包含阴离子空位晶格的哈伯-博施催化剂
文献发布时间:2023-06-19 09:44:49
技术领域
本发明涉及用于哈伯-博施(Haber-Bosch)方法的催化剂。具体地,涉及催化组合物、包含所述组合物的盒、所述组合物在哈伯-博施方法中催化氨生产的用途以及其中所述组合物被提供为催化剂的哈伯-博施方法。
背景技术
哈伯-博施方法是20世纪发现的最重要的化学反应之一。氨是几乎所有化学上有用的含氮化合物的基础,并且是依靠金属催化剂从氢气和相对惰性的氮气的混合物产生。哈伯-博施方法的重要性进一步体现在授予以他们的名字命名该方法的两位先驱者诺贝尔化学奖。
氢气和氮气在加压容器中组合并加热。在合适的催化剂存在下,氢和氮分子在催化剂表面反应形成氨,氨随后从催化剂中解吸。
进行反应的确切机理尚不完全清楚,但据信,不受理论的束缚,氮气分子吸附在催化剂表面上并解离形成更能与氢气分子反应的高反应性的氮物种。
在过去的100年中,已经研究了许多催化剂并提出了对该技术的许多修改。例如,已经测试将助催化材料与传统的哈伯-博施催化剂组合,以尝试提高催化活性。实例包括K
由于保持连续、高温、高压反应条件是昂贵的,因此已经尝试摆脱常规的哈伯-博施方法。已探索的一种技术是氨的电化学生产,如在"Ammonia synthesis at atmosphericpressure in a BaCe
考虑到昂贵的操作成本,需要改进的催化剂材料以允许反应在较温和的条件下以可比的(comparable)速率进行并在可比的条件下提高反应速率。
本发明旨在解决或至少缓解这些问题。
发明内容
在本发明的第一方面中,提供用于催化哈伯-博施方法的组合物,所述组合物包含阴离子空位晶格和哈伯-博施催化剂。
术语“哈伯-博施方法”旨在指在非均相催化剂存在下由氢气和氮气的混合物生产氨,其中氢气和氮气在催化剂表面上一起反应。换句话说,方法类似于基于Fritz Haber和Carl Bosch提出的反应的方法。该方法通常在本领域技术人员所熟悉的高温和高压下进行。例如,术语“哈伯-博施方法”不被认为涵盖氨的电化学合成,因为氢源和氮源在不同的腔室中提供并且该方法被认为是经由完全不同的机制进行,除其他方面外尤其需要活性中间物种经由电极进行扩散。
术语“哈伯-博施催化剂”旨在指在哈伯-博施方法中催化氨生产的任何材料。历史上,许多不同的材料被用作催化剂(甚至锇和铀曾经被认为是有效的催化剂)。随后的研究表明了其他更容易获得的材料(例如钴、铁、镍和钌)的有效性。据信,这些材料对于哈伯-博施方法很好地充当催化剂,因为它们吸附氮气并促进反应性氮物种的形成。据信,这些反应性氮物种使氨的形成迅速发生。因此,如本文所指的“哈伯-博施催化剂”旨在涵盖以这种能力操作的所有材料。
为了适合在哈伯-博施方法中作为催化剂,组合物必须在该方法操作的整个条件范围内保持足够稳定。通常,哈伯-博施方法在高达700℃的温度和超过20MPa的压力下进行。
术语“阴离子空位晶格”旨在描述具有包含阴离子的结构(例如晶体结构)的材料,其中那些阴离子中的一些会丢失,从而产生阴离子空位。这主要使用掺杂来实现。优选的是包含氧和氮阴离子的材料,且因此通常采用氧和氮空位晶格。该材料可以处于结晶或非晶态。术语“氧空位晶格”或“氮空位晶格”旨在描述这样的晶格,其分别具有作为晶格结构的关键组分的氧或氮并且固有地或由于暴露到某些反应条件而从其结构缺失氧或氮离子从而在晶格内留下空位(尺寸分别相当于氧和氮离子)。在一些材料中,氧空位和氮空位二者可能共存,例如掺杂的氮氧化铈。对本发明中使用的晶格的类型没有特别限制。该材料也可以处于非晶态。晶格可以是7种常规类型晶格中的任何一种:三斜晶系、单斜晶系、斜方晶系、四方晶系、三方晶系、六方晶系和立方晶格。通常,晶格可以是斜方的、四方的、六方的或立方的。通常,晶格将是立方或伪立方的。用于本发明的晶体结构的典型实例包括钙钛矿和萤石。阴离子空位晶格充当助催化剂,与哈伯-博施催化剂结合提高反应速率。
发明人惊奇地发现,具有阴离子空位的晶格非常好地充当常规哈伯-博施催化剂的助催化剂,导致与常规催化剂相比,催化剂活性的显著改进。不受理论的束缚,据信氮气分子将解离吸附在助催化剂中的哈伯-博施催化剂上,导致助催化剂组合物的“阴离子空位晶格”的表面上的活性氢物种与所述氮物种反应的趋势增加。阴离子空位晶格内的阴离子没有特别限制,但通常选自氧、氮、氟、氯、溴、碘、硫、硒或它们的组合。最典型地,阴离子空位晶格中的阴离子是氧和/或氮。
通常,将组合物配置用于催化哈伯-博施方法。哈伯-博施方法是一种非均相反应,其中气体吸附到固体催化剂表面上,反应且然后解吸。因此,组合物通常是为此目的配制的。这可以包括提供固体组合物的最小表面积以确保有效的反应速率。例如,该组合物可以作为以下形式提供:粉末、高表面积载体上的涂层;支撑颗粒上的涂层;浸渍在多孔介质内;或它们的组合。
尽管对哈伯-博施催化剂的选择没有特别限制,典型情况下,哈伯-博施催化剂包含选自由Co、Ni、Fe、Ru或它们的组合组成的组的金属化合物。更典型地,金属化合物是Fe、Ru或它们的组合且甚至更典型地,金属化合物是Fe。仍更典型地,哈伯-博施催化剂是铁氧化物(例如Fe
通常,阴离子空位晶格中的阴离子空位通过掺杂母体阴离子晶格(例如氧化物或氮化物)产生。在哈伯-博施方法中加热或加压时,某些晶格会自然地从其结构中丢失阴离子(例如氧或氮),从而原位形成空位。然而,为了辅助该方法和/或为了产生或最大化阴离子空位的数量,可以使用掺杂剂离子产生电荷失配,从而将空位引入到主要呈规则形式的晶格中。这也是有利的,不仅因为它增加空位的数量,而且还因为(取决于电荷失配的大小)它可以增加空位内的氮三键所感觉到的作用的幅度。掺杂剂(相对富电子或相对贫电子)的选择可以改变阴离子空位周围的环境的特性,特别是对氮三键的影响的幅度。因此,掺杂允许针对不同场景创建量身定制的环境。
本发明的关键优点是具有阴离子空位(无论是内部或外部空位)的任何材料都可以用作基于Fe、Co和Ru的氨合成催化剂的促进剂。典型的阴离子空位是氧空位和氮空位或两者的组合,如一些氮氧化物中存在的空位。催化剂不仅限于Fe、Co、Ni或Ru中的一种,它可以是这三种元素即Fe、Ru、Ni和/或Co中的混合物或合金,例如Fe/Ni合金。
尽管对待掺杂的氧空位晶格的选择没有特别限制,但氧空位晶格通常是氧化物。最典型地,氧空位晶格是萤石或钙钛矿结构(但不限于这些结构),例如铈氧化物、锆氧化物、铋氧化物、钛氧化物、铝氧化物、镁氧化物、铁氧化物或它们的组合(所有这些可以被掺杂)。其中,铈氧化物、锆氧化物和钛氧化物通常是最常使用的材料类别。合适的氧空位晶格材料的典型实例包括但不限于:BaZrO
通常情况下,可以向其中添加掺杂剂的氧空位晶格选自:CeO
氧化物之间或之中可能存在固溶体。例如,CeO
Ce
Ce
在从x=0至x=0.50的(Ce
同样的固溶体可以在特定的组成范围内在CeO
Bi
可以将更通用的式提供为:
A
其中A、B、C和D中的至少一个是电荷(化合价)高于3(+3)的元素,例如形成相的固溶体的Ce、Zr、Ti、Sn、Bi、Si、V、W、Nb、Ta、Hf或镧系元素。例如,Zr
化合价由IUPAC定义为:可以与所考虑元素的原子或片段结合或该元素的原子可以被取代的单价原子(最初为氢或氯原子)的最大数量。
A
典型的+4价的元素是Ce、Zr和Ti。典型的+3价的元素是Al、Sc、Cr、Mn、Fe、Co、Ni、Y、Bi。典型的+2价的元素是Ba、Sr、Ca、Mg。典型的+1价的元素是Na、K。
在一个实施方案中,A是Ce;和/或B是Zr;和/或C是Ti;和/或D是Ca或Y。
用作促进剂的卤氧化物中的氧空位
除了简单的氧化物和复杂的钙钛矿氧化物外,晶格中具有卤化物的材料(例如金属卤氧化物)也可用作氨合成催化剂的促进剂。典型的材料是卤氧化铋,例如BiOCI、BiOBr和BiOI。其他卤氧化物包括卤氧化铁,例如FeOCl、FeOBr、FeOI,以及卤化钴,例如CoOCI、CoOBr、CoOI。本领域技术人员将理解,当在哈伯-博施方法中使用这些卤氧化物时,必须考虑它们的熔点,即熔点必须高于用于由H
用作催化剂促进剂的氮化物中的阴离子空位
已经报告一些氮化物,例如Fe
除这些已知的氮化物外,包含氮空位或可以在氨合成条件下产生氮空位的任何氮化物,例如氮化铁、氮化镍、氮化钴、氮化锰、氮化钒、氮化铬、氮化钛、氮化锆、氮化硅、氮化铝、氮化锡或具有这些元素的组合的氮化物,也可用作氨合成催化剂的促进剂。
用作催化剂促进剂的氮氧化物中的阴离子空位
发明人提出金属氮氧化物(例如Ce
氮氧化物可以是“纯净的”或“纯”或掺杂的,并且包括:CeO
氮氧化物可以是固溶体,例如Ce
氮氧化物可以是掺杂的固溶体,例如Ti
阴离子空位晶格内包含的掺杂剂的量自然会根据所需的空位数量和材料保持其总体结构的能力而变化。存在于阴离子空位晶格内的掺杂剂可以是少数组分,即被替换的材料比替换它的掺杂剂更多。然而,发明人已经确定掺杂剂不必是少数组分。实际上,较高的掺杂剂水平可以提供更多的空位和更大的活性。通常,掺杂剂以全部阴离子空位晶格的1mol%至90mol%,例如1mol%至70mol%,例如1mol%至60mol%,例如1mol%至30mol%范围的量存在,有时以全部阴离子空位晶格的5mol%至30mol%或30mol%至60mol%,例如5mol%至20mol%或40至60mol%范围且通常是10mol%至40mol%,例如全部阴离子空位晶格的10mol%至30mol%范围的量存在。掺杂水平受到在制备条件下离子在母体晶格中的溶解度极限的限制。多个低化合价元素的共掺杂可扩展溶解度极限且因此最大化掺杂水平且因此最大化阴离子空位。
通常,掺杂水平越高,阴离子空位的浓度越高,并且可用的活性位点越多,导致活性越高。因此,接近固溶体的掺杂极限将使阴离子浓度水平最大化以实现最高活性。然而,由于催化方法的复杂性,最高活性可能偏离最高掺杂水平。
当母体氧化物掺杂有较低化合价离子(例如Ce
对于超出固溶体的溶解度极限的任何掺杂水平,掺杂剂将不能进入晶格,因此不能产生氧空位。因此,掺杂至溶解度极限可使掺杂水平最大化。最高的促进效果可能不是由最高的掺杂水平导致的。可以注意到,溶解度极限不仅与材料有关,而且与烧制温度有关。
在式I中示出本发明中使用的氧空位晶格的典型实例;
Ba
其中:“a”表示0到0.2的值,并且“x”、“y”和“z”中的每一个独立地在0.01至0.99的范围内,典型地在0.05至0.95的范围内,前提是“x”、“y”和“z”加起来等于1。发明人已经发现铈和钇掺杂的钡锆氧化物(BZCYO)不仅在标准的哈伯-博施方法操作条件下是稳定的,而且与市场上现有的催化剂相比也表现非常好。典型地,“x”、“y”和“z”中的每一个独立地在0.1至0.8的范围内,并且最典型地,氧空位晶格包括BaZr
在本发明的另一个实施方案中,氧空位晶格可以是根据式II的化合物;
Ce
其中,M是化合价小于4的元素,通常是铈以外的镧系元素或稀土元素,例如Sm、Pr、Eu、Gd或它们的组合,或Sm、Eu、Gd或它们的组合,或Sm、La、Pr、Gd或它们的组合。“a”和“b”独立地在0.05至0.95的范围内,前提是“a”和“b”加起来等于1(大约)。在某些实施方案中,a是0.6或更多、0.7或更多或0.8或更多和/或0.7或更少、0.6或更少或0.5或更少。通常,M是Sm。发明人已经发现掺杂钐的铈氧化物与合适的哈伯-博施催化剂一起在促进哈伯-博施方法方面显示良好的结果。通常,“a”和“b”各自独立地在0.1至0.8的范围内,并且可以是氧空位晶格包含Ce
掺杂水平与元素有关。例如,PrO
已经发现所有这些材料在标准的哈伯-博施方法条件下是稳定的,这是特别有利的,因为在工业上,这种方法通常是连续进行的。因此,催化剂的寿命对于防止该方法的频繁启动和停止是重要的。
在本发明的另一个实施方案中,阴离子空位晶格可以是根据式III的化合物;
Ce
其中M是化合价小于4的元素,通常是铈以外的镧系元素或稀土元素,例如Sm、Pr、Eu、Gd或它们的组合;或Sm、Pr、La、Gd或它们的组合。“a”和“b”独立地在0.05至0.95的范围内,前提是“a”和“b”加起来等于1(大约)。0 X大于0且小于2。Y大于零且小于或等于X。例如,X可以是0.1至1.9。X表示被氮“替代”的氧的量。Y可以等于X。或者,Y可以小于X。在一系列实施方案中,Y是至少0.5X、至少0.6X或2/3X(0.66X)。 通常,M是Sm或Pr或La,或它们的组合。在某些实施方案中,a是0.3或更多、0.4或更多、0.5或更多或0.6或更多和/或a是0.9或更少、0.8或更少、0.7或更少、0.6或更少或0.5或更少。 这些示例描述了Ce 阴离子空位晶格可以是Ce 阴离子空位晶格可以是Ce 在本发明的另一个实施方案中,阴离子空位晶格可以描述为根据式IV的化合物;
其中M是化合价小于4的元素,通常是铈以外的镧系元素或稀土元素,例如Sm、Pr、Eu、Gd或它们的组合或Sm、Pr、La、Gd或它们的组合。“a”和“b”独立地在0.05至0.95的范围内,前提是“a”和“b”加起来等于1。0 如上解释,据信阴离子空位晶格活化氢分子,使得它们更易于与催化剂表面上的活性氮物种反应。然而,仍然需要哈伯-博施催化剂来驱动反应的氮部分的解离吸附。因此,期望具有哈伯-博施催化剂和阴离子空位晶格助催化剂两者的平衡。 在本发明的另一个实施方案中,该组合物包含根据式V的阴离子空位晶格; Zr 其中M是钛和/或铈和/或化合价小于4的元素,通常是镧系元素或稀土元素,例如Sm、Pr、La、Gd或它们的组合。“a”和“b”独立地在0.05至0.95的范围内,前提是“a”和“b”加起来等于1(大约)。0 在本发明的另一个实施方案中,组合物包含根据式VI的阴离子空位晶格; Ti 其中M是锆;和/或铈;和/或化合价小于4的元素,通常是镧系元素或稀土元素,例如Sm、Pr、La、Gd或它们的组合。“a”和“b”的范围分别在0.05至0.95的范围内,前提是“a”和“b”加起来等于1(大约)。0 通常情况下,存在的阴离子空位晶格的量在全部组合物的1重量%至70重量%范围内。更通常地,组合物中存在的阴离子空位晶格的量是在全部组合物的2重量%至60重量%范围内,并且通常在全部组合物的3重量%至40重量%范围内。更通常,组合物中存在的阴离子空位晶格的量在全部组合物的3重量%至30重量%范围内,并且通常在全部组合物的3重量%至20重量%范围内。通常存在的阴离子空位晶格的量在全部组合物的4重量%至6重量%范围内,最典型的是全部组合物的约5%。组合物中存在的阴离子空位晶格的量可以在全部组合物的5重量%至30重量%内或全部组合物的10重量%至20重量%范围内。发明人已经确定,包含20重量%的阴离子空位晶格的组合物促进哈伯-博施方法的催化。 通常情况下,存在的阴离子空位晶格的量在全部组合物的1mol%至70mol%范围内。更通常地,组合物中存在的阴离子空位晶格的量在全部组合物的2mol%至60mol%范围内,并且通常在全部组合物的3mol%至40mol%范围内。更通常,组合物中存在的阴离子空位晶格的量在全部组合物的10mol%至35mol%范围内。通常存在的阴离子空位晶格的量在全部组合物的15mol%至30mol%范围内,最典型的是全部组合物的约25%。组合物中存在的阴离子空位晶格的量可以在全部组合物的5mol%至30mol%内或在全部组合物的15mol%至25mol%的范围内。 在本发明的第二方面,还提供一种用于哈伯-博施方法的催化剂盒(cartridge),该盒包含根据本发明的第一方面的组合物。在哈伯-博施方法的工业应用中,反应在反应容器内进行(通常在高压下)。催化剂通常悬浮在支架(cradle)或支撑结构中的反应容器内以确保混合的氢气和氮气充分暴露到催化剂。与将粉末简单地倒入反应器中相比,这还允许容易地引入和去除催化剂。因此,催化剂组合物通常以盒形式提供,该盒形式可以在操作之前简单地插入反应器中,并且一旦催化剂分解或降至低于阈值活性就被丢弃。因此,本文所用的术语“盒”旨在涵盖这样的容器,其被构造成容纳并允许与容纳在其中的多相催化剂的部分进行气体相互作用。盒通常适合于容易地从反应器中插入和移除。 由于其提供的大表面积,该组合物通常以粉末或颗粒的形式提供。然而,只要载体在典型的哈伯-博施方法条件下是稳定的,则用于多相催化的任何大表面积布置或制剂将是合适的(例如上述的那些)。或者,可以将催化剂与粘合剂或其他材料混合,以形成特定尺寸和分布的颗粒。催化剂也可以提供在通常具有高表面积的载体(例如多孔载体)上。 在本发明的第三方面,还提供一种用于生产氨的哈伯-博施方法,其包括以下步骤:i)提供根据本发明的第一方面的组合物,和ii)将所述组合物暴露到氮气和氢气的混合物中。 该方法的条件可以根据所需的反应速度和系统的操作要求变化。技术人员将熟悉在哈伯-博施反应中发生的平衡过程以及控制温度和压力以最有效地促进氨形成的重要性。对于本发明的催化剂,已经发现需要较少的能量密集条件以提供与现有技术相当的结果。因此,该方法的反应条件通常比工业标准温和,并且通常低于600℃且低于25MPa或20MPa。 所述组合物可以在600℃或更低,500℃或更低,400℃或更低或300℃或更低的温度下暴露到氮气和氢气的混合物中和/或所述组合物可以在250℃或更高,300℃或更高,350℃或更高,400℃或更高或450℃或更高的温度下暴露到氮气和氢气的混合物。应当理解,在反应器上可能存在温度梯度,因此对600℃的温度的提及可以涉及反应器中的平均(均值)温度。 所述组合物可以在25MPa或更低,15MPa或更低,10MPa或更低,8MPa或更低,5MPa或更低的压力下暴露到氮气和氢气的混合物中和/或所述组合物可以在1MPa或更高,3MPa或更高,5MPa或更高,8MPa或更高,或10MPa或更高,或15MPa或更高,或20MPa或更高的压力下暴露到氮气和氢气的混合物中。 所述组合物可以在400℃或更低的温度和15MPa或更低的压力下暴露到氮气和氢气的混合物。 尽管哈伯-博施方法是连续流技术,发明人已经确定本发明可应用于连续方法和间歇方法。本发明的组合物(哈伯-博施催化剂和阴离子空位晶格的组合)允许使用较低的温度。具体地,它允许该方法在产生比通常更高的氨比例的条件下进行。这样,更容易将氨与未反应的氢和氮(如果有的话)分离,从而使分批方法可行。典型的哈伯-博施方法包括适合于容纳加压气体的反应器,在反应器内保持催化剂以确保反应气最大程度暴露到反应器的区域,以及用于在反应器内提供和提取气氛的装置。这种反应器通常装备有外部分离装置以收集氨并将未反应的氢和氮返回到试剂源流中。可以采用各种系统来确保通过此方法的最大的热量保留。 有趣的是,发明人发现,当将本发明的组合物用于催化该方法时,可能不需要对进入的氢气和氮气流进行彻底的(intensive)纯化。可以预期,当使用更纯净的反应气体(混合的H 典型情况下,催化剂使用固态反应、沉淀、共沉淀、球磨、渗透、溶胶-凝胶法、燃烧合成或溶剂热合成或任何现有方法制备。 另一选择是将氧化物或氮化物或氮氧化物与现有的工业催化剂以一定的重量比混合以进一步提高活性。可以使用包括常规熔融方法的任何方法,例如W.Arabczyk等人在Studies in Surface and Catalysts,91,1995,677-682中描述的那些,将氧化物促进剂或其前体直接添加到用于制备现有的基于Fe或Ru的市售氨合成催化剂的前体中。 在本发明的第四方面,提供根据本发明的第一方面的组合物在哈伯-博施方法中生产氨的用途。 在本发明的第五方面,提供如上定义的根据式III、V或VI的阴离子空位晶格。上面关于根据式III、V或VI的阴离子空位晶格的评论在此同样适用。 应当注意,掺杂剂M不限于一种元素。共掺杂是指母相被多于一种元素掺杂。例如,Ce 具体地,本发明在于根据式III的阴离子空位晶格,其中M是Sm且a是0.1至0.9,或0.3至0.9,或0.5至0.9,包括Ce 具体地,本发明在于根据式III的阴离子空位晶格,其中M是Pr且a是0.1至0.9,或0.2至0.8,包括Ce 具体地,本发明在于根据式III的阴离子空位晶格,其中M是La且a是0.1至0.9,或0.2至0.8,包括Ce 现在将参考附图和具体实施例描述本发明。 附图简要说明 图1显示BZCY质子传导载体和负载的Ni催化剂在稳定性测试之前和之后的XRD图像。 图2显示还原之前和之后Ni-BZCY催化剂的UV-Vis光谱。 图3显示未还原的催化剂(a)、在稳定性测试之前的被还原的催化剂(b)和在稳定性测试之后的被还原的催化剂(c)的SEM图像。放大倍数为10000。 图4显示被还原的催化剂在稳定性测试之前的SEM图像,其中突出显示元素映射(mapping)区域(a)、Ni的EDS映射(b)、Ba的EDS映射(c)、Zr的EDS映射(d)、Ce的EDS映射(e)、Y的EDS映射(f)、O的EDS映射(g)。 图5显示(a):N 图6显示使用Ni-BZCY催化剂在不同温度下的氨合成速率(120ml min 图7显示使用Ni-BZCY催化剂在不同流速下的氨合成速率(620℃,H 图8显示不同流速下的氨出口浓度(620℃,H 图9显示使用Ni-BZCY催化剂在不同进料摩尔比下的氨合成速率(200ml min 图10显示使用60%NiO/40%MgO-CeO 图11显示使用Ni-BZCY催化剂在干的和湿的稳定性测试下的氨合成速率(620℃,200ml min 图12显示纯Fe、具有CeO 图13显示纯Fe、具有CeO 图14显示纯Fe和具有SrFe 图15显示具有BCZY(60重量%)的Fe催化剂在各种压力下的催化活性(总流速80mlmin 图16显示具有Sr 图17至19显示具有14至26%的载体重量百分比的Fe-Ce 图20显示80%Fe-20%Ce 图21显示提出的在催化剂上的反应路径,其中氮解离吸附在Fe表面上并进行氢化。氢气在Ce 图22显示与Fe-20%CeO 图23显示在3MPa下具有20%的载体重量百分数的Fe-CeO 图24显示在1MPa下具有20%的载体重量百分数的Fe-CeO 图25显示Fe-CeO 图26显示Fe-20%Ce 图27显示Fe-20%Ce 图28显示Fe-Ce 图29显示在3MPa和1MPa下在不同温度下10%Ru-Ce 图30显示纯净的CeO 图31显示在3MPa下80%Fe-20%ZrO 图32显示Ce 图33显示纯氮氧化铈和Sm掺杂的氮氧化铈的晶格参数。 图34显示Ce 实施例 实施例1
为了合成BaZr
使用X射线衍射(XRD)和扫描电子显微镜(SEM)二者对催化剂进行表征。使用Panalytical X’Pert Pro多功能衍射仪(MPD)在45kV和40mA下工作的Cu K alpha 1辐射确定晶体结构。用在10kV下操作的ZEISS SUPRA 55-VP获得SEM图像。预还原的Ni-BZCY催化剂的热重分析-差示扫描量热法(TG-DSC)分析在NETSCH F3热分析仪上在到800℃的流动N2与70ml min
为了测量催化活性,将0.48g催化剂装载到氧化铝反应器中并负载在玻璃纤维的中央。然后将催化剂在700℃下在H 稀H 为了计算氨的合成速率,使用以下方程:
其中[NH
在图1所示的XRD结果中,可以看到,在与NiO混合之前和之后存在对于BaZr
为了鉴定BaCO
图3a&b显示未还原的NiO-BZCY催化剂的SEM照片。大颗粒是BCZY氧化物,其中小NiO颗粒均匀分布在氧化物基质中。在还原后(图3c&d),粒径略有变大。被还原的Ni-BZCY的元素映射在图4中显示。Ni的分布(图4b)是均匀的。
为了找出水分对还原的Ni-BZCY催化剂的性质的影响,对干燥和湿润的还原的Ni-BZCY催化剂进行TG-DSC分析。对于湿润的催化剂,在TG-DSC测量之前,将还原的Ni-BZCY催化剂暴露到室温下流动的空气1小时。两种样品的TG-DSC数据分别显示在下面的图5(a)和(b)中。对于干燥的催化剂,低于100℃(~0.12重量%)的初始重量损失是由于吸收水的损失。观察到冷却时重量略有增加,峰值在~270℃(~0.03重量%),可能是由于蒸汽被BZCY吸附。当使用湿润的被还原的Ni-BZCY时,初始重量损失在高得多的温度下持续,直到~250℃且重量损失较大(~0.34重量%),表明BZCY可以将水保持在更高的温度。观察到肩部重量增加在大约450℃达到峰值,这是由于水的吸收,在质子传导氧化物中也观察到水的吸收。冷却时,观察到更多的水吸收(~0.18重量%),表明BZCY可以在较低温度下强烈吸收水。
当恒定流速保持在120ml min
然后在620℃的恒定温度下测试总流速的影响,结果在图7中显示。可以看出,活性随着流速的增加而增加。为了确认该氨出口浓度相对于总气体流速作图,预期活性的这种增加仅仅是由于反应气体的增加。 如图8所示,当将总流速相对于氨出口浓度作图时,在平稳之前,它升至120mlmin
为了确定最佳进料比,将气体入口摩尔比调节为2.6至3.4(H
为了检验催化剂载体的质子传导特性的促进作用,在相同条件下测试负载在非质子导体上的Ni催化剂。MgO-CeO
氨合成催化剂在氧化剂存在下的稳定性是很大的挑战。在620℃下在144小时内在H 除BZCY促进的催化剂外,还在总流速为200ml min 在研究作为氨合成催化剂的潜在载体的材料时,强烈考虑载体的电负性。在这项工作中,我们已经表明,载体材料的另一个期望效果可以是其传导质子的能力。质子传导载体的这种促进能力可以通过进料到反应器的H 实施例2 催化剂制备方法 i)Fe-SrFe 在玛瑙研钵和研杵中将18.4538g SrCO ii)Fe-BaZr 称量化学计量的BaCO iii)Fe-Ce 将0.001mol 0.3487g Sm iv)Fe-CeO 将0.01mol的4.3423g Ce(NO v)Fe-Sr 将Sr(NO
Fe-SDC在3MPa和1MPa下在高温下对氨合成的活性分别在图17和18中显示。通常,具有20重量%SDC的催化剂在所测试的不同SDC促进剂比率中表现出最高活性。在该比率下,Fe上的N 表1提供所选高活性氨合成催化剂的比较。在最佳压力和温度下测量活性。还比较所用气体供应的纯度。
(1)C.J.H.Jacobsen,Chemical Communications,1057-1058(2000). (2)M.Kitano,Y.Inoue,M.Sasase,K.Kishida,Y.Kobayashi,K.Nishiyama,T.Tada,S.Kawamura,T.Yokoyama,M.Hara,H.Hosono,Angewandte Chemie InternationalEdition 57,2648-2652(2018). (3)M.Kitano,Y.Inoue,Y.Yamazaki,F.Hayashi,S.Kan巴a,S.Matsuishi,T.Yokoyama,S.-W.Kim,M.Hara,H.Hosono,Nature Chemistry 4,934-940(2012). (4)J.Z.Wu,Y.T.Gong,T.Inoshita,D.C.Fredrickson,J.J.Wang,Y.F.Lu,M.Kitano,H.Hosono,Adv.Mater.29,(2017). (5)K.Sato,K.Imamura,Y.Kawano,S.Miyahara,T.Yamamoto,S.Matsumura,K.Nagaoka,Chemical Science 8,674-679(2017). (6)P.Wang,F.Chang,W.Gao,J.Guo,G.Wu,T.He,P.Chen,Nature Chemistry 9,64-70(2016). 在1MPa和450℃下,用20重量%SDC促进的Fe催化剂的活性为8.7mmolg 发明人提出,该改进与通过在CeO 为了进一步证实外在氧空位在促进基于Fe的催化剂中的关键作用,纯ZrO 然而,SDC的还原比CeO 在图22中突出显示催化剂的最佳组成在450℃和3MPa下的活性,以及工业铁催化剂在相同反应条件下的活性。由此可以观察到,80%Fe-20%SDC的活性几乎是基于磁铁矿Fe的工业催化剂的3倍。对于通过SDC促进的催化剂,随着压力降低,活性的降低也不太明显。值得注意的是,煅烧的催化剂(中间的条)的活性低于未煅烧的等效催化剂。这支持阴离子空位在促进反应中的作用。 除了氧化铈和掺杂的氧化铈对铁催化剂合成氨具有明显的促进作用外,还应注意对催化剂中毒的耐受性。我们的实验中使用的两种反应气体的纯度均为99.995%,无需进一步纯化,氧气和水均存在于50ppm的杂质中。可以看出,我们测得的工业上使用的市售的促进的Fe催化剂的活性低于其他地方报告的活性,这种较低的活性表明我们的实验中进料气中的含氧化合物具有负面影响。然而,对于氧化铈和掺杂的氧化铈负载的催化剂获得的高活性表明对进料气体中的杂质具有优异的耐受性。如在图20中显示,在450℃和3MPa下进行的200小时测试期间,尽管使用纯度较低的H 其中氧使氨合成催化剂中毒的机理通过经由进行的连续氧化和还原循环形成的大铁晶体的生长而发生。基于CeO 简而言之,向铁催化剂中加入具有外在氧空位的氧化物促进剂(例如SDC或YSZ)已显示比常规熔融铁催化剂在活性和气体杂质容忍性两方面均得到改进,从而产生令人兴奋的新种类的氨合成催化剂。这为开发新型氨合成催化剂提供新的策略,该氨合成催化剂具有很高的活性和对含氧化合物的高容忍性以用于实际应用,特别是用于使用可再生电力作为能源的低碳氨合成。 实施例3
在陶瓷蒸发皿中将0.02mol的8.6844g Ce(NO 将获得的CeO 通过XRF分析确认氮和x和y的值。结果在下表中给出。这分别给出稳定性测试之前和之后的CeO 为了计算给定的组成,假定Ce的化合价为+4,O的化合价为-2,N的化合价为-3,总电荷为零(中性)。由此可以构建以下电荷平衡。 (+4)(a)+(-2)(2-x)+(-3)(y)=O 其中a是组合物中Ce的摩尔数。对于CeO 这给出:
从XRF数据,可以通过N与Ce的直接摩尔比获得y值,从而从上式计算x。 为了确认在XRF结果中观察到的所有氮都在CeO 在包括纯净的市售CeO 由于CO 这分别给出在稳定性测试之前和之后CeO
在陶瓷蒸发皿中将0.016mol的6.9475g Ce(NO 将获得的Ce 通过XRF分析确认氮和x和y的值。结果在下表中给出。这给出Ce 由此可以构建以下电荷平衡。 (+4)(a)+(+3)(b)+(-2)(2-x)+(-3)(y)=O 其中a是Ce的摩尔数并且b是组合物中Sm的摩尔数。 这给出:
从XRF数据,可以通过N与Ce和Sm的直接摩尔比获得y值,从而可以从上式计算x。
在陶瓷蒸发皿中,将0.01mol的4.3422g Ce(NO 将获得的Ce 通过XRF分析确认氮和x和y的值。这给出分别在稳定性测试之前和之后的Ce
如上所述,由硝酸铈、硝酸钐和尿素合成CeO
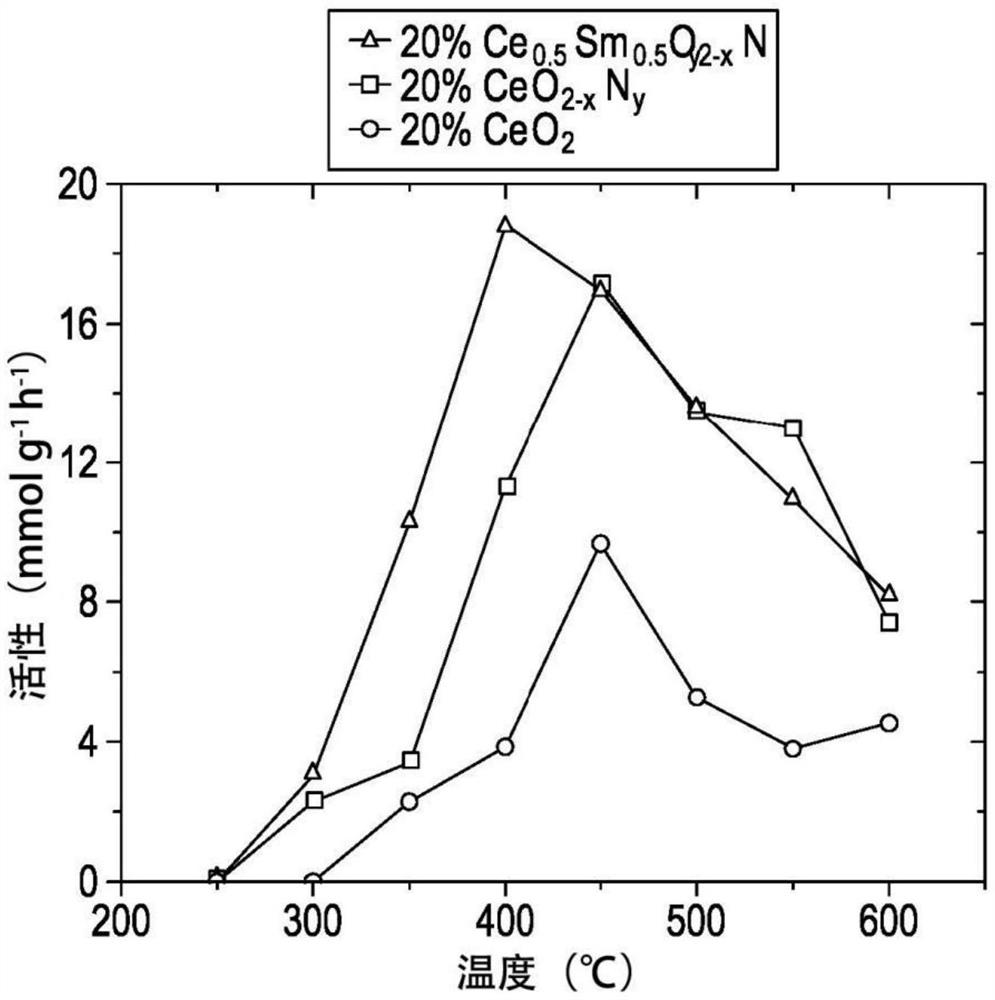
可以观察到,煅烧(在空气中加热)增加氧气的比例并且减少氮的比例,而在哈伯-博施条件(氢气和氮气气氛)下的稳定性测试中却没有观察到。据信这是由于前体中存在大量的N 纯净的和Sm-掺杂的CeO 使用×5物镜和Renishaw CCD检测器,在配备有DPSS激光器的Renishaw inViaReflex拉曼显微镜(Gonzo)上在激发波长λ=532nm下记录氧化物和氮氧化物样品的拉曼光谱。 还收集并绘制这些样品的拉曼光谱(图30)。纯净的CeO 在570cm 随着Sm掺杂水平提高,该峰变得更强,表明氧空位的浓度更高。样品Ce 图23和24表明,Ce Ce Ce Ce Ce Ce 通常,具有测得的Ce Fe-Ce
Ce 在陶瓷蒸发皿中将0.0046mol Ce(NO 将获得的Ce
在陶瓷蒸发皿中,将0.01mol的4.3422g Ce(NO 将0.1054g Ru Ru-Ce
- 包含阴离子空位晶格的哈伯-博施催化剂
- 包含弱配位阴离子盐的施胶组合物及其用途