一种水相合成含三氟甲基C-2膦酰基吲哚的方法
文献发布时间:2023-06-19 11:19:16
技术领域
本发明属于化学合成技术领域,具体涉及到一种水相合成含三氟甲基C-2膦酰基吲哚的方法。
背景技术
吲哚是自然界中最常见的含氮杂环化合物。杂环化合物因其良好生物活性被广泛应用于医药领域、农药材料等各个领域。另一方面,有机膦化合物不仅作为配体和有机中间体,还在医药、农药、材料等领域中起关键作用。如作为非核苷逆转录酶抑制剂在治疗HIV-1方面表现出优异的效果;此外C2-磷酰基化合物不仅作为配体广泛应用于有机催化领域,而且在材料领域也具有十分重要的应用。
有机氟化合物具有高稳定性、脂溶性以及疏水性。引入氟原子可有效改善和提高药物的药效学和药物动力学,或者改变其物理特性、化学特性或生物活性,而三氟甲基官能团是最重要的含氟基团之一,在医药、材料、农药等化学化工领域得到广泛应用。如在染料分子中引入氟原子或者三氟甲基可以改善其染色鲜度、着色值和耐光度。含氟基团还可以广泛用于药物成分当中,如治疗乳腺癌的药物Panomifene和非核苷逆转录酶抑制剂A、B(Org.Biomol.Chem.2014,12.8308-8317)
目前2-膦酰基吲哚的合成方法有很多,比如2-异氰基苯乙烯与二苯氧膦在光催化条件下的环化反应(Org.Lett.2018,20,2382-2385),吲哚与二苯氧膦类化合物在银催化剂以及过量氧化剂作用下自由基加成C2-磷酰化反应(Adv.Synth.Catal.2016,358,1753–1758),邻碘苯胺与氧膦取代的炔在过渡金属钯催化剂催化下的偶联环化反应(Org.Lett.2010,12,1476-1479)等,但是已知的方法仍存在诸多问题,如使用价格不菲的金属催化剂、过量的氧化剂,反应需要严格的无水无氧操作等。而合成含三氟甲基C-2膦酰基吲哚类化合物的合成研究很少,目前仅有一例文献报道可实现含三氟甲基C-2膦酰基吲哚类化合物的合成(Adv.Synth.Catal.2019,361,5311-5316),该文献使用三氟甲基吲哚甲醇和二苯氧磷为原料,使用樟脑磺酸为催化剂,在二氯乙烷溶剂中加热可制备含三氟甲基C-2膦酰基吲哚类化合物,但是该文献仅报道了一个例子,另外该方法使用有机溶剂以及樟脑磺酸磺酸为催化剂,同时存在区域选择性问题,不够绿色环保。
因此,如何使用一种绿色环保,简单高效的合成方法合成含三氟甲基C-2膦酰基吲哚类化合物具有重大的研究价值和应用前景。
发明内容
本部分的目的在于概述本发明的实施例的一些方面以及简要介绍一些较佳实施例。在本部分以及本申请的说明书摘要和发明名称中可能会做些简化或省略以避免使本部分、说明书摘要和发明名称的目的模糊,而这种简化或省略不能用于限制本发明的范围。
鉴于上述和/或现有技术中存在的问题,提出了本发明。
因此,本发明的目的是,克服现有技术中的不足,提供一种水相合成含三氟甲基C-2膦酰基吲哚的方法。
为解决上述技术问题,本发明提供了如下技术方案:一种水相合成含三氟甲基C-2膦酰基吲哚的方法,包括,
在十二烷基苯磺酸水溶液中加入三氟甲基吲哚甲醇类化合物、二取代氧磷类化合物,在100℃条件下反应24小时,反应结束后用乙酸乙酯萃取,经过真空浓缩,并通过胶柱层析,即得目标产物(I):
式(I)中,R
作为本发明所述水相合成含三氟甲基C-2膦酰基吲哚的方法的一种优选方案,其中:所述三氟甲基吲哚甲醇类化合物的结构式如式(II)所示:
式(II)中R
作为本发明所述水相合成含三氟甲基C-2膦酰基吲哚的方法的一种优选方案,其中:所述二取代氧磷化合物的结构式如式(III)所示:
式(III)中R
作为本发明所述水相合成含三氟甲基C-2膦酰基吲哚的方法的一种优选方案,其中:所述十二烷基苯磺酸水溶液,其为十二烷基苯磺酸与水以质量比0.32wt%混合配制而成的水溶液。
作为本发明所述水相合成含三氟甲基C-2膦酰基吲哚的方法的一种优选方案,其中:所述反应中,三氟甲基吲哚甲醇类化合物与二取代氧磷类化合物的摩尔比为1:1~1.5。
作为本发明所述水相合成含三氟甲基C-2膦酰基吲哚的方法的一种优选方案,其中:所述三氟甲基吲哚甲醇类化合物与二取代氧磷类化合物的摩尔比为1:1.5。
作为本发明所述水相合成含三氟甲基C-2膦酰基吲哚的方法的一种优选方案,其中:所述十二烷基苯磺酸水溶液,其用量为每0.1毫摩尔三氟甲基吲哚甲醇类化合物对应1~3mL十二烷基苯磺酸水溶液。
作为本发明所述水相合成含三氟甲基C-2膦酰基吲哚的方法的一种优选方案,其中:所述每0.1毫摩尔三氟甲基吲哚甲醇类化合物对应2mL十二烷基苯磺酸水溶液。
作为本发明所述水相合成含三氟甲基C-2膦酰基吲哚的方法的一种优选方案,其中:所述胶柱层析,其中,洗脱剂为乙酸乙酯:石油醚按照体积比40:60组合的混合液。
本发明有益效果:
(1)本发明提供了一种水相合成含三氟甲基C-2膦酰基吲哚类化合物的绿色合成方法。
(2)本发明以水为反应介质,十二烷基苯磺酸为非金属催化剂,反应无需无水无氧条件、具有操作简单、反应条件温和、用底物范围广,产率高、绿色环保适等优点。
(3)本发明的提供的含三氟甲基C-2膦酰基吲哚类化合物结构复杂多样,即可作为配体又可以作为有机合成中间体,具有广阔的应用前景。
附图说明
为了更清楚地说明本发明实施例的技术方案,下面将对实施例描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动性的前提下,还可以根据这些附图获得其它的附图。其中:
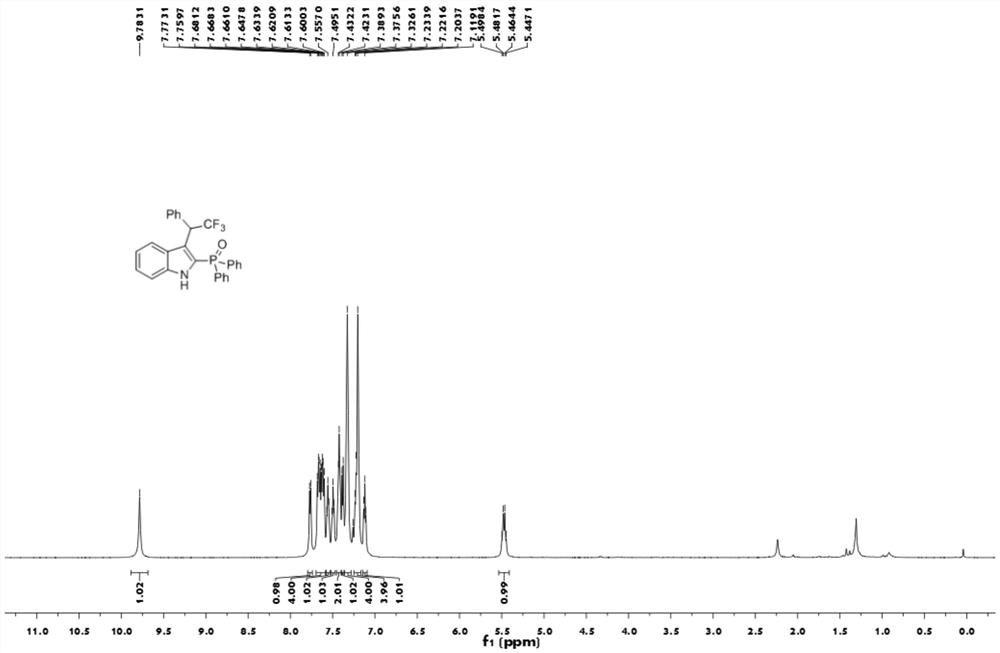
图1为本发明实施例1制备的化合物1aa的核磁共振氢谱图;
图2为本发明实施例1制备的化合物1aa的核磁共振碳谱图;
图3为本发明实施例1制备的化合物1aa的核磁共振氟谱图;
图4为本发明实施例1制备的化合物1aa的核磁共振磷谱图;
图5为本发明实施例1制备的化合物1ba的单晶衍射图。
具体实施方式
为使本发明的上述目的、特征和优点能够更加明显易懂,下面结合说明书实施例对本发明的具体实施方式做详细的说明。
在下面的描述中阐述了很多具体细节以便于充分理解本发明,但是本发明还可以采用其他不同于在此描述的其它方式来实施,本领域技术人员可以在不违背本发明内涵的情况下做类似推广,因此本发明不受下面公开的具体实施例的限制。
其次,此处所称的“一个实施例”或“实施例”是指可包含于本发明至少一个实现方式中的特定特征、结构或特性。在本说明书中不同地方出现的“在一个实施例中”并非均指同一个实施例,也不是单独的或选择性的与其他实施例互相排斥的实施例。
实施例1
反应式为:
一种含三氟甲基C-2膦酰基吲哚类化合物的合成方法:取25mL圆底烧瓶,在空气氛围中,依次加入三氟甲基吲哚甲醇2a(0.3mmol),二苯氧磷3a(0.45mmol),最后加入0.32wt%的DBSA水溶液(6mL),在100℃搅拌反应24小时,待反应结束,冷却至室温,用乙酸乙酯萃取三次,合并有机相,无水硫酸镁干燥,过滤、浓缩,粗品通过硅胶柱层析分离(洗脱剂为乙酸乙酯:石油醚=40:60),即得目标产物1aa(128.4mg,白色固体,产率90%)。
实施例1制备的化合物1aa的核磁共振氢谱图如图1所示;实施例1制备的化合物1aa的核磁共振碳谱图如图2所示;实施例1制备的化合物1aa的核磁共振氟谱图如图3所示;实施例1制备的化合物1aa的核磁共振磷谱图如图4所示。
1H NMR(600MHz,CDCl3)δ9.78(s,1H),7.77(d,J=8.0Hz,1H),7.64(ddd,J=28.5,12.2,7.8Hz,4H),7.56(t,J=7.0Hz,1H),7.50(t,J=7.0Hz,1H),7.43(t,J=5.5Hz,2H),7.38(d,J=8.2Hz,1H),7.33(s,4H),7.25-7.16(m,4H),7.12(t,J=7.4Hz,1H),5.47(dd,J=20.6,10.2Hz,1H);13C NMR(150MHz,CDCl3)δ138.4(d,J=9.3Hz),134.7,132.6(d,J=2.3Hz),132.6(d,J=2.3Hz),131.9(d,J=4.5Hz),131.8(d,JC-P=68.6Hz),131.8(d,J=4.5Hz),131.1(d,JC-P=66.9Hz),128.8(d,J=12.6Hz),128.7,128.6(d,J=13.2Hz),128.2,127.3,126.8(q,JC-F=280.0Hz),126.7(d,J=10.4Hz),126.5(d,JC-P=118.5Hz),124.5,122.4,120.9,119.4(d,J=11.1Hz),112.3,46.4(q,JC-F=28.6Hz);19F NMR(565MHz,CDCl3)δ-63.5(d,J=10.7Hz,3F);31P NMR(243MHz,CDCl3)δ22.3.
实施例2
反应式为:
一种含三氟甲基C-2膦酰基吲哚类化合物的合成方法:取25mL圆底烧瓶,在空气氛围中,依次加入三氟甲基吲哚苄醇2b(0.3mmol),二苯氧磷3a(0.45mmol),最后加入0.32wt%的DBSA水溶液(6mL),在100℃搅拌反应24小时,待反应结束,冷却至室温,用乙酸乙酯萃取三次,合并有机相,无水硫酸镁干燥,过滤,浓缩,粗品通过硅胶柱层析分离(洗脱剂为乙酸乙酯:石油醚=40:60),即得目标产物1ba(128.9mg,白色固体,产率85%)。
实施例2制备的化合物1ba的单晶衍射图如图5所示。
1H NMR(400MHz,CDCl3)δ8.58(s,1H),7.70-7.51(m,6H),7.46(td,J=7.7,3.0Hz,2H),7.39(td,J=7.7,3.0Hz,2H),7.35-7.30(m,2H),7.24(d,J=9.2Hz,1H),7.22-7.17(m,3H),7.12(d,J=1.4Hz,1H),6.94(dd,J=9.0,1.9Hz,1H),5.51(q,J=10.7Hz,1H),3.74(s,3H);13C NMR(100MHz,CDCl3)δ154.6,134.7,133.5(d,J=9.5Hz),132.68(d,J=2.3Hz),132.60(d,J=2.3Hz),131.8(d,J=9.9Hz),131.7(d,JC-P=107.9Hz),131.3(d,JC-P=108.5Hz),128.8(d,J=19.9Hz),128.7(d,J=13.2Hz),128.2,127.3,127.2,127.0(d,JC-P=118.7Hz),126.6(q,JC-F=278.9Hz),119.2(d,J=13.4Hz),116.0,112.8,102.9(q,JC-F=4.2Hz),55.6,46.5(q,JC-F=29.3Hz);19F NMR(376MHz,CDCl3)δ-63.4(d,J=10.7Hz,3F);31P NMR(161MHz,CDCl3)δ21.9.
实施例3
反应式为:
一种含三氟甲基C-2膦酰基吲哚类化合物的合成方法:取25mL圆底烧瓶,在空气氛围中,依次加入三氟甲基吲哚苄醇2c(0.3mmol),二苯氧磷3a(0.45mmol),最后加入0.32wt%的DBSA水溶液(6mL),在100℃搅拌反应24小时,待反应结束,冷却至室温,用乙酸乙酯萃取三次,合并有机相,无水硫酸镁干燥,过滤,浓缩,粗品通过硅胶柱层析分离(洗脱剂为乙酸乙酯:石油醚=40:60),即得目标产物1ca(138.0mg,白色固体,产率83%)。
1H NMR(600MHz,CDCl3)δ9.46(s,1H),7.87(s,1H),7.68-7.55(m,5H),7.52(td,J=7.5,1.0Hz,1H),7.47(td,J=7.8,3.0Hz,2H),7.37-7.32(m,3H),7.27(d,J=8.8Hz,1H),7.24-7.18(m,5H),5.27(q,J=10.5Hz,1H);13C NMR(150MHz,CDCl3)δ137.2(d,J=9.9Hz),134.0,132.88(d,J=2.6Hz),132.75(d,J=2.4Hz),131.9(d,J=7.3Hz),131.8(d,J=7.5Hz),131.2(d,JC-P=108.5Hz),130.8(d,JC-P=109.1Hz),128.9(d,J=12.7Hz),128.8(d,J=12.8Hz),128.6,128.3,128.2(d,J=10.2Hz),128.1(d,JC-P=117.3Hz),127.6,127.5,126.3(q,JC-F=280.5Hz),124.5(q,JC-F=4.4Hz),117.7(d,J=10.1Hz),114.3,114.0,46.4(q,JC-F=29.0Hz);19F NMR(565MHz,CDCl3)δ-63.5(d,J=10.6Hz,3F);31PNMR(243MHz,CDCl3)δ21.8.
实施例4
反应式为:
氟甲基C-2膦酰基吲哚类化合物的合成方法:取25mL圆底烧瓶,在空气氛围中,依次加入三氟甲基吲哚苄醇2d(0.3mmol),二苯氧磷3a(0.45mmol),最后加入0.32wt%的DBSA水溶液(6mL),在100℃搅拌反应24小时,待反应结束,冷却至室温,用乙酸乙酯萃取三次,合并有机相,无水硫酸镁干燥,过滤,浓缩,粗品通过硅胶柱层析分离(洗脱剂为乙酸乙酯:石油醚=50:50),即得目标产物1da(76.8mg,白色固体,产率48%)。
1H NMR(600MHz,CDCl3)δ10.14(s,1H),8.53(s,1H),7.93(d,J=8.7Hz,1H),7.66(dd,J=12.6,7.6Hz,2H),7.62-7.54(m,3H),7.51(t,J=7.3Hz,1H),7.47(td,J=7.4,2.2Hz,2H),7.43(d,J=8.7Hz,1H),7.33(td,J=7.3,2.0Hz,2H),7.25-7.16(m,5H),5.23(q,J=10.3Hz,1H),3.88(s,3H);13C NMR(150MHz,CDCl3)δ167.6,140.8(d,J=9.3Hz),134.1,132.9(d,J=2.5Hz),132.8(d,J=2.5Hz),131.9(d,J=6.2Hz),131.8(d,J=6.3Hz),131.2(d,JC-P=108.7Hz),130.7(d,JC-P=109.2Hz),129.0(d,J=12.7Hz),128.8(d,J=12.8Hz),128.7,128.4,128.1,127.6,126.3(d,J=10.4Hz),126.2(q,JC-F=279.6Hz),125.5(d,J=3.8Hz),125.4,123.1,120.0(d,J=12.0Hz),112.3,52.0,46.6(q,JC-F=24.4Hz);19F NMR(565MHz,CDCl3)δ-63.4(d,J=10.4Hz,3F);31P NMR(243MHz,CDCl3)δ22.2.
实施例5
反应式为:
一种含三氟甲基C-2膦酰基吲哚类化合物的合成方法:取25mL圆底烧瓶,在空气氛围中,依次加入三氟甲基吲哚苄醇2e(0.3mmol),二苯氧磷3a(0.45mmol),最后加入0.32wt%的DBSA水溶液(6mL),在100℃搅拌反应24小时,待反应结束,冷却至室温,用乙酸乙酯萃取三次,合并有机相,无水硫酸镁干燥,过滤,浓缩,粗品通过硅胶柱层析分离(洗脱剂为乙酸乙酯:石油醚=50:50),即得目标产物1ea(130.7mg,白色固体,产率89%)。
1H NMR(600MHz,CDCl3)δ8.66(s,1H),7.67-7.51(m,7H),7.45(td,J=7.8,3.0Hz,2H),7.37(td,J=7.9,3.0Hz,2H),7.34-7.30(m,2H),7.25(s,1H),7.21-7.17(m,3H),7.10(dd,J=8.5,0.9Hz,1H),5.50(q,J=10.7Hz,1H),2.41(s,3H);13C NMR(150MHz,CDCl3)δ136.9(d,J=9.4Hz),134.7(d,J=4.8Hz),132.5(d,J=2.4Hz),132.4(d,J=2.5Hz),131.8(d,J=4.2Hz),131.75(dd,JC-P=107.9Hz,JC-F=4.0Hz),131.7(d,J=4.4Hz),131.0(d,J=1.8Hz),130.1,128.7,128.6(d,J=7.1Hz),128.5,128.1,127.2,126.9(d,J=10.3Hz),126.5(q,JC-F=278.1Hz),126.4,126.3(d,JC-P=119.4Hz),121.4,118.8(d,J=10.5Hz),112.0,46.4(q,JC-F=28.8Hz),21.6;19F NMR(565MHz,CDCl3)δ-63.2(d,J=10.7Hz,3F);31P NMR(243MHz,CDCl3)δ22.4.
实施例6
反应式为:
一种含三氟甲基C-2膦酰基吲哚类化合物的合成方法:取25mL圆底烧瓶,在空气氛围中,依次加入三氟甲基吲哚苄醇2f(0.3mmol),二苯氧磷3a(0.45mmol),最后加入0.32wt%的DBSA水溶液(6mL),在100℃搅拌反应24小时,待反应结束,冷却至室温,用乙酸乙酯萃取三次,合并有机相,无水硫酸镁干燥,过滤,浓缩,粗品通过硅胶柱层析分离(洗脱剂为乙酸乙酯:石油醚=40:60),即得目标产物1fa(115.5mg,白色固体,产率78%)。
1H NMR(600MHz,CDCl3)δ10.58(s,1H),7.69-7.61(m,3H),7.60-7.55(m,3H),7.49(td,J=7.5,0.9Hz,1H),7.45(td,J=7.8,2.9Hz,2H),7.31(td,J=7.8,2.9Hz,2H),7.23-7.17(m,5H),7.09(dd,J=9.3,2.2Hz,1H),6.87(td,J=9.2,2.3Hz,1H),5.14(q,J=10.5Hz,1H);13C NMR(150MHz,CDCl3)δ160.9(d,JC-F=242.3Hz),138.8(dd,J=12.6,9.8Hz),134.4,132.8(d,J=2.6Hz),132.7(d,J=2.6Hz),131.9(d,J=5.8Hz),131.8(d,J=5.8Hz),131.2,130.9(d,JC-P=109.0Hz),128.9(d,J=12.7Hz),128.8(d,J=12.7Hz),128.6,128.3,127.4,127.2(dd,JC-P=118.9Hz,JC-F=3.6Hz),126.4(q,JC-F=279.8Hz),123.5(dd,J=9.0,3.3Hz),123.3(d,J=10.4Hz),118.8(dd,J=11.3,1.7Hz),110.3(d,J=25.0Hz),98.2(d,J=25.2Hz),46.3(q,JC-F=28.9Hz);19F NMR(565MHz,CDCl3)δ-63.4(d,J=10.6Hz,3F),-117.4(td,J=9.2,5.3Hz);31P NMR(243MHz,CDCl3)δ22.2.
实施例7
反应式为:
一种含三氟甲基C-2膦酰基吲哚类化合物的合成方法:取25mL圆底烧瓶,在空气氛围中,依次加入三氟甲基吲哚苄醇2g(0.3mmol),二苯氧磷3a(0.45mmol),最后加入0.32wt%的DBSA水溶液(6mL),在100℃搅拌反应24小时,待反应结束,冷却至室温,用乙酸乙酯萃取三次,合并有机相,无水硫酸镁干燥,过滤,浓缩,粗品通过硅胶柱层析分离(洗脱剂为乙酸乙酯:石油醚=40:60),即得目标产物1ga(128.1mg,白色固体,产率77%)。
1H NMR(600MHz,CDCl3)δ8.44(s,1H),7.74-7.66(m,5H),7.61(t,J=7.1Hz,1H),7.57(t,J=7.1Hz,1H),7.53-7.48(m,2H),7.46-7.41(m,3H),7.33(d,J=3.1Hz,2H),7.21(d,J=2.9Hz,3H),7.02(t,J=7.9Hz,1H),5.63(q,J=10.4Hz,1H);13C NMR(150MHz,CDCl3)δ136.6(d,J=9.2Hz),134.4,132.9(d,J=2.7Hz),132.9(d,J=2.5Hz),131.8(d,J=1.4Hz),131.7(d,J=1.0Hz),131.4(d,JC-P=108.3Hz),130.0(d,JC-P=109.1Hz),129.0(d,J=12.6Hz),128.9(d,J=12.7Hz),128.7,128.3,127.9,127.7(d,JC-P=65.6Hz),127.5,127.0,126.4(q,JC-F=278.8Hz),122.3,121.9(q,JC-F=3.1Hz),121.4(d,J=10.2Hz),105.4,46.7(q,JC-F=28.5Hz);19F NMR(565MHz,CDCl3)δ-63.5(s,3F);31P NMR(243MHz,CDCl3)δ21.3.
实施例8
反应式为:
一种含三氟甲基C-2膦酰基吲哚类化合物的合成方法:取25mL圆底烧瓶,在空气氛围中,依次加入三氟甲基吲哚苄醇2h(0.3mmol),二苯氧磷3a(0.45mmol),最后加入0.32wt%的DBSA水溶液(6mL),在100℃搅拌反应24小时,待反应结束,冷却至室温,用乙酸乙酯萃取三次,合并有机相,无水硫酸镁干燥,过滤,浓缩,粗品通过硅胶柱层析分离(洗脱剂为乙酸乙酯:石油醚=40:60),即得目标产物1ha(142.1mg,白色固体,产率94%)。
1H NMR(600MHz,CDCl3)δ9.24(s,1H),7.84(d,J=8.3Hz,1H),7.69(dd,J=12.1,7.7Hz,3H),7.60-7.54(m,3H),7.51(td,J=7.5,1.0Hz,1H),7.45(td,J=7.8,2.9Hz,2H),7.40(d,J=8.3Hz,1H),7.33(td,J=7.8,2.9Hz,2H),7.27(t,J=7.5Hz,1H),7.23-7.18(m,1H),7.16(t,J=7.6Hz,1H),7.08-7.02(m,1H),6.87-6.82(m,1H),5.74(q,J=10.1Hz,1H);13C NMR(150MHz,CDCl3)δ160.6(d,JC-F=249.8Hz),138.1(d,J=9.0Hz),132.7(d,J=2.6Hz),132.6(d,J=2.6Hz),132.0(d,J=10.7Hz),131.8(d,J=10.7Hz),131.5(d,JC-P=108.6Hz),131.1(d,JC-P=108.3Hz),130.3,129.5(d,J=8.4Hz),128.8(d,J=12.7Hz),128.7(d,J=12.7Hz),127.1,127.0,126.8(d,JC-P=117.4Hz),124.5,123.9(d,J=3.5Hz),122.3,122.1(q,JC-F=3.2Hz),121.2,117.9(d,J=10.9Hz),115.6(d,J=22.4Hz),112.2,40.5(q,JC-F=29.8Hz);19F NMR(565MHz,CDCl3)δ-64.5(t,J=9.0Hz,3F),-112.5–-112.9(m);31P NMR(243MHz,CDCl3)δ22.1.
实施例9
反应式为:
一种含三氟甲基C-2膦酰基吲哚类化合物的合成方法:取25mL圆底烧瓶,在空气氛围中,依次加入三氟甲基吲哚苄醇2i(0.3mmol),二苯氧磷3a(0.45mmol),最后加入0.32wt%的DBSA水溶液(6mL),在100℃搅拌反应24小时,待反应结束,冷却至室温,用乙酸乙酯萃取三次,合并有机相,无水硫酸镁干燥,过滤,浓缩,粗品通过硅胶柱层析分离(洗脱剂为乙酸乙酯:石油醚=40:60),即得目标产物1ia(127.4mg,白色固体,产率77%)。
1H NMR(600MHz,CDCl3)δ9.02(s,1H),7.83(d,J=8.2Hz,1H),7.66(td,J=12.6,8.0Hz,4H),7.58(t,J=7.3Hz,1H),7.53(d,J=7.1Hz,3H),7.48-7.36(m,11H),7.34(t,J=7.2Hz,1H),7.30-7.24(m,1H),7.16(t,J=7.6Hz,1H),5.59(q,J=10.2Hz,1H);13C NMR(150MHz,CDCl3)δ140.4,140.0,138.4(d,J=9.8Hz),133.8,132.6(d,J=5.9Hz),131.9(d,J=2.7Hz),131.8(d,J=2.7Hz),131.6(d,JC-P=107.5Hz),131.3(d,JC-P=108.5Hz),129.2,128.8(d,J=12.5Hz),128.7,128.6(d,J=12.4Hz),127.3,126.9,126.8,126.6,126.4(d,JC-P=75.7Hz),126.5(q,JC-F=279.1Hz),124.5,122.3,120.9,119.4(d,J=9.5Hz),112.3,46.2(q,JC-F=28.2Hz);19F NMR(565MHz,CDCl3)δ-63.3(d,J=9.4Hz,3F);31P NMR(243MHz,CDCl3)δ22.4.
实施例10
反应式为:
一种含三氟甲基C-2膦酰基吲哚类化合物的合成方法:取25mL圆底烧瓶,在空气氛围中,依次加入三氟甲基吲哚噻吩醇2k(0.3mmol),二苯氧磷3a(0.45mmol),最后加入0.32wt%的DBSA水溶液(6mL),在100℃搅拌反应24小时,待反应结束,冷却至室温,用乙酸乙酯萃取三次,合并有机相,无水硫酸镁干燥,过滤,浓缩,粗品通过硅胶柱层析分离(洗脱剂为乙酸乙酯:石油醚=40:60),即得目标产物1ka(95.3mg,白色固体,产率66%)。
1H NMR(600MHz,CDCl3)δ8.99(s,1H),7.76(d,J=8.3Hz,1H),7.71-7.63(m,4H),7.60(td,J=7.4,1.3Hz,1H),7.56(td,J=7.4,1.3Hz,1H),7.48(td,J=7.8,3.0Hz,2H),7.42(td,J=7.9,3.1Hz,2H),7.39(d,J=8.4Hz,1H),7.28(t,J=7.6Hz,1H),7.16(dd,J=5.1,1.2Hz,1H),7.15-7.12(m,1H),6.92(d,J=3.4Hz,1H),6.86(dd,J=5.1,3.6Hz,1H),5.61(q,J=9.8Hz,1H);13C NMR(150MHz,CDCl3)δ138.5(d,J=9.6Hz),136.7,132.7(d,J=2.4Hz),132.6(d,J=2.5Hz),131.88(d,J=10.9Hz),131.8(d,J=10.8Hz),131.6(d,JC-P=108.3Hz),131.2(d,JC-P=108.8Hz),128.8,128.7(d,J=26.7Hz),127.37,126.7(d,J=118.3Hz),125.8(q,JC-F=279.5Hz),126.29,126.16(d,J=10.1Hz),125.42,124.53,122.50,120.65,118.26(d,J=11.9Hz),112.39,42.65(q,JC-F=30.7Hz);19F NMR(565MHz,CDCl3)δ-65.6(d,J=9.9Hz,3F);31P NMR(243MHz,CDCl3)δ21.9.
实施例11
反应式为:
一种含三氟甲基C-2膦酰基吲哚类化合物的合成方法:取25mL圆底烧瓶,在空气氛围中,依次加入三氟甲基吲哚甲醇2a(0.3mmol),二(3,5-二甲基苯基)氧膦3b(0.45mmol),最后加入0.32wt%的DBSA水溶液(6mL),在100℃搅拌反应24小时,待反应结束,冷却至室温,用乙酸乙酯萃取三次,合并有机相,无水硫酸镁干燥,过滤,浓缩,粗品通过硅胶柱层析分离(洗脱剂为乙酸乙酯:石油醚=40:60),即得目标产物1ab(130.8mg,白色固体,产率82%)。
1H NMR(600MHz,CDCl3)δ9.19(s,1H),7.83(d,J=7.9Hz,1H),7.46(d,J=8.1Hz,1H),7.30(d,J=8.8Hz,5H),7.25-7.15(m,7H),7.13(s,1H),5.23(q,J=10.3Hz,1H),2.32(s,6H),2.17(s,6H);13C NMR(150MHz,CDCl3)δ138.7(d,J=13.3Hz),138.5(d,J=13.2Hz),138.1(d,J=9.3Hz),134.6,134.4(d,J=2.6Hz),134.3(d,J=2.3Hz),131.4(d,JC-P=107.1Hz),130.9(d,JC-P=107.8Hz),129.4,129.3(d,J=22.9Hz),128.9,128.2,127.4,127.0(d,J=9.7Hz),126.4(q,JC-F=273.2Hz),124.3,122.4,120.9,117.8(d,J=11.1Hz),112.3,46.9(q,JC-F=28.8Hz),21.2,21.0;19F NMR(565MHz,CDCl3)δ-63.5(d,J=9.8Hz,3F);31P NMR(243MHz,CDCl3)δ22.8
实施例12
反应式为:
一种含三氟甲基C-2膦酰基吲哚类化合物的合成方法:取25mL圆底烧瓶,在空气氛围中,依次加入三氟甲基吲哚甲醇2a(0.3mmol),异丙基(苯基)氧膦3c(0.45mmol),最后加入0.32wt%的DBSA水溶液(6mL),在100℃搅拌反应24小时,待反应结束,冷却至室温,用乙酸乙酯萃取三次,合并有机相,无水硫酸镁干燥,过滤,浓缩,粗品通过硅胶柱层析分离(洗脱剂为乙酸乙酯:石油醚=40:60),即得目标产物1ac(99.3mg,白色固体,产率75%)。
1H NMR(600MHz,CDCl3)δ10.32(s,1H),10.09(s,1H),7.92(dd,J=18.6,7.5Hz,4H),7.69(d,J=8.2Hz,1H),7.51(td,J=14.6,7.4Hz,3H),7.46-7.38(m,6H),7.34(t,J=8.7Hz,2H),7.25-7.19(m,5H),7.20-7.13(m,5H),7.08(t,J=7.6Hz,1H),7.01(t,J=7.6Hz,1H),6.07(q,J=10.6Hz,1H),5.81(q,J=10.5Hz,1H),2.92(qd,J=11.2,6.9Hz,1H),2.78(dq,J=13.7,6.9Hz,1H),1.33-1.22(m,9H),1.17(dd,J=17.2,7.1Hz,3H);13CNMR(151MHz,CDCl3)δ138.4(d,J=8.6Hz),138.3(d,J=8.6Hz),135.3,134.9,132.3(d,J=2.5Hz),132.2(d,J=2.3Hz),131.9(d,JC-P=97.5Hz),131.8(d,JC-P=98.3Hz),130.7(d,J=9.5Hz),130.7(d,J=9.4Hz),129.1(d,J=1.7Hz),129.0(d,J=1.9Hz),128.6,128.4,128.3,128.2,127.3,127.1,126.9(d,J=44.9Hz),126.6(d,J=17.4Hz),126.4(d,J=26.7Hz),126.9(q,JC-F=263.7Hz),126.6(q,JC-F=279.8Hz),124.3,124.1,122.2(d,J=3.2Hz),122.2(d,J=2.6Hz),120.7,120.5,119.6,119.5,118.5,118.4,112.0,46.1(q,JC-F=30.0Hz),46.0(q,JC-F=28.9Hz),27.5(d,JC-P=74.6Hz),27.2(d,JC-P=74.5Hz),15.3(dd,J=12.0,2.6Hz),14.8(dd,J=5.1,2.6Hz);19F NMR(565MHz,CDCl3)δ-63.4(d,J=10.7Hz,3F),-63.6(d,J=10.6Hz,3F);31P NMR(243MHz,CDCl3)δ34.5,34.4.
实施例13
反应式为:
一种含三氟甲基C-2膦酰基吲哚类化合物的合成方法:取25mL圆底烧瓶,在空气氛围中,依次加入三氟甲基吲哚甲醇2a(0.3mmol),异丙基(苯基)氧膦3d(0.45mmol),最后加入0.32wt%的DBSA水溶液(6mL),在100℃搅拌反应24小时,待反应结束,冷却至室温,用乙酸乙酯萃取三次,合并有机相,无水硫酸镁干燥,过滤,浓缩,粗品通过硅胶柱层析分离(洗脱剂为乙酸乙酯:石油醚=40:60),即得目标产物1ad(100.9mg,白色固体,产率72%)。
1H NMR(600MHz,CDCl3)δ9.47(s,1H),9.29(s,1H),7.83-7.74(m,4H),7.57(t,J=7.3Hz,1H),7.54-7.47(m,4H),7.45-7.36(m,6H),7.30-7.20(m,6H),7.18-7.10(m,4H),7.02(dd,J=16.2,7.7Hz,3H),5.40(q,J=10.6Hz,1H),5.23(q,J=10.4Hz,1H),2.94-2.87(m,1H),2.62-2.54(m,1H),2.21-1.93(m,4H),1.88-1.69(m,5H),1.69-1.58(m,5H),1.41-1.34(m,2H);13C NMR(150MHz,CDCl3)δ138.3(d,J=9.5Hz),138.2(d,J=8.5Hz),135.2,134.7,133.5,113.4,132.8,132.7,132.3,132.2,130.79,130.71,130.64,128.97,128.89,128.73,128.40,128.28,128.14,127.48,127.11,126.66,126.4(q,JC-F=275.2Hz),124.1,124.0,122.1,120.7,120.4,117.5(d,J=10.7Hz),112.2,112.1,46.6(q,JC-F=30.3Hz),46.4(q,JC-F=26.8Hz),37.3(d,JC-p=77.7Hz),37.2(d,JC-p=77.7Hz),31.6,27.1(d,JC-p=8.3Hz),26.9(d,JC-p=8.2Hz),26.8(d,JC-p=8.0Hz),26.6(d,JC-p=9.2Hz),26.11(d,JC-p=20.3Hz),22.6,14.1;19F NMR(565MHz,CDCl3)δ-63.7(d,J=10.5Hz,3F),-63.8(d,J=10.5Hz,3F);31P NMR(243MHz,CDCl3)δ31.1,31.0.
实施例14
试验发现反应的区域选择性与三氟甲基的存在密切相关,当把三氟甲基替换成普通甲基时,无法得到预期的C2膦酰基吲哚类化合物。
反应式为:
一种C-2膦酰基吲哚类化合物的合成方法:取25mL圆底烧瓶,在空气氛围中,依次加入甲基吲哚苄醇4(0.3mmol),二苯氧膦3a(0.45mmol),最后加入0.32wt%的DBSA水溶液(6mL),在100℃搅拌反应24小时,待反应结束,冷却至室温,用乙酸乙酯萃取三次,合并有机相,无水硫酸镁干燥,过滤,浓缩,粗品通过硅胶柱层析分离(洗脱剂为乙酸乙酯:石油醚=40:60),即得目标产物5(116.3mg,白色固体,产率92%)。
1H NMR(600MHz,CDCl3)δ8.96(s,1H),7.84(s,1H),7.67(t,J=9.1Hz,2H),7.49(q,J=8.5Hz,3H),7.40(t,J=7.4Hz,1H),7.36-7.31(m,3H),7.28-7.22(m,3H),7.20(d,J=8.0Hz,2H),7.14(t,J=7.6Hz,2H),7.05(t,J=7.5Hz,1H),6.72(t,J=7.5Hz,1H),6.59(d,J=8.2Hz,1H),2.13(d,J=15.8Hz,3H).
13C NMR(100MHz,CDCl3)δ140.2(d,JC-P=3.2Hz),136.5,133.3(d,JC-P=7.9Hz),131.5(d,JC-P=2.3Hz),131.4(d,JC-P=91.0Hz),131.3(d,JC-P=2.4Hz),130.4(d,JC-P=4.5Hz),130.2,127.9(d,JC-P=10.9Hz),127.5(d,JC-P=11.1Hz),127.4(d,JC-P=2.2Hz),127.0(d,JC-P=2.8Hz),126.0(d,JC-P=8.9Hz),125.5(d,JC-P=5.7Hz),121.5(d,JC-P=8.2Hz),118.7,116.4(d,JC-P=2.0Hz),111.4,48.0(d,JC-P=64.1Hz),24.5.
31P NMR(162MHz,CDCl3)δ36.7.
实施例15
在实施例1的基础上,对反应条件进行优化,优化结果见表1。
表1
由此可见,本发明所提供的一种含三氟甲基C-2膦酰基吲哚类化合物的合成方法可以实现将用途广泛的三氟甲基与膦酰基同时引入到吲哚中,反应利用一锅法方式进行,以水为溶剂,十二烷基苯磺酸为催化剂,反应无需无水无氧条件以及金属催化剂,具有操作简便、绿色环保、产率高并且反应底物范围广等优点。
应说明的是,以上实施例仅用以说明本发明的技术方案而非限制,尽管参照较佳实施例对本发明进行了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的精神和范围,其均应涵盖在本发明的权利要求范围当中。
- 一种水相合成含三氟甲基C-2膦酰基吲哚的方法
- 一种水相合成含三氟甲基C-2膦酰基吲哚的方法