一种测定茶叶中氟吡草酮残留量的方法
文献发布时间:2023-06-19 19:30:30
[技术领域]
本发明属于分析化学技术领域,具体地说是一种测定茶叶中氟吡草酮残留量的方法。
[背景技术]
随着杂草对传统除草剂逐渐产生抗药性,相对安全而且高效除草的替代产品是当前农业生产的迫切需求。HPPD(对羟基苯基丙酮酸双氧化酶)是目前最重要的除草剂作用靶点之一,HPPD抑制类除草剂是目前全球农药行业的研发热点。近年来各类HPPD抑制除草剂方兴未艾,以三酮类为代表的新型除草剂的生物安全性逐渐受到各国卫生部和环保署的关注。
氟吡草酮属于三酮类HPPD抑制剂,通过抑制HPPD的活性,阻止HPPA(对羟基苯基丙酮酸)转化为HGA(尿黑酸),从而阻碍杂草的生育酚和质体醌的正常合成,影响类胡萝卜素的生物合成,促使植物分生、新生组织白化,导致杂草的植株死亡。为了提高除草剂对作物的安全性,经常需要将除草剂与安全剂解草嗪或解草酯等混配施用。新的除草剂氟吡草酮自2015年上市以来,市场增长迅速,上市首年即实现销售额1亿美元,2015-2019年复合增长率为47%,目前在世界范围内广泛使用。氟吡草酮属于相对低毒的植物源农药,在哺乳动物体内吸收分布广泛,主要分布在肝脏和肾脏中,通过尿液排出。该除草剂对水生生物毒性极大并具有长期持续影响。高剂量的氟吡草酮会有严重的肝毒性和肾毒性,可能对生育能力或胎儿造成伤害。2017年加拿大卫生部有害生物管理局对原生农产品等拟定了氟吡草酮最大残留限量,2021年美国环保署规定了柠檬草等作物中氟吡草酮的残留限量。世界各国对氟吡草酮的限量要求和含量检测越来越重视。2020年,我国在GB2763-2021中规定了谷物中氟吡草酮的临时限量。
随着各国对氟吡草酮这一农药的持续关注,茶叶中氟吡草酮残留风险评估以及最大残留限量值(MRL)的制定势在必行。然而.现阶段鲜有成熟且稳定的茶叶中氟吡草酮残留检测方法,这使得茶叶中氟吡草酮残留检测工作难以开展,风险评估也难以进行。氟吡草酮相对分子质量较小,微溶于甲醇,较难提取且在分析过程中容易受到样品中杂质的干扰。而茶叶中的干扰物多,分析难度本就较大,常规的茶叶检测方法显然不适用于氟吡草酮,因此建立一个准确、稳定的茶叶中氟吡草酮含量的检测方法迫在眉睫。
[发明内容]
本发明的目的就是要解决上述的不足而提供一种经提取、净化后用液相色谱-串联质谱仪测定茶叶中氟吡草酮残留量的方法,该测定方法准确、稳定、抗干扰能力强且适用性广,能够满足多种测试需求。
为实现上述目的设计一种测定茶叶中氟吡草酮残留量的方法,包括以下步骤:1)样品预处理:样品用粉碎机粉碎后,混合均匀装入洁净干燥的自封袋中,密封备用;2)样品的提取:取适量样品,使用1%乙酸-乙腈溶液和混合盐进行提取,得样品提取液;3)样品的净化:将步骤2)所得的样品提取液经GCB/NH2固相萃取柱净化,得上机分析溶液;4)基质校准溶液制备:称取不含氟吡草酮的阴性样品,根据步骤1)、2)、3)制备不加入内标的阴性样品提取液,用于配制氟吡草酮基质校准溶液;5)采用液相色谱串联质谱仪测定步骤3)所得的上机分析溶液和步骤4)所得的基质校准溶液中的氟吡草酮的浓度;6)根据步骤5)所得的样品上机分析溶液和基质校准溶液中氟吡草酮浓度,计算得到样品中的氟吡草酮浓度。
进一步地,步骤1)中,样品用粉碎机粉碎后,若样品颗粒大小仍不均匀,则先将经粉碎后的样品2mm筛后再混合均匀。
进一步地,步骤2)中,在称样前再次混匀样品,准确称取2g样品于50mL离心管中,加入0.2mL的1mg/L磷酸三苯酯TPP内标溶液,加入10mL水,涡旋使样品分散到水溶液中,浸泡20min;加入1%乙酸-乙腈溶液20mL,将离心管固定在水平振荡器上,以250-300次/分钟的速度,振荡提取15min后,加入6g无水硫酸镁和1.5g乙酸钠组成的混合盐,迅速混匀1min,防止结块,然后以8000r/min的转速在4℃离心5min。
进一步地,步骤3)中,用10mL体积比为3:1的乙腈-甲苯溶液活化GCB/NH2固相萃取柱,不保留流出液;准确移取10mL步骤2)中的上清液至离心管中,加入3.33mL甲苯,混匀后转移至小柱上,并收集流出液;待全部样液流出后,用15mL体积比为3:1的乙腈-甲苯溶液洗脱并收集,合并上述两次流出液;在40℃条件下,将样液旋蒸至近干,然后氮吹至干,用丙酮复溶并定容至1mL后,以10000r/min的转速离心3min;取0.1mL上清液与80%甲醇-水溶液0.4mL混合均匀,供液相色谱串联质谱仪检测。
进一步地,步骤4)中,若样品为阳性,根据步骤1)、2)、3)制备不加入内标的阴性样品提取液,再根据样品中目标物残留量的浓度,吸取适量的氟吡草酮标准中间溶液和磷酸三苯酯TPP内标溶液,配制成等同浓度的基质标准溶液上机分析,用于计算样品中目标物的实际残留量。
进一步地,步骤5)中,液相色谱-串联质谱仪的测定条件为:
a)液相色谱分析柱采用Poroshell 120EC-C18柱,规格为柱长10cm,柱内径3.0mm,粒径2.7μm;
b)液相色谱保护柱采用Poroshell 120 EC-C18柱,规格为柱长5mm,柱内径3.0mm,粒径2.7μm;
c)液相色谱柱温度设定为40℃;
d)进样量:20μL;
e)流速:0.4L/min;
f)流动相:
g)运行时间:15min;
h)质谱参数见下表:
。
进一步地,步骤6)中,由液相色谱串联质谱仪所得样品溶液中氟吡草酮的量,采用内标法定量,内标物为磷酸三苯酯;同时通过基质校准溶液中氟吡草酮的量检测结果进行基质校准换算,计算得到样品中氟吡草酮的实际含量。
进一步地,步骤6)中,通过比较样液中色谱峰保留时间与标准溶液中氟吡草酮的保留时间和各离子对的相对丰度,确定样品中是否检出氟吡草酮;再根据基质校准溶液进行基质效应的校准,采用内标法定量。
进一步地,所述氟吡草酮是指CAS号为352010-68-5,分子式为C
本发明同现有技术相比,填补了市场上茶叶中氟吡草酮检测方法的空缺,提供了一种样品经1%乙酸-乙腈溶液,混合盐辅助提取,固相萃取柱进净化后,使用液相色谱串联质谱仪进行检测,内标法定量,基质校准溶液进行实际含量校正的测定茶叶中氟吡草酮含量的方法,该测定方法准确、稳定、抗干扰能力强、快速且适用性广,适用于绿茶、红茶、乌龙茶等多种茶叶,最低定量浓度较低为0.01mg/kg,能够满足多种测试需求,值得推广应用。
[附图说明]
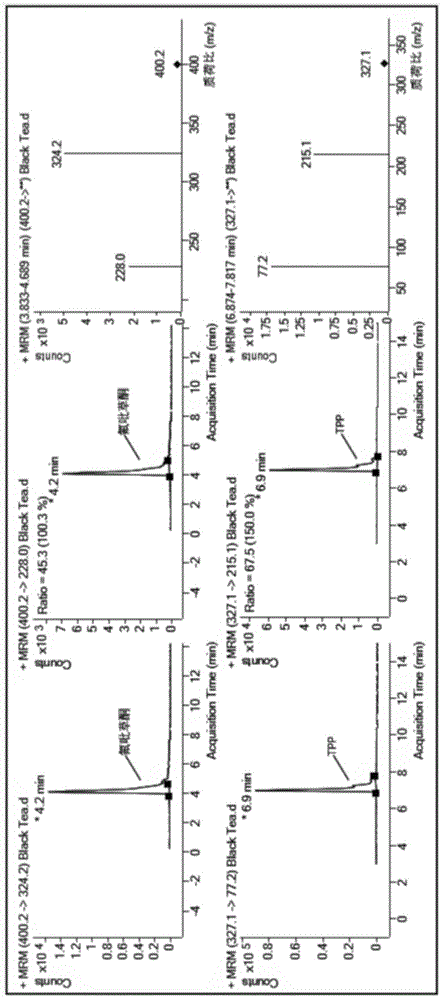
图1是本发明实施例2中使用液相色谱-串联质谱仪检测红茶样品中氟吡草酮含量,所得氟吡草酮及内标物磷酸三苯酯的质谱图。
[具体实施方式]
本发明属于分析化学领域中对茶叶中氟吡草酮含量的准确测定,其原理为:样品经1%乙酸-乙腈溶液,混合盐辅助提取,固相萃取柱进净化后,使用液相色谱串联质谱仪进行检测,内标法定量,基质校准溶液进行实际含量校正。
该测定方法包括以下步骤:1)样品预处理:样品用粉碎机粉碎后,若样品颗粒大小仍不均匀,则先将经粉碎后的样品2mm筛后混合均匀装入洁净干燥的自封袋中,密封备用;2)样品的提取:取2g样品,使用1%乙酸-乙腈溶液和混合盐进行提取,得样品提取液;3)样品的净化:样品提取液经GCB/NH2固相萃取柱净化,得上机分析溶液;4)基质校准溶液制备:称取不含氟吡草酮的阴性样品,根据步骤1)、2)、3)制备不加入内标的阴性样品提取液,用于配制氟吡草酮基质校准溶液;5)采用液相色谱串联质谱仪测定步骤3)所得的上机分析溶液和步骤4)所得的基质校准溶液中的氟吡草酮的浓度;6)根据步骤5)所得的样品上机分析溶液和基质校准溶液中氟吡草酮浓度,计算得到样品中的氟比草酮浓度。
其中,步骤2)中,在称样前再次混匀样品,准确称取2g样品于50mL离心管中,加入0.2mL的1mg/L磷酸三苯酯(TPP)内标溶液,加入10mL水,涡旋使样品分散到水溶液中,浸泡20min;加入1%乙酸-乙腈溶液20mL,将离心管固定在水平振荡器上,以250-300次每分钟的速度,振荡提取15min后,加入混合盐(6g无水硫酸镁和1.5g乙酸钠),迅速混匀1min,防止结块,然后以8000r/min的转速在4℃离心5min。
步骤3)中,用10mL乙腈-甲苯(体积比3:1)溶液活化GCB/NH2固相萃取柱,不保留流出液;准确移取10mL上清液至离心管中,加入3.33mL甲苯,混匀后转移至小柱上,并收集流出液;待全部样液流出后,用15mL乙腈-甲苯(体积比3:1)溶液洗脱并收集,合并上述两次流出液;在40℃条件下,将样液旋蒸至近干,然后氮吹至干,用丙酮复溶并定容至1mL后,以10000r/min的转速离心3min;取0.1mL上清液与80%甲醇-水溶液0.4mL混合均匀,供液相色谱串联质谱仪检测。
步骤4)中,若样品为阳性,根据步骤1)、2)、3)制备不加入内标的阴性样品提取液,再根据样品中目标物残留量的浓度,吸取适量的氟吡草酮标准中间溶液和磷酸三苯酯(TPP)内标溶液,配制成等同浓度的基质标准溶液上机分析,用于计算样品中目标物的实际残留量,现配现用。
步骤5)中,液相色谱-串联质谱仪的测定条件为:
a)液相色谱分析柱采用Poroshell 120 EC-C18柱,规格为柱长10cm,柱内径3.0mm,粒径2.7μm;
b)液相色谱保护柱采用Poroshell 120 EC-C18柱,规格为柱长5mm,柱内径3.0mm,粒径2.7μm;
c)液相色谱柱温度设定为40℃;
d)进样量:20μL;
e)流速:0.4L/min;
f)流动相:
g)运行时间:15min;
h)质谱参数见下表:
步骤6)中,由液相色谱串联质谱仪所得样品溶液中氟吡草酮的量,采用内标法定量,内标物为磷酸三苯酯,通过基质校准溶液中的氟吡草酮的量进行基质校准换算,计算得到样品中氟吡草酮的实际含量。氟吡草酮是指CAS号为352010-68-5,分子式为C
下面结合具体实施例对本发明作以下进一步说明:
实施例1:绿茶中氟吡草酮残留量的测定
1.样品溶液的制备:
将绿茶用粉碎机打碎,过2mm筛,混匀,装入洁净干燥的自封袋中,密封备用。在称样前再次混匀样品,准确称取2g样品于50mL离心管中,加入0.2mL的1mg/L磷酸三苯酯(TPP)内标溶液,加入10mL水,涡旋使样品分散到水溶液中,浸泡20min。加入1%乙酸-乙腈溶液20mL,将离心管固定在水平振荡器上,以250-300次/分钟的速度,振荡提取15min后,加入混合盐(6g无水硫酸镁和1.5g乙酸钠),迅速混匀1min,防止结块,然后以8000r/min的转速在4℃离心5min;用10mL乙腈-甲苯(体积比3:1)溶液活化GCB/NH2固相萃取柱,不保留流出液。准确移取10mL上清液至离心管中,加入3.33mL甲苯,混匀后转移至小柱上,并收集流出液。待全部样液流出后,用15mL乙腈-甲苯(体积比3:1)溶液洗脱并收集,合并上述两次流出液。在40℃条件下,将样液旋蒸至近干,然后氮吹至干,用丙酮复溶并定容至1mL后,以10000r/min的转速离心3min。取0.1mL上清液与80%甲醇-水溶液0.4mL混合均匀,供液相色谱串联质谱仪检测。
2.设定仪器参数:
a)液相色谱分析柱采用Poroshell 120EC-C18柱,规格为柱长10cm,柱内径3.0mm,粒径2.7μm;
b)液相色谱保护柱采用Poroshell 120EC-C18柱,规格为柱长5mm,柱内径3.0mm,粒径2.7μm;
c)液相色谱柱温度设定为40℃;
d)进样量:20μL;
e)流速:0.4L/min;
f)流动相:
g)运行时间:15min;
h)质谱参数见下表:
3.定性和定量
1)定性:通过比较样液中色谱峰保留时间与标准溶液中氟吡草酮的保留时间和各离子对的相对丰度,确定样品中是否检出氟吡草酮。
2)定量:根据基质校准溶液进行基质效应的校准,采用内标法定量。
4.计算
根据样品提取溶液中氟吡草酮的浓度,计算样品中氟吡草酮的含量。经检测,该绿茶样品中未检出有氟吡草酮。
实施例2:红茶中氟吡草酮残留量的测定
1.样品溶液的制备
将红茶用粉碎机打碎过2mm筛,混匀,装入洁净干燥的自封袋中,密封备用。在称样前再次混匀样品,准确称取2g样品于50mL离心管中,加入0.2mL的1mg/L磷酸三苯酯(TPP)内标溶液,加入10mL水,涡旋使样品分散到水溶液中,浸泡20min。加入20mL 1%乙酸-乙腈溶液,将离心管固定在水平振荡器上,以250-300次/分钟的速度,振荡提取15min后,加入混合盐(6g无水硫酸镁和1.5g乙酸钠),迅速混匀1min,防止结块,然后以8000r/min的转速在4℃离心5min;用10mL乙腈-甲苯(体积比3:1)溶液活化GCB/NH2固相萃取柱,不保留流出液。准确移取10mL上清液至离心管中,加入3.33mL甲苯,混匀后转移至小柱上,并收集流出液。待全部样液流出后,用15mL乙腈-甲苯(体积比3:1)溶液洗脱并收集,合并上述两次流出液。在40℃条件下,将样液旋蒸至近干,然后氮吹至干,用丙酮复溶并定容至1mL后,以10000r/min的转速离心3min。取0.1mL上清液与80%甲醇-水溶液0.4mL混合均匀,供液相色谱串联质谱仪检测。
2.设定仪器参数:
a)液相色谱分析柱采用Poroshell 120EC-C18柱,规格为柱长10cm,柱内径3.0mm,粒径2.7μm;
b)液相色谱保护柱采用Poroshell 120EC-C18柱,规格为柱长5mm,柱内径3.0mm,粒径2.7μm;
c)液相色谱柱温度设定为40℃;
d)进样量:20μL;
e)流速:0.4L/min;
f)流动相:
g)运行时间:15min;
h)质谱参数见下表:
3.定性和定量
1)定性:通过比较样液中色谱峰保留时间与标准溶液中氟吡草酮的保留时间和各离子对的相对丰度,确定样品中是否检出氟吡草酮。
2)定量:根据基质校准溶液进行基质效应的校准,采用内标法定量。
4.计算
根据样品提取溶液中氟吡草酮的浓度,计算样品中氟吡草酮的含量。经检测,于该红茶样品中氟吡草酮的含量为0.021mg/kg。
本发明并不受上述实施方式的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。