗体。标准重组DNA方法用于获得抗体重链和轻链基因,将这些基因整合到重组表达载体中,并将载体引入宿主细胞,诸如MolecularCloning;A Laboratory Manual,第2版(Sambrook、Fritsch和Maniatis(编辑),ColdSpring Harbor,N.Y.,1989),Current Protocols in Molecular Biology(Ausubel,F.M.等人,编辑,Greene Publishing Associates,1989)和美国专利第4,816,397号中描述的那些。
在一个实施方案中,Fc变体抗体与其野生型等同物相似,但其Fc结构域发生了变化。为了产生编码此类Fc变体抗体的核酸,可以合成编码野生型抗体的Fc结构域(称为“野生型Fc结构域”)或Fc结构域的部分的DNA片段,并将其用作诱变的模板,以使用常规诱变技术产生本文所述的抗体;或者,可以直接合成编码抗体的DNA片段。
一旦获得编码野生型Fc结构域的DNA片段,就可通过标准重组DNA技术进一步操作这些DNA片段,例如,以将恒定区基因转化为全长抗体链基因。在这些操作中,编码CH的DNA片段与编码另一种蛋白质(诸如抗体可变区或柔性接头)的另一个DNA片段可操作地连接。如本说明书中所用,术语“可操作地连接的”旨在指连接两个DNA片段,使得由两个DNA片段编码的氨基酸序列保持在框内。
为了表达Fc变体抗体,将如上所述获得的编码部分或全长轻链和重链的DNA插入表达载体,使得基因可操作地连接至转录和翻译控制序列。在本说明书中,术语“可操作地连接的”旨在指将抗体基因连接至载体中,使得载体中的转录和翻译控制序列发挥其调控抗体基因的转录和翻译的预期功能。选择与所用表达宿主细胞相容的表达载体和表达控制序列。变体抗体轻链基因和抗体重链基因可被插入到不同的载体中,或者更常见地,两个基因被插入到同一表达载体中。
通过标准方法(例如,抗体基因片段和载体上互补限制性位点的连接,或者如果不存在限制性位点,则为平端连接)将抗体基因插入表达载体。在插入变体Fc结构域序列之前,表达载体可以已经携带抗体可变区序列。另外地或可选地,重组表达载体可编码促进抗体链从宿主细胞分泌的信号肽。可将抗体链基因克隆到载体中,使得信号肽与抗体链基因的氨基端框内连接。信号肽可以是免疫球蛋白信号肽或异源信号肽(即,来自非免疫球蛋白蛋白质的信号肽)。
除了抗体链基因以外,重组表达载体还携带控制抗体链基因在宿主细胞中表达的调控序列。术语“调控序列”旨在包括控制抗体链基因转录或翻译的启动子、增强子和其它表达控制元件(例如,多腺苷酸化信号)。此类调控序列描述于例如Goeddel,GeneExpression Technology:Methods in Enzymology 185(Academic Press,San Diego,Calif.,1990)中。本领域技术人员将会理解,表达载体的设计,包括调控序列的选择,可取决于诸如待转化的宿主细胞的选择、所需蛋白质的表达水平等因素。哺乳动物宿主细胞表达的合适调控序列包括在哺乳动物细胞中指导高水平蛋白质表达的病毒元件,诸如源自巨细胞病毒(CMV)(诸如CMV启动子/增强子)、猿猴病毒40(SV40)(诸如SV40启动子/增强子)、腺病毒(例如,腺病毒主要晚期启动子(AdMLP))和多瘤病毒的启动子和/或增强子。关于病毒调控元件及其序列的进一步描述,参见例如Stinski的美国专利第5,168,062号、Bell等人的美国专利第4,510,245号专利和Schaffner的美国专利第4,968,615号。
除了抗体链基因和调控序列以外,重组表达载体可携带额外的序列,诸如调控载体在宿主细胞中复制的序列(例如,复制起始点)和选择标记基因。选择标记基因有助于选择已引入了载体的宿主细胞(参见,例如,美国专利第4,399,216号、第4,634,665号和第5,179,017号,均属于Axel等人)。例如,通常地,选择标记基因为已引入了载体的宿主细胞赋予对药物诸如G418、嘌呤霉素、灭瘟素(blasticidin)、潮霉素或甲氨蝶呤的抗性。合适的选择标记基因包括二氢叶酸还原酶(DHFR)基因(以用于利用甲氨蝶呤选择/扩增的DHFR-宿主细胞)和neo基因(以用于G418选择)。为了表达轻链和重链,通过标准技术将编码重链和轻链的表达载体转染到宿主细胞中。术语“转染”的各种形式旨在涵盖通常用于将外源DNA引入原核或真核宿主细胞的多种技术,例如电穿孔、脂转染、磷酸钙沉淀、DEAE-葡聚糖转染等。
在原核或真核宿主细胞中表达抗体是可能的。在某些实施方案中,在真核细胞,例如哺乳动物宿主细胞中进行抗体的表达,以最佳地分泌适当折叠的具有免疫活性的抗体。用于表达重组抗体的示例性哺乳动物宿主细胞包括中国仓鼠卵巢(CHO)细胞(包括DHFR-CHO细胞,描述于Urlaub和Chasin,1980,Proc.Natl.Acad.Sci.USA 77:4216-4220中,与DHFR选择标记一起使用,例如,如Kaufman和Sharp,1982,Mol.Biol.159:601-621中所述)、NS0骨髓瘤细胞、COS细胞、293细胞和SP2/0细胞。当将编码抗体基因的重组表达载体引入哺乳动物宿主细胞时,通过将宿主细胞培养足以允许抗体在宿主细胞中表达或将抗体分泌到宿主细胞生长的培养基中的一段时间来产生抗体。可使用标准的蛋白质纯化方法从培养基中回收抗体。还可将宿主细胞用来产生完整抗体的一部分,诸如Fab片段或scFv分子。
在一些实施方案中,ADC的抗体可以是双功能抗体。此类抗体(其中一条重链和一条轻链对一种抗原是特异的,而另一条重链和轻链对第二种抗原是特异的)可通过标准化学交联方法将抗体与第二种抗体交联来生产。双功能抗体也可通过表达经工程化以编码双功能抗体的核酸来制备。
在某些实施方案中,双特异性抗体,即使用同一结合部位结合一种抗原和第二种无关抗原的抗体,可通过突变轻链和/或重链CDR中的氨基酸残基来产生。示例性第二抗原包括促炎细胞因子(诸如,淋巴毒素、干扰素-γ或白细胞介素-1)。双特异性抗体可以例如通过突变抗原结合部位周围的氨基酸残基来产生(例如,参见Bostrom等人,2009,Science323:1610-1614)。双功能抗体可通过表达经工程化以编码双特异性抗体的核酸来制备。
抗体还可以通过化学合成(例如,通过Solid Phase Peptide Synthesis,第2版,1984The Pierce Chemical Co.,Rockford,Ill中描述的方法)产生。抗体还可使用无细胞平台(参见,例如,Chu等人,Biochemia No.2,2001(Roche Molecular Biologicals))产生。
Fc融合蛋白的重组表达方法描述于Flanagan等人,Methods in MolecularBiology,378卷:Monoclonal Antibodies:Methods and Protocols中。
一旦通过重组表达产生了抗体,就可通过本领域已知的用于纯化免疫球蛋白分子的任何方法,例如,通过色谱法(例如,离子交换、亲和(特别是通过在选择蛋白质A或蛋白质G后对抗原的亲和),以及分级柱色谱(sizing column chromatography))、离心法、差异溶解度法(differential solubility),或通过用于纯化蛋白质的任何其它标准技术来纯化其。
一旦分离,如果需要,可将抗体进一步纯化,例如通过高效液相色谱法(参见,例如,Fisher,Laboratory Techniques In Biochemistry And Molecular Biology(Work和Burdon,编辑,Elsevier,1980)),或通过Superdex
治疗方法
缀合物的靶向部分可被细胞识别,从而提供所谓的靶标导向治疗。
在一些实施方案中,本文所述的缀合物包含本文所述的活性剂,例如式(I)的活性剂。在一些此类实施方案中,活性剂是:
本文公开的化合物和缀合物可用于诱导细胞凋亡的方法中。
细胞凋亡失调与多种疾病相关,所述疾病包括例如自身免疫性疾病(例如,系统性红斑狼疮、类风湿性关节炎、移植物抗宿主病、重症肌无力或
尽管绝对治愈在任何治疗方案中总是可取的,但获得治愈并不需要提供治疗益处。治疗益处可包括停止或减缓疾病的进展,使疾病消退而不治愈,和/或改善或减缓疾病症状的进展。与统计平均值相比的延长的生存期和/或提高的生活质量也可被视为治疗益处。
癌症是一类特殊的疾病,其涉及失调的细胞凋亡,并且是全球范围内的重大健康负担。在具体的实施方案中,本文公开的化合物和组合物可用于治疗癌症。癌症可以是例如实体瘤或血液肿瘤。可用本文公开的化合物和组合物治疗的癌症包括但不限于膀胱癌、脑癌、乳腺癌、骨髓癌、宫颈癌、慢性淋巴细胞性白血病、结直肠癌、食道癌、肝细胞癌、淋巴母细胞性白血病、滤泡性淋巴瘤、T细胞或B细胞来源的淋巴样恶性肿瘤、黑色素瘤、骨髓性白血病、骨髓瘤、口腔癌、卵巢癌、非小细胞肺癌、慢性淋巴细胞性白血病、骨髓瘤、前列腺癌、小细胞肺癌和脾癌。本文公开的化合物和组合物在癌症治疗方面可能特别有益,因为抗体可用于特异性靶向肿瘤细胞,从而潜在地避免或改善可能与未缀合的抑制剂的全身性施用相关的不良副作用和/或毒性。一个实施方案涉及治疗涉及失调的内在细胞凋亡的疾病的方法,所述方法包括向患有涉及失调的细胞凋亡的疾病的受试者施用有效地提供治疗益处的量的本文公开的化合物和组合物,其中本文公开的化合物和组合物的配体结合其内在细胞凋亡失调的细胞上的细胞表面受体。一个实施方案涉及治疗癌症的方法,所述方法包括以有效提供治疗益处的量向患有癌症的受试者施用本文公开的化合物和组合物,其中所述配体能够结合细胞表面受体或在癌细胞表面上表达的肿瘤相关抗原。
在致瘤性癌症的情况下,与所治疗癌症的类型和分期的统计平均值相比,治疗益处除了包括上述效果之外,还可特别地包括停止或减缓肿瘤生长的进程、使肿瘤生长消退、根除一个或多个肿瘤和/或提高患者存活率。在一个实施方案中,被治疗的癌症是致瘤性癌症。
本文公开的化合物和缀合物可以作为单一疗法施用以提供治疗益处,或者可以作为其它化学治疗剂和/或放射疗法的辅助施用或与所述其它化学治疗剂和/或放射疗法一起施用。本文公开的化合物和组合物可用作其辅助疗法的化学治疗剂可以是靶向的(例如,ADC、蛋白激酶抑制剂等)或非靶向的(例如,非特异性细胞毒性剂诸如放射性核苷酸、烷化剂和嵌入剂)。本文公开的化合物和组合物可以辅助给药的非靶向化学治疗剂包括但不限于甲氨蝶呤、紫杉醇、L-天冬酰胺酶、巯基嘌呤、硫鸟嘌呤、羟基脲、阿糖胞苷、环磷酰胺、异环磷酰胺、亚硝脲类、顺铂、卡铂、丝裂霉素、达卡巴嗪、丙卡巴嗪(procarbizine)、拓扑替康、氮芥、环磷酰胺(Cytoxan)、依托泊苷、5-氟尿嘧啶、BCNU、伊立替康、喜树碱、博莱霉素、多柔比星、伊达比星、柔红霉素、更生霉素、普卡霉素、米托蒽醌、asperaginase、长春碱、长春新碱、长春瑞滨、紫杉醇、加利车霉素和多西他赛。
本文公开的化合物和缀合物可能不能有效地作为单一疗法来治疗癌症,可以作为其它化学治疗剂或放射疗法的辅助施用或与其它化疗剂或放射疗法一起施用,以提供治疗益处。一个实施方案涉及其中以有效地使肿瘤细胞对标准化学疗法和/或放射疗法敏感的量施用本文公开的化合物或组合物的方法。因此,在治疗癌症的背景下,“治疗益处”包括在尚未开始这种疗法或已经始这种疗法但尚未表现出抗性体征的患者中,将本文公开的化合物和组合物作为化学治疗剂和/或放射疗法的辅助施用或与化学治疗剂和/或放射疗法一起施用,或者在已开始表现出抗性体征的患者中,将所述化合物和组合物作为使肿瘤对化学疗法和/或放射疗法敏感的手段施用。
药物组合物及其施用
本文公开的化合物和缀合物可用于治疗有此需要的个体。在某些实施方案中,个体是哺乳动物,诸如人,或非人类哺乳动物。当向动物诸如人施用时,组合物或化合物优选以包含例如公开的化合物和药学上可接受的载体的药物组合物的形式施用。
药学上可接受的载体是本领域公知的,包括例如水溶液,诸如水或生理缓冲盐水或其它溶剂或媒介物,诸如乙二醇、甘油、油诸如橄榄油或可注射的有机酯。在优选实施方案中,当此类药物组合物用于人施用,特别是用于侵入性施用途径(即绕过上皮屏障转运或扩散的途径,诸如注射或植入)时,水溶液是无热原的,或基本上无热原的。可以选择赋形剂,例如,以实现药物的延迟释放或选择性地靶向一种或多种细胞、组织或器官。药物组合物可呈剂量单位形式,诸如片剂、胶囊(包括喷洒胶囊和明胶胶囊)、颗粒剂、用于复原的冻干剂、粉剂、溶液、糖浆剂、栓剂、注射剂等。组合物也可存在于经皮肤递送系统中,例如皮肤贴剂。组合物也可存在于适合局部施用的溶液中,诸如软膏或霜剂。
药学上可接受的载体可包含生理上可接受的剂,所述剂例如用于稳定化合物诸如本发明的化合物、增加其溶解度或增加其吸收。此类生理上可接受的剂包括,例如,碳水化合物,诸如葡萄糖、蔗糖或葡聚糖、抗氧化剂,诸如抗坏血酸或谷胱甘肽、螯合剂,低分子量蛋白质或其它稳定剂或赋形剂。药学上可接受的载体(包括生理上可接受的剂)的选择取决于例如组合物的给药途径。药物组合物的制剂可以是自乳化药物递送系统或自微乳化药物递送系统。药物组合物(制剂)也可以是脂质体或其它聚合物基质,其可在其中掺入例如本发明的化合物。例如,包含磷脂或其它脂质的脂质体是无毒的、生理上可接受的和可代谢的载体,其制备和施用相对简单。
短语“药学上可接受的”在本文中用于指在合理的医学判断范围内,适合用于与人类和动物的组织接触而没有过度毒性、刺激、过敏反应或其它问题或并发症,与合理的益处/风险比相称的那些化合物、材料、组合物和/或剂型。
如本文中所用,短语“药学上可接受的载体”是指药学上可接受的材料、组合物或媒介物,诸如液体或固体填料、稀释剂、赋形剂、溶剂或包封材料。每种载体必须在与制剂的另外的成分相容并且对患者无害的意义上是“可接受的”。可用作药学上可接受的载体的材料的一些实例包括:(1)糖类,诸如乳糖、葡萄糖和蔗糖;(2)淀粉,诸如玉米淀粉和马铃薯淀粉;(3)纤维素及其衍生物,诸如羧甲基纤维素钠、乙基纤维素和醋酸纤维素;(4)粉末黄芪胶;(5)麦芽;(6)明胶;(7)滑石;(8)赋形剂,诸如可可脂和栓剂蜡;(9)油类,诸如花生油、棉籽油、红花油、芝麻油、橄榄油、玉米油和大豆油;(10)二醇类,诸如丙二醇;(11)多元醇类,诸如甘油、山梨醇、甘露醇和聚乙二醇;(12)酯类,诸如油酸乙酯和月桂酸乙酯;(13)琼脂;(14)缓冲剂类,诸如氢氧化镁和氢氧化铝;(15)藻酸;(16)无热原水;(17)等渗盐水;(18)林格氏溶液;(19)乙醇;(20)磷酸盐缓冲溶液;和(21)药物制剂中使用的其它无毒相容性物质。
可通过多种施用途径中的任一种向受试者施用药物组合物(制剂),所述施用途径包括例如口服(例如,以水溶液或非水溶液或悬浮液、片剂、胶囊(包括喷洒胶囊和明胶胶囊)、药丸剂、粉剂、颗粒剂、涂于舌头的糊剂的形式给药);通过口腔粘膜的吸收(例如,舌下);经肛门、经直肠或经阴道(例如,作为阴道栓剂、霜剂或泡沫);胃肠外(包括肌内、静脉内、皮下或鞘内,例如无菌溶液或悬浮液);经鼻;腹膜内;皮下;经皮肤(例如作为涂于皮肤的贴片);和局部(例如,作为涂在皮肤上的霜剂、软膏或喷雾剂,或作为滴眼剂)。所述化合物也可以配制用于吸入。在某些实施方案中,可以仅将化合物溶解或悬浮在无菌水中。合适的施用途径和适合所述施用途径的组合物的细节可见于例如美国专利第6,110,973号、第5,763,493号、第5,731,000号、第5,541,231号、第5,427,798号、第5,358,970号和第4,172,896号,以及其中引用的专利中。
制剂可以方便地以单位剂量形式存在,并且可通过制药领域公知的任何方法来制备。可与载体材料结合以产生单一剂型的活性成分的量将根据被治疗的宿主、特定的施用方式而变化。可与载体材料结合以产生单一剂型的活性成分的量通常是产生治疗效果的化合物的那个量。通常,以百分数计,该量的范围为约1%至约99%的活性成分,优选约5%至约70%,最优选约10%至约30%。
制备这些制剂或组合物的方法包括将活性化合物诸如本发明的化合物与载体和任选的一种或多种辅助成分结合的步骤。一般而言,制剂是通过将本发明的化合物与液体载体或细碎固体载体或两者均匀而密切地结合,然后,如果需要,将产品成型来制备的。
适于口服施用的本发明的制剂可呈胶囊(包括喷洒胶囊和明胶胶囊)、扁囊剂、丸剂、片剂、锭剂(使用调味基质,通常为蔗糖和阿拉伯胶或黄蓍胶)、亲液剂、粉剂、颗粒的形式,或作为水溶液或非水溶液中的溶液或悬浮液,或作为水包油或油包水的液体乳液,或作为酏剂或糖浆剂,或作为软锭剂(pastille)(使用惰性基质,诸如明胶和甘油,或蔗糖和阿拉伯胶)和/或作为漱口剂等,每种含有预定量的本发明化合物作为活性成分。化合物、缀合物或其组合物还可作为药丸剂、干药糖剂或糊剂施用。
为了制备用于口服施用的固体剂型(胶囊(包括喷洒胶囊和明胶胶囊)、片剂、丸剂、锭剂、粉剂、颗粒剂等),将活性成分与一种或多种药学上可接受的载体(诸如柠檬酸钠或磷酸二钙)和/或以下的任一种混合:(1)填充剂或增充剂,诸如淀粉、乳糖、蔗糖、葡萄糖、甘露醇和/或硅酸;(2)粘合剂,诸如羧甲基纤维素、藻酸盐类、明胶、聚乙烯吡咯烷酮、蔗糖和/或阿拉伯胶;(3)保湿剂,诸如甘油;(4)崩解剂,诸如琼脂、碳酸钙、马铃薯或木薯淀粉、藻酸、某些硅酸盐和碳酸钠;(5)溶液阻溶剂,诸如石蜡;(6)吸收促进剂,诸如季铵化合物类;(7)润湿剂,例如鲸蜡醇和单硬脂酸甘油酯;(8)吸附剂,诸如高岭土和膨润土;(9)润滑剂,诸如滑石、硬脂酸钙、硬脂酸镁、固体聚乙二醇、十二烷基硫酸钠及其混合物;(10)络合剂,诸如改性和未改性的环糊精;和(11)着色剂。在胶囊(包括喷洒胶囊和明胶胶囊)、片剂和丸剂的情况下,药物组合物还可包含缓冲剂。类似类型的固体组合物也可用作软填充和硬填充明胶胶囊中的填充剂,使用诸如乳糖或乳糖的赋形剂,以及高分子量聚乙二醇等。
片剂可通过压缩或模塑制成,任选地具有一种或多种辅助成分。压缩片剂可使用粘合剂(例如,明胶或羟丙基甲基纤维素)、润滑剂、惰性稀释剂、防腐剂、崩解剂(例如,淀粉羟乙酸钠或交联的羧甲基纤维素钠)、表面活性剂或分散剂来制备。模制片剂可通过在合适的机器中对用惰性液体稀释剂润湿的粉末状化合物的混合物进行模制来制备。
片剂和药物组合物的其它固体剂型,诸如锭剂、胶囊(包括喷洒胶囊和明胶胶囊)、丸剂和颗粒剂,可以任选地用包衣和壳,诸如肠溶包衣和药物配制领域中熟知的其它包衣进行刻痕或制备。还可使用例如不同比例的羟丙基甲基纤维素(以提供所需的释放曲线)、其它聚合物基质、脂质体和/或微球将它们配制成提供其中的活性成分的缓慢或受控释放。它们可以通过例如通过保菌过滤器过滤来灭菌,或者通过以无菌固体组合物的形式加入灭菌剂来灭菌,可在使用前立即将所述无菌固体组合物溶解在无菌水或一些其它无菌注射介质中。这些组合物还可任选地包含遮光剂,并且可以是这样的组合物,即它们仅或优先在胃肠道的特定部分释放(任选地以延迟的方式)一种或多种活性成分。可使用的埋植组合物(embedding composition)的实例包括聚合物质和蜡类。如果合适的话,活性成分还可以与一种或多种上述赋形剂一起为微囊形式。
用于口服施用的液体剂型包括药学上可接受的乳剂、用于复原的冻干剂(lyophiles)、微乳剂、溶液、悬浮液、糖浆剂和酏剂。除了活性成分之外,液体剂型还可包含本领域常用的惰性稀释剂,例如水或其它溶剂、环糊精及其衍生物、增溶剂和乳化剂,诸如乙醇、异丙醇、碳酸乙酯、乙酸乙酯、苯甲醇、苯甲酸苄酯、丙二醇、1,3-丁二醇、油(特别是棉籽油、花生油、玉米油、胚芽油、橄榄油、蓖麻油和芝麻油)、甘油、四氢糠醇、聚乙二醇及脱水山梨糖醇的脂肪酸酯,以及其混合物。
除了惰性稀释剂以外,口服组合物还可以包括佐剂,例如润湿剂、乳化剂和助悬剂、甜味剂、调味剂、着色剂、芳香剂和防腐剂。
除了活性化合物以外,悬浮液还可包含助悬剂,例如乙氧基化异硬脂醇、聚氧乙烯山梨醇和脱水山梨糖醇酯、微晶纤维素、偏氢氧化铝、膨润土、琼脂和黄蓍胶,以及其混合物。
用于直肠、阴道或尿道施用的药物组合物的制剂可作为栓剂提供,其可通过将一种或多种活性化合物与一种或多种合适的非刺激性赋形剂或载体混合来制备,所述赋形剂或载体包括例如可可脂、聚乙二醇、栓剂蜡或水杨酸酯,并且其在室温下是固体,但在体温下是液体,因此将在直肠或阴道腔中熔化并释放活性化合物。
用于口腔施用的药物组合物的制剂可作为漱口水或口腔喷雾剂或口腔软膏提供。
可选地或加外地,组合物可被配制成通过导管、支架、线或其它管腔内装置递送。通过此类装置的递送对于递送至膀胱、尿道、输尿管、直肠或肠可以是特别有用的。
适于阴道施用的制剂还包括阴道栓剂、棉塞、霜剂、凝胶、糊剂、泡沫或喷雾制剂,其含有本领域已知的合适的此类载体。
局部或经皮肤施用的剂型包括粉剂、喷雾剂、软膏、糊剂、霜剂、洗剂、凝胶、溶液、贴剂和吸入剂。可将活性化合物在无菌条件下与药学上可接受的载体以及可能需要的任何防腐剂、缓冲剂或喷射剂混合。
除了活性化合物以外,软膏、糊剂、霜剂和凝胶还可包含赋形剂,诸如动物和植物脂肪、油、蜡、石蜡、淀粉、黄蓍胶、纤维素衍生物、聚乙二醇、硅酮、膨润土、硅酸、滑石和氧化锌,或其混合物。
除了活性化合物以外,粉剂和喷雾剂还可包含赋形剂,诸如乳糖、滑石、硅酸、氢氧化铝、硅酸钙和聚酰胺粉末,或这些物质的混合物。喷雾剂还可包含常规喷射剂,诸如氯氟烃和挥发性未取代的烃,诸如丁烷和丙烷。
透皮贴剂具有额外的优点,即向身体提供本发明化合物的受控递送。此类剂型可通过将活性化合物溶解或分散在合适的介质中来制备。吸收促进剂也可用来增加化合物穿过皮肤的流动。这种通动的速率可通过提供速率控制膜或将化合物分散在聚合物基质或凝胶中来控制。
眼科制剂、眼膏、粉剂、溶液等也被认为在本发明的范围内。示例性眼科制剂描述于美国公布第2005/0080056号、第2005/0059744号、第2005/0031697号和第2005/004074号以及美国专利第6,583,124号中,所述公布和专利的内容通过引用并入本文。如果需要,液体眼科制剂具有与泪液、房水或玻璃体液相似的性质,或者与此类流体相容。
如本文中所用,短语“胃肠外施用”和“胃肠外地施用”意指除了肠内和局部施用以外的施用方式,通常是通过注射,并且包括但不限于静脉内、肌内、动脉内、鞘内、囊内、眶内、心内、真皮内、腹膜内、经气管、皮下、关节内、包囊下、蛛网膜下、椎管内和胸骨内注射和输注。
适于胃肠外施用的药物组合物包含一种或多种活性化合物与一种或多种药学上可接受的无菌等渗水性或非水性溶液、分散体、悬浮液或乳液或无菌粉末的组合,可在即将使用时将所述无菌粉末复原为无菌注射溶液或分散体,所述无菌注射溶液或分散体可包含抗氧化剂、缓冲剂、抑菌剂、使制剂与预期受者的血液等渗的溶质或助悬剂或增稠剂。
可用于本发明的药物组合物的合适的水性和非水性载体的实例包括水、乙醇、多元醇(诸如甘油、丙二醇、聚乙二醇等)及其合适的混合物、植物油(诸如橄榄油)和可注射的有机酯(诸如油酸乙酯)。例如,可以通过使用包衣材料诸如卵磷脂,通过在分散体的情况下保持所需的粒度,以及通过使用表面活性剂来保持适当的流动性。
这些组合物还可包含佐剂,诸如防腐剂、润湿剂、乳化剂和分散剂。通过包含各种抗细菌和抗真菌剂,例如对羟基苯甲酸酯、氯丁醇、苯酚山梨酸等,可以确保防止微生物的作用。还可能期望在将等渗剂,诸如糖、氯化钠等包含到组合物中。另外,可注射药物形式的延长吸收可通过包含延迟吸收的试剂诸如单硬脂酸铝和明胶来实现。
在一些情况下,为了延长药物的作用,需要减缓药物从皮下或肌内注射中的吸收。这可通过使用水溶性差的晶体或无定形材料的液体悬浮液来实现。因此药物的吸收速率取决于其溶解速率,而溶解速率又取决于晶体尺寸和晶体形式。或者,胃肠外施用的药物形式的延迟吸收通过将药物溶解或悬浮在油媒介物中来实现。
可注射储库形式是通过在生物可降解聚合物诸如聚丙交酯-聚乙交酯(polylactide-polyglycolide)中形成主题化合物的微囊化基质来制备的。根据药物与聚合物的比例以及所用特定聚合物的性质,可以控制药物释放的速率。其它生物可降解聚合物的例子包括聚(原酸酯)和聚(酸酐)。还通过将药物包裹在与身体组织相容的脂质体或微乳剂中来制备可注射储库制剂。
为了在本发明的方法中使用,可将活性化合物本身单独施用或将其作为药物组合物施用,所述药物组合物包含例如0.1%至约99.5%(更优选约0.5%至约90.0%)的与药学上可接受的载体组合的活性成分。
在本发明的一些实施方案中,可将本发明的化合物与一种或多种另外的化合物/剂联合施用。
在某些此类实施方案中,联合施用是同时的。在某些此类实施方案中,将本发明的化合物与一种或多种另外的化合物共同配制。在某些其它此类实施方案中,将本发明的化合物与一种或多种另外的化合物分开但同时施用。在某些此类实施方案中,联合施用是连续的,在施用一种或多种另外的化合物之前或之后数分钟或数小时,施用本发明的化合物。
引入本发明化合物的方法还可由可再装填或可生物降解的装置提供。近年来,已经开发了各种缓释聚合物装置,并在体内测试了其药物(包括蛋白质生物药物)的受控递送。各种生物相容性聚合物(包括水凝胶),包括生物可降解和不可降解的聚合物,可用于形成植入物,用于在特定的靶部位持续释放化合物。
药物组合物中活性成分的实际剂量水平可以变化,以获得对特定患者、组合物和施用方式实现所需治疗反应有效而对患者无毒的活性成分的量。
所选择的剂量水平将取决于多种因素,包括所使用的特定化合物、缀合物或化合物和/或缀合物的组合、或其酯、盐或酰胺的活性、施用途径、施用时间、所使用的一种或多种特定化合物的排泄速率、治疗持续时间、与所使用的一种或多种特定化合物结合使用的其它药物、化合物和/或材料、所治疗患者的年龄、性别、体重、状况、一般健康状况和既往病史,以及医学领域中公知的类似因素。
具有本领域普通技术的医生或兽医可以容易地确定和开出所需药物组合物的治疗有效量。例如,医生或兽医可以将药物组合物或化合物的起始剂量设定在低于达到所需治疗效果所需的水平,并逐渐增加剂量,直到达到所需效果。“治疗有效量”是指足以引起所需治疗效果的化合物浓度。一般认为,化合物的有效量将根据受试者的体重、性别、年龄和病史而变化。影响有效量的其它因素可包括但不限于患者疾患的严重程度、所治疗的病症、化合物的稳定性,以及如果需要,与本发明的化合物一起施用的另一种类型的治疗剂。可通过多次施用剂来递送更大的总剂量。确定功效和剂量的方法是本领域技术人员已知的(Isselbacher等人(1996)Harrison’s Principles of Internal Medicine第13版,1814-1882,通过引用并入本文)。
一般而言,用于本发明的组合物和方法的活性化合物的合适日剂量将是有效产生治疗效果的最低剂量的化合物的那个量。这种有效剂量通常取决于上述因素。
如果需要,可将活性化合物或缀合物的有效日剂量以合适的间隔在一天中以1个、2个、3个、4个、5个、6个或更多个分开施用的亚剂量施用,任选地以单位剂型施用。在本发明的某些实施方案中,可以每天施用活性化合物两次或三次。在优选实施方案中,将每天施用活性化合物一次。
接受这种治疗的患者是任何有需要的动物,包括灵长类动物,特别是人,以及其它哺乳动物诸如马、牛、猪和绵羊;以及家禽和宠物。
在某些实施方案中,可将本文公开的化合物或缀合物单独使用或与另一种类型的治疗剂联合施用。如本文中所用,短语“联合施用”是指两种或更多种不同治疗性化合物或缀合物的任何施用形式,所述施用形式使得在先前施用的治疗性化合物或缀合物在体内仍然有效的同时施用第二种化合物或缀合物(例如,两种化合物或缀合物在患者体内同时有效,这可包括两种化合物或缀合物的协同作用)。例如,可将不同的治疗性化合物或缀合物在同一制剂中或在单独的制剂中同时或依次施用。在某些实施方案中,可在1小时内、12小时内、24小时内、36小时内、48小时内、72小时内、一周内或一周以上的时间内施用不同的治疗性化合物或缀合物。因此,接受这种治疗的个体可以受益于不同治疗性化合物或缀合物的联合作用。
本发明包括本文公开的化合物或缀合物的药学上可接受的盐的用途。在某些实施方案中,本发明的设想的盐包括但不限于烷基、二烷基、三烷基或四烷基铵盐。在某些实施方案中,本发明的设想的盐包括但不限于L-精氨酸、苯乙苄胺(benenthamine)、苄星、甜菜碱、氢氧化钙、胆碱、地阿诺(deanol)、二乙醇胺、二乙胺、2-(二乙基胺基)乙醇、乙醇胺、乙二胺、N-甲基葡糖胺、海巴明(hydrabamine)、1H-咪唑、锂、L-赖氨酸、镁、4-(2-羟乙基)吗啉、哌嗪、钾、1-(2-羟乙基)吡咯烷、钠、三乙醇胺、氨丁三醇和锌盐。在某些实施方案中,本发明的设想的盐包括但不限于Na、Ca、K、Mg、Zn或其它金属盐。
药学上可接受的酸加成盐还可以以各种溶剂化物的形式(诸如与水、甲醇、乙醇、二甲基甲酰胺等一起)存在。还可制备此类溶剂化物的混合物。这种溶剂化物的来源可来自结晶的溶剂、制备或结晶的溶剂中固有的、或者外加入这类溶剂中的。
湿润剂、乳化剂和润滑剂,诸如十二烷基硫酸钠和硬脂酸镁,以及着色剂、脱模剂、包衣剂、甜味剂、调味剂和芳香剂、防腐剂和抗氧化剂也可以存在于组合物中。
药学上可接受的抗氧化剂的实例包括:(1)水溶性抗氧化剂,诸如抗坏血酸、半胱氨酸盐酸盐、硫酸氢钠、偏亚硫酸氢钠、亚硫酸钠等;(2)油溶性抗氧化剂,诸如抗坏血酸棕榈酸酯、丁基化羟基苯甲醚(BHA)、丁基化羟基甲苯(BHT)、卵磷脂、没食子酸丙酯、α-生育酚等;和(3)金属螯合剂,诸如柠檬酸、乙二胺四乙酸(EDTA)、山梨糖醇、酒石酸、磷酸等。
现已总体描述了本发明,通过参考以下实施例,将更易于理解本发明,将所述实施例包括在内仅仅是为了阐明本发明的某些方面和实施方案,并不意图限制本发明。
合成方案
缩写
AcO: 乙酰基
AcOH: 乙酸
EA: 乙酸乙酯
DCM: 二氯甲烷
m-CPBA: 间氯过氧苯甲酸
TBDMSOTf: 三氟甲磺酸叔丁基二甲基甲硅烷基酯
TBDMS: 叔丁基二甲基甲硅烷基
DMF: 二甲基甲酰胺
EDCI: 1-乙基-3-(3-二甲氨基丙基)碳二亚胺
HOBt: 1-羟基苯并三唑水合物
ACN: 乙腈
TBDMS-Cl: 叔丁基二甲基甲硅烷基氯
DBU: 1,8-二氮杂双环[5.4.0]十一碳-7-烯
THF: 四氢呋喃
DCC: N,N’-二环己基碳二亚胺
DMAP: 4-二甲基氨基吡啶
NHS: N-羟基琥珀酰亚胺
DIPEA: 二异丙基乙胺
TEA: 三乙胺
DEAD: 偶氮二甲酸二乙酯
Boc: 叔丁氧羰基
LAH: 氢化锂铝
CDI: 1,1’-羰基二咪唑
BEMP: 2-叔丁基亚氨基-2-二乙基氨基-1,3-二甲基全氢-1,3,2-二氮杂膦
TPSCl: 三苯基氯硅烷
tfa: 三氟乙酰基
PyBop: 苯并三唑-1-基-氧基三吡咯烷子基鏻六氟磷酸盐
HBTU: N,N,N’,N’-四甲基-O-(1H-苯并三唑-1-基)脲鎓六氟磷酸盐
TFA: 三氟乙酸
DIC: N,N’-二异丙基碳二亚胺
DMPA: 2,2-二甲氧基-2-苯基苯乙酮
TBAF: 四正丁基氟化铵
AgOTf: 三氟甲磺酸银
(BimC4A)
表A
接头的制备
在-78℃和N
在N
在室温和N
在0℃和N
将L-2-1(100mg,0.72mmol)和HBr(1.5mL,在AcOH中48%)的混合物在120℃搅拌3小时。用NaHCO
在室温和N
将化合物L-3-1(10mg,0.05mmol)和HBr(2.0mL,在水中48%)的混合物在微波中于150℃下反应3小时。在混合物减压浓缩后,将化合物L-3直接用于下一步骤而无需进一步纯化。(20mg)。EI-MS m/z:309(M
向香草酸(50.0g,0.30mol)在MeOH(700mL)中的溶液中滴加SOCl
在N
在0℃和N
向化合物Int-1-3(85.5g,0.27mol)在THF(800mL)和MeOH(300mL)中的溶液中加入2N NaOH(404mL,0.81mol)。在65℃下搅拌5小时后,将反应物冷却至室温,并通过加入2NHCl溶液调节至pH 2,然后用蒸馏水(100mL)和EA(300mL X 2)萃取。将有机层经无水Na
向化合物Int-1-4(100mg,0.33mmol)在无水THF(500μL)和无水DCM(1.5mL)中的溶液中缓慢滴加草酰氯(42.4μL),并在0℃和N
在室温和N
在N
1H NMR(400Hz,CDCl
在N
在室温下,将5%Pd/C(1.04g,0.49mmol)加入到搅拌的L-5-2(1.0g,3.25mmol)在EtOH(5mL)中的溶液中。将氢气鼓泡通过反应混合物4小时。将混合物通过硅藻土过滤以除去Pd/C,并减压浓缩。在将残留物溶解在DCM(25mL)中后,向其中加入BOC
EI-MS m/z:382(M
通过与Journal of Polymer Science,Part A:Polymer Chemistry,2012,50(19),3986-3995(通过引用并入本文)中所述类似的合成途径合成了化合物L-6。
收率为30%
收率68%
收率为63%
收率为76%
在N
EI-MS m/z:462(M
在N
使反应物冷却,用MeOH(5mL)猝灭,并减压浓缩。将残留物通过柱色谱纯化,得到化合物L-7-2(1.91g,93%)。
EI-MS m/z:610(M
在0℃和N
EI-MS m/z:584(M
在-20℃和N
向在冰浴中冷却的CBr
在室温和N
EI-MS m/z:303(M
在0℃和N
EI-MS m/z:671(M
在0℃和N
EI-MS m/z:645(M
在0℃和N
EI-MS m/z:571(M
在室温和N
在室温和N
在N
在50mL的圆瓶装烧瓶中加入L-10-3(500mg,2.24mmol)、10mL MeOH、5%Pd/C(715mg,0.34mmol,0.15当量)和Boc
在室温于N
EI-MS m/z:284(M
在室温和N
EI-MS m/z:850(M
在室温和N
EI-MS m/z:749(M
在室温和N
在室温和N
在室温和N
在室温和N
在室温和N
EI-MS m/z:367(M
在室温和N
EI-MS m/z:483(M
在室温和N
EI-MS m/z:382(M
在室温和N
EI-MS m/z:319(M
在室温和N
EI-MS m/z:293(M
在0℃和N
EI-MS m/z:245(M
在-78℃和N
在室温和N
在室温和N
在0℃和N
在0℃和N
在室温和N
在室温和N
在0℃和N
EI-MS m/z:574(M
在0℃和N
在N
在室温和N
EI-MS m/z:799(M
在室温和N
在室温和N
向化合物Int-TG1(100mg,0.13mmol)和化合物Int-TG3a(26mg,0.13mmol)在无水ACN(3mL)中的溶液中加入DBU(4μL,25μmol)。将混合物在室温下搅拌1小时,用蒸馏水(10mL)洗涤,并用EA(15mL×2)萃取。将有机层经无水Na
EI-MS m/z:869(M
在0℃和N
EI-MS m/z:871(M
在0℃和N
EI-MS m/z:934(M
在室温和N
在室温和N
在室温和N
EI-MS m/z:874(M
在室温和N
EI-MS m/z:869(M
在0℃和N
EI-MS m/z:945(M
在0℃和N
EI-MS m/z:1024(M
在室温和N
EI-MS m/z:1008(M
通过与化合物Int-5的制备方法类似的方式合成化合物Int-6。
收率为72%,无色油状物
EI-MS m/z:1226(M
收率为82%,无色油状物
EI-MS m/z:1296(M
收率为75%,无色油状物
EI-MS m/z:1298(M
收率为82%,无色油状物
EI-MS m/z:1376(M
收率为82%,无色油状物
EI-MS m/z:1361(M
在室温和N
在室温下,向MPS-D1-1(6.11g,20.52mmol)在EtOH(40mL)和MeOH(26mL)中的溶液中加入4-甲氧基苯硫醇(2.55g,20.52mmol)和哌啶(0.3mL,3.08mmol)。在100℃下搅拌16小时后,将混合物冷却至0℃,并另外搅拌1小时。将固体过滤并用醚(30mL×2)洗涤,得到化合物MPS-D1-2(5.56g,90%)。
在0℃和N
在N
化合物MPS-D3通过与实施例22中所述类似的合成途径来合成。
收率为72%
EI-MS m/z:899(M
在0℃和N
在N
在N
在0℃下,向化合物POS-D1-2(5g,25.75mmol)在THF(100mL)中的溶液中滴加Et
在0℃和N
向POS-D1-4(310mg,1.29mmol)和L-8-1(660mg,2.84mmol)在THF(8mL)和DMF(0.8mL)中的溶液中加入PPh
EI-MS m/z:455(M
在室温和N
EI-MS m/z:430(M
在室温和N
EI-MS m/z:1064(M
通过与实施例22中所述类似的合成途径合成了化合物MPS-D6、MPS-D7、MPS-D8和MPS-D9。
收率为53%,浅黄色固体
收率为52%,黄色固体
收率为53%,黄色油状物
EI-MS m/z:753(M
收率为84%,浅黄色油状物
EI-MS m/z:621(M
在室温和N
EI-MS m/z:262(M
在室温和N
EI-MS m/z:301(M
在0℃和N
EI-MS m/z:333(M
在室温和N
EI-MS m/z:430(M
在室温和N
EI-MS m/z:1064(M
在室温和N
EI-MS m/z:697(M
在室温和N
EI-MS m/z:546(M
将在室温和N
在室温和N
经由与实施例21的化合物MPS-D1的制备方法类似的方式合成化合物mMPS-D5-3、mMPS-D5-4和mMPS-D5。
收率为30%,白色固体。
EI-MS m/z:264(M
收率为11%,白色固体。
收率为43%,白色固体。
EI-MS m/z:334(M
通过与实施例27的化合物MPS-D9的制备方法类似的方式合成化合物mMPS-D6。
收率为13%,黄色油状物。
EI-MS m/z:622(M
在室温和N
通过与实施例22的化合物MPS-D2的制备方法类似的方式合成化合物MPS-D10-1和MPS-D11-1。
收率为71%,浅黄色油状物
收率为71%,浅黄色油状物
通过与实施例10的化合物L-9的制备方法类似的方式合成化合物MPS-D10-2和MPS-D11-2。
收率为99%,浅黄色油状物。
收率为定量,浅黄色油状物。
EI-MS m/z:463(M
在室温和N
通过与化合物MPS-D10的制备方法类似的方式合成化合物MPS-D11。
收率为82%,浅黄色油状物。
EI-MS m/z:639(M
单体的制备
通过以EP20071813614中描述的类似方法进行反应获得PBD单体。
在N
在0℃下向化合物Int-1(9.07g,28.22mmol)在无水THF(50mL)中的溶液中加入化合物M-1-1(6.42g,28.22mmol)在THF(100mL)和TEA(7.9mL,56.43mmol)中的溶液。在于室温下搅拌2小时后,将反应物用蒸馏水(500mL)稀释,并用EA(800mL)萃取。将有机层经无水Na
在-78℃和N
在室温下,向化合物M-1-3(3g,6.72mmol)在THF(130mL)和蒸馏水(86mL)中的溶液中加入Na
在0℃下,向化合物M-1-4(1g,2.51mmol)在无水DCM(10mL)中的溶液中加入甲磺酸(5mL)在DCM(10mL)中的溶液。在0℃下搅拌3小时后,将混合物用NaHCO
通过以WO2010091150中描述的类似合成方法进行反应获得IBD单体。
在室温和N
在0℃和N
在0℃和N
1H NMR(400MHz,CDCl
在室温和N
在N
在0℃和N
在N
在0℃和N
在-78℃和N
在-10℃和N
在室温和N
在室温和N
在-78℃和N
在室温和N
在0℃下向化合物Int-1(10.24g,31.85mmol)在无水THF(20ml)中的溶液中加入在THF(20mL)中的化合物M-2-5(6.7g,28.95mmol)和DIPEA(15.1mL,86.86mmol)。室温下搅拌5小时后,用蒸馏水(50mL)和EA(2X 150mL)稀释反应物。将有机层经无水Na
在H
在0℃下将氯甲酸2,2,2-三氯乙酯(555uL,4.03mmol)在无水DCM(20mL)中的溶液滴加到M-2-16(2.0g,5.04mmol)和吡啶(2.0mL,25.2mmol)在无水DCM(30mL)中的溶液中。在0℃搅拌1小时后,用CuSO
在-78℃下,将DMSO(485.2uL,6.45mmol)在无水DCM(5mL)中的溶液滴加到草酰氯(369uL,4.3mmol)在无水DCM(20mL)中的溶液中。在-78℃下搅拌1小时后,将M-2-17(1.23g,2.15mmol)在无水DCM(20mL)中的溶液滴加到反应混合物中,并在-78℃搅拌2小时。2小时后,在反应混合物中加入TEA(3mL,21.5mmol),并在室温下搅拌1小时。用NH
通过WO2005/085251和Tetrahedron Letters 56(2015)4512-4515(所述两篇文献均通过引用完全并入本文)中描述的类似合成途径合成PBD化合物。
向(S)-2-氨基-3-(4-羟基-3,5-二碘苯基)丙酸(8.0g,18.48mmol)在浓HCl水溶液(90mL)中的溶液中加入1,2-二甲氧基乙烷(7.5mL)和多聚甲醛(在H
在室温和H
在N
在0℃下向化合物Int-1(640.9mg,1.86mmol)在无水THF(4.0mL)中的溶液中加入于DMF(5.0mL)中的化合物M-3-3(350mg,1.43mmol),然后加入DIPEA(750uL,4.3mmol)。在室温下搅拌2小时后,用蒸馏水(10mL)和EA(2X 50mL)洗涤混合物。将有机层经无水Na
在0℃下,将叔丁基二甲基甲硅烷基氯(188.5mg,1.25mmol)加入到M-3-4(616mg,1.25mmol)和咪唑(102.2mg,1.50mmol)在无水DCM(6.0mL)中的溶液中。在室温下搅拌反应混合物过夜。将反应物用2N HCl(5mL)和盐水(10mL)猝灭,并用DCM(10mL x 2)萃取。将有机层经无水Na
在-78℃和N
在室温下,向化合物M-3-6(280.4mg,0.49mmol)在THF(6.0mL)和蒸馏水(86mL)中的溶液中加入Na
在N
在室温和N
EI-MS m/z:458(M
通过WO2016/115191A1中描述的类似合成途径合成Ref-1。在N
EI-MS m/z:719(M
在室温和N
在室温和N
EI-MS m/z:20(M
在N
EI-MS m/z:734(M
通过与实施例44中所述类似的合成途径合成了化合物D-4(收率为85%)。
EI-MS m/z:720(M
在室温和N
EI-MS m/z:812(M
在室温和N
EI-MS m/z:776(M
在50℃和N
通过与实施例43中所述类似的合成途径合成了化合物D-7。EI-MS m/z:707(M
通过与实施例47中所述类似的合成途径合成了化合物D-8(收率为50%)。EI-MSm/z:748(M
在室温和N
EI-MS m/z:700(M
在室温和N
通过与实施例47中所述类似的合成途径合成了化合物D-10。
收率为23%,为白色固体。EI-MS m/z:658(M
在室温和N
EI-MS m/z:1146(M
在室温和N
EI-MS m/z:759(M
异二聚体的制备
表B
在室温和N
EI-MS m/z:585(M
在室温和N
EI-MS m/z:943(M
在室温和N
EI-MS m/z:907(M
在0℃和N
EI-MS m/z:792(M
在室温和N
在室温和N
在室温和N
在N
在室温和N
在室温和N
收率为63%
EI-MS m/z:1834(M
在N
收率为83%
EI-MS m/z:1666(M
在室温和N
在N
在室温和N
在N
在室温和N
EI-MS m/z:1162(M
在0℃和N
EI-MS m/z:1072(M
通过与实施例55所述类似的合成途径合成了化合物T-Int-6-3。
收率为64%,为白色固体。
EI-MS m/z:1448(M
在0℃和N
EI-MS m/z:1280(M
在室温和N
EI-MS m/z:1704(M
通过与实施例60的化合物T-Int-6的制备方法类似的方式合成化合物T-Int-7-2。
收率为81%,白色固体
EI-MS m/z:1536(M
在0℃和N
EI-MS m/z:1436(M
通过与化合物T-Int-7的制备方法类似的方式合成了T-Int-8。
收率为84%,黄色固体
EI-MS m/z:2057(M
收率为84%,无色油状物
EI-MS m/z:1889(M
收率为74%,乳白色固体
EI-MS m/z:1789(M
在室温和N
EI-MS m/z:1687(M
通过与实施例62中所述类似的合成途径合成T-Int-9。
收率为72%,乳白色固体
EI-MS m/z:1789(M
通过与化合物T-Int-9的制备方法类似的方式合成了T-Int-10。
收率为75%,乳白色固体
EI-MS m/z:2040(M
收率为60%,乳白色固体
EI-MS m/z:1940(M
在室温和N
EI-MS m/z:585(M
通过与实施例54的化合物D-12-2的制备方法类似的方式合成化合物T-Int-11-2。
收率为60%,乳白色固体
EI-MS m/z:1539(M
通过与实施例56的化合物T-Int-1的制备方法类似的方式合成化合物T-Int-11。
收率为72%,白色固体
EI-MS m/z:1371(M
通过与实施例59所述类似的合成途径合成了化合物T-Int-12。
收率为54%,乳白色固体
EI-MS m/z:1544(M
收率为74%,黄色固体
EI-MS m/z:1386(M
通过与实施例59所述类似的合成途径合成了化合物T-Int-13。
收率为53%,白色固体
EI-MS m/z:1530(M
收率为81%,黄色固体
EI-MS m/z:1362(M
在室温和N
EI-MS m/z:1612(M
通过如实施例66所述的类似合成途径合成了化合物T-Int-15和T-Int16。
收率为35%,白色固体;EI-MS m/z:1763(M
收率为11%,白色固体;EI-MS m/z:2116(M
在室温和氮气气氛下加入DMSO(8643μL),并将化合物T-Int-1(15mg,0.012mmol)溶解在DMSO(1237μL)中并加入其中。以4207μL的量向其中加入经制备浓度为10mmol的(BimC
EI-MS m/z:1668(M
将化合物T-Int-1与MPS-D2以与实施例68的化合物T-1的制备方法类似的途径反应,从而获得化合物T-2(收率为7%)。
EI-MS m/z:1759(M
通过如实施例68所述的类似合成途径合成了化合物T-3和T-4。
收率为9.0%;EI-MS m/z:1860(M
收率为26%;EI-MS m/z:1282(M/2
通过与实施例68中所述类似的合成途径合成了化合物T-5。
收率为27%
EI-MS m/z:1945(M
通过与实施例68中所述类似的合成途径合成了化合物T-6和T-7。
收率为25%
EI-MS m/z:1004(M/2
收率为62%
EI-MS m/z:1180(M/2
通过与实施例68中所述类似的合成途径合成了化合物T-8。
收率为27%
EI-MS m/z:1174(M/2
在室温和N
EI-MS m/z:2178(M
通过与实施例74中所述类似的合成途径合成了为白色固体的化合物T-10(收率为56%)。
EI-MS m/z:2269(M
通过与实施例74中所述类似的合成途径合成了为白色固体的化合物T-11(收率为34%)。
EI-MS m/z:2377(M
通过与实施例74中所述类似的合成途径合成了为白色固体的化合物T-12(收率为58%)。
EI-MS m/z:2377(M
通过与实施例74中所述类似的合成途径合成了为白色固体的化合物T-13(收率为58%)。
EI-MS m/z:2434(M
通过与实施例74中所述类似的合成途径合成了为白色固体的化合物T-14(收率为72%)。
EI-MS m/z:2011(M
通过与实施例74中所述类似的合成途径合成了为淡黄色固体的化合物T-15(收率为48%)。
EI-MS m/z:2284(M
通过与实施例74中所述类似的合成途径合成了为淡黄色固体的化合物T-16(收率为69%)。
EI-MS m/z:2360(M
通过与实施例74中所述类似的合成途径合成了为白色固体的化合物T-17(收率为40%)。
EI-MS m/z:1759(M
通过与实施例74中所述类似的合成途径合成了为乳白色固体的化合物T-18(收率为71%)。
EI-MS m/z:1908(M
通过与实施例47中所述类似的合成途径合成了化合物D-9(收率为50%)。
EI-MS m/z:800(M
生物测试
将A-498、HCT-116、JIMT-1、MIA-PaCa-2和HEK-293E以10x10
表1A.体外生物学和药代动力学数据
表1B.体外生物学和药代动力学数据
在37℃下用约20-50倍过量的TCEP(三(2-羧乙基)膦盐酸盐或DTT(二硫苏糖醇)在含有1mM EDTA的4mM Tris pH 7.3中还原半胱氨酸工程化的单克隆抗体,持续1小时。将还原的硫代单抗(thiomab)稀释并加载到PBS中的PD-10柱上。用pH 7.3的10mM PBS洗脱该柱。通过空气氧化重新建立洗脱的还原的硫代单抗。硫醇/Ab值通过从溶液在280nm处的吸光度确定还原的抗体浓度来检查,并且硫醇浓度通过与DTNB(Aldrich,CAS No D8130)反应和并确定412nm处的吸光度来检查。
缀合方法1:还原和再氧化反应后,将抗体溶解在PBS中。用还原的再氧化抗体(1650uL,0.083mmol)处理化合物T-2(329.6uL,3.0mmol,作为接头-毒素中间体)在DMSO中的溶液,并在室温下轻轻搅拌3小时。将羟胺(329.6uL,1,500mmol)加入到反应混合物的溶液中,并在37℃孵育8小时,以阻断可逆的解缀合(deconjugation)反应。加载缀合混合物,并通过PD-10柱洗脱,以除去过量的药物-接头中间体和其它杂质。将混合物通过离心超滤浓缩,并将缀合物用HIC NPR柱(TOSOH#0007656TSKgel Phenyl-5PW,21.5x 150mm,13μm)纯化,并用从40%至100%B的线性梯度以0.8ml/min洗脱(A缓冲液1.5M的于50mM磷酸钠(pH7.0)中的硫酸铵;B缓冲液:20%的于50mM磷酸钠(pH 7.0)中的乙腈)。通过HIC分析缀合抗体的DAR(药物与抗体的比率),并且分析结果如表2所示。
将实施例70、72、73、74、76、77、78、79、80、81和82中获得的化合物T-3、T-7、T-8、T-9、T-11、T-12、T-13、T-14、T-15、T-16和T-17用于与曲妥珠单抗(或人血清白蛋白等)的工程化半胱氨酸的巯基基团进行缀合反应,从而参照文献中给出的方法分别制备T-3-AB、T-7-AB、T-8-AB、T-9-AB、T-11-AB、T-12-AB、T-13-AB、T-14-AB、T-15-AB、T-16-AB和T-17-AB作为硫代单抗药物缀合物(TDC)。[参见Nature Biotechnology,2008,26,925-932,Bioconjugate Chem.,2013,24,1256-1263,Bioconjugate Chem.,2016,27,1324-1331,Bioconjugate Chem.2014,25,460-469]。
缀合方法2:将实施例34中获得的MPS-D10、实施例27中获得的MPS-D9和实施例32中获得的mMPS-D6中的每一种化合物用于与抗体(例如曲妥珠单抗)的工程化半胱氨酸的巯基基团进行第一步缀合反应。
在实施例84的还原和再氧化程序之后,用DMSO中的每种化合物(6.62uL,3.0mmol)处理PBS中的抗体。3小时后,向缀合的溶液中加入硼氢化钠(6.62ul,300mmol),以在室温下阻断可逆的解缀合反应1小时。并且用PD-10柱纯化第一缀合的抗体。对于第二缀合,将实施例56中获得的T-Int-1(13.24uL,3.0mmol)或实施例57中获得的T-Int-2(20.0uL,3.0mmol)(含官能团诸如在Cu(I)催化剂不存在的情况下促进环加成的N
表2.抗体-药物缀合物(ADC)
将NCI-N87、JIMT-2、SK-BR3和SK-OV3癌细胞以每孔2,000至8,000个细胞的密度接种在96孔板中的100μL培养基中,并培养24小时。将实施例46中获得的四种化合物用从600nM至0.009nM的1:4的系列稀释液处理,并将抗体药物缀合物T-DM1用从200nM至0.003nM的1:4的系列稀释液处理。以与测试实施例1类似的方式进行实验,以在96小时后定量活细胞,并且结果如下表2中所示。另外,化合物T-2-AB、T-3-AB、T-7-AB、T-8-AB、T-9-AB、T-11-AB、T-12-AB、T-13-AB、T-14-AB、T-15-AB、T-16-AB、T-17-AB、T-Int-1-AB、T-Int-2-AB、T-Int-14-AB和mT-Int-1-AB的体外分析结果如图10、图11、图12和表3所示。
表3.抗体-药物缀合物的细胞毒性
(*T-DM1:Roche CAS编号;1018448-65-1)
T-2-AB和T-7-AB采用15mg规模的反应来制备。通过HIC柱纯化后,LC/MS分析证实硫代单抗(曲妥珠单抗A121C)的分子量为145,324Da。对制备的抗体药物缀合物T-2-AB的LC/MS分析显示存在约148,497Da的质量。这些质量偏移与两个D-1分子的缀合一致。并且T-7-AB显示出存在约149,671Da的质量,与两个D-4分子相似。
通过在小鼠中进行肿瘤异种移植研究来测量本发明的抗体-药物缀合物的体内功效。在雄性BALB/c nu/nu的右侧腹部皮下注射分别在PBS中的5X10
除了初始肿瘤尺寸为150mm
这些体内实验由CACT(阿桑医疗中心癌症治疗促进中心,项目代码:HI15C0972)进行
本文提及的所有出版物和专利在此通过引用整体并入,就如同每个单独的出版物或专利被具体地和单独地指示通过引用并入。在发生冲突的情况下,以本申请(包括本文的任何定义)为准。
虽然已经讨论了本主题发明的具体实施方案,但以上说明书是说明性的而非限制性的。通过阅读本说明书和下面的权利要求书,本发明的许多变化对于本领域技术人员来说将变得显而易见。本发明的全部范围应该通过参考权利要求书连同其等同物的全部范围、以及说明书连同此类变化来确定。
- 8-氮杂二环3.2.1辛烷-,8-氮杂二环3.2.1辛-6-烯,9-氮杂二环3.3.1壬烷-,9-氮杂-3-氧杂二环3.3.1壬烷-,9-氮杂-3-硫杂二环3,3,1壬烷衍生物,其制备方法及其作为杀虫剂的用途
- 二环苯衍生物取代的5,6-二氢-4H-吡咯并1,2-A1,4苯并二氮杂*和6H-吡咯并1,2-A1,4苯并二氮杂*新抗真菌剂