一种热致延迟荧光性能的有机发光材料及其制备方法和应用
文献发布时间:2023-06-19 09:33:52
技术领域
本发明涉及有机发光材料技术领域,具体涉及一种基于环芴酮二噻吩为骨架的双极性热致延迟荧光性能有机发光材料及其制备方法和应用。
背景技术
有机电致发光器件(organic light-emitting diodes,OLEDs)由于超薄、重量轻、能耗低、主动发光、视角宽、响应快等优点,在显示和照明领域有极大的应用前景,越来越受到人们的重视。
近三十年来,广大科研工作者从未停止对于高激子利用率的有机光电功能材料的探索,1998年,Forrest等人利用金属配合物发现电致磷光现象以来,基于重金属配合物的磷光客体材料取得了迅速的发展(Highly efficient phosphorescent emission fromorganic electroluminescent devices.Nature,1998,395,151-154.);由于重原子效应,具有较强的自旋-轨道耦合作用,该类磷光材料理论上可以实现100%的内量子效率,突破了基于传统的荧光材料为客体的电致荧光器件外量子效率为5%的极限,然而该类金属配合物合成路线较复杂,过渡金属原料昂贵且资源稀缺,不适宜可持续发展战略。
2009年,日本九州大学的Adachi课题组首次发现了锡配体的化合物具有延迟荧光的现象,即在外界热能的作用下,最低三线态激子T
公开号为CN108048077B公开了一种以1,4-二氰基并环戊二烯为中心的小分子有机电致发光材料,该类材料具有D-A型分子结构和非常小的ΔE
公开号为CN109678861A公开了一种双极性热激活延迟荧光化合物及其应用,所述的双极性热激活延迟荧光化合物,作为电致发光材料的单线态与三线态能级差小于300meV,有利于提高三线态激子向单线态激子的反向间隙串越概率。此外,在保持较低的ΔE
总体来说,现有的部分热致延迟荧光性能有机发光材料还存在不足,比如,基于TADF材料为客体的有机电致荧光器件,普遍存在器件结构复杂,效率衰减快、不稳定的现象,在实际应用过程中简化器件制备程序,稳定器件效率,对发光材料的设计提出了更高的要求。
发明内容
本发明提供一种热致延迟荧光性能的有机发光材料及其制备方法和应用,该类有机发光材料可以用于有机电致发光器件、防伪、化学生物检测以及生物成像等领域。
一种热致延迟荧光性能的有机发光材料,所述的有机发光材料以环芴酮二噻吩为骨架,通过连接不同的给体单元与受体单元,形成双极性有机小分子发光材料。
所述的热致延迟荧光性能的有机发光材料,具有式I-1~I-4所示的结构:
其中,Ar为给体单元,所述的给体单元为芳胺或杂芳胺;R为受体单元,所述的受体单元为卤素、氰基、含取代基的吡啶或二苯基磷氧基。
所述的给体单元Ar为芳胺或杂芳胺,具体为式II-1~II-7所示结构:
其中,R
所述的受体单元R为卤素、氰基、含取代基的吡啶或二苯基磷氧基;所述的含取代基的吡啶或二苯基磷氧基,具有式III-1~III-4所示结构:
其中,R
本发明还提供了一种热致延迟荧光性能的有机发光材料的合成方法,包括:
步骤1,将化合物A与卤素源加入有机溶剂,加热反应,得到式V-1所示结构的环芴酮二噻吩中间体;或,将式V-1所示结构的环芴酮二噻吩中间体与对卤素苯硼酸在有机溶剂反应得到式V-2所示结构的环芴酮二噻吩中间体;
所述的化合物A具有式IV所示结构:
所述的环芴酮二噻吩中间体具有式V-1或V-2所示结构:
其中,X1与X2各自独立为卤素;
步骤2,将摩尔比为1~1.5:1的有机胺与步骤1得到的环芴酮二噻吩中间体置于有机溶剂中,加热反应,得到式VI-1或VI-2所示结构的有机胺单取代的有机发光材料;或,将摩尔比为2~3:1的有机胺与步骤1得到的环芴酮二噻吩中间体置于有机溶剂中,加热反应,得到式I-1或I-2所示结构的有机发光材料;
其中,Ar的定义与式I结构中的一致;x为卤素;
步骤3,将式VI-1或VI-2所示结构的有机胺单取代的有机发光材料在催化剂的作用下在有机溶液中通过取代反应,得到式I-3或I-4所示结构的热致延迟荧光性能的有机发光材料。
优选的,步骤1中,所述的式V-1所示结构的环芴酮二噻吩中间体具体为式V
所述的式V-1所示结构的环芴酮二噻吩中间体的制备方法为方法1、方法2、方法3、方法4、方法5中的一种:
方法1,将摩尔比为1:2~3的化合物A和N-溴代琥珀酰亚胺加入含有乙酸的有机溶剂中,于室温下反应,淬灭,分离纯化,得到式V
方法2,将摩尔比为1:2~3的化合物A和液溴加入含有四氢呋喃的有机溶剂中,加热反应,淬灭,分离纯化,得到式V
方法3,将摩尔比为1:1~2的化合物A、N-碘代丁二酰亚胺和三氟乙酸加入含有乙氰的有机溶剂中,于室温下反应,淬灭,分离纯化,得到式V
方法4,将方法1或方法2中得到的双边溴取代的环芴酮二噻吩中间体B、四甲基氟化胺置于有机溶剂中,加热反应,得到式V
方法5,将方法1或方法2中得到的双边溴取代的环芴酮二噻吩中间体B、氯化锂、三氯化铁置于有机溶剂中,室温反应,得到式V
步骤2中,所述的式VI-1或VI-2所示结构的有机胺单取代的有机发光材料的制备方法为方法1、方法2中的一种:
方法1,将步骤1得到的环芴酮二噻吩中间体、有机胺置于有机溶剂中,通过乌尔曼(Ullmann)偶联反应,得到式VI-1或VI-2所示结构的有机胺单取代的有机发光材料;
方法2,在无水无氧条件下,将步骤1得到的环芴酮二噻吩中间体、有机胺、碱、钯催化剂、三叔丁基膦四氟硼酸盐加入到有机溶剂中,加热反应24-60小时,淬灭,分离纯化,得到式VI-1或VI-2所示结构的有机胺单取代的有机发光材料。
步骤3中,所述的得到式I-3或I-4所示结构的热致延迟荧光性能的有机发光材料的制备方法为方法1~3中的一种:
方法1,在无水无氧条件下,将步骤2中制得的有机胺单取代的有机发光材料、氰化亚铜在有机溶液中通过取代反应,得到I-3或I-4所示结构的热致延迟荧光性能的有机发光材料;
方法2,在无水无氧条件下,将步骤2中制得的有机胺单取代的有机发光材料、吡啶硼酸或吡啶硼酸频哪醇酯通过Suzuki偶联反应,得到I-3或I-4所示结构的热致延迟荧光性能的有机发光材料;
方法3,在无水无氧条件下,将步骤2制得的有机胺单取代的有机发光材料溶于有机溶剂中,低温环境加入二苯基氯化膦,搅拌,加入双氧水进行氧化反应,得到I-3或I-4所示结构的热致延迟荧光性能的有机发光材料。
所述的有机胺的具体结构如式VII-1~VII-7所示:
其中,R
所述的有机溶剂为优选的,所述的有机溶剂为氯仿、四氢呋喃、N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、乙腈、乙醚和甲苯中的一种或几种。
本发明还提供一种热致延迟荧光性能的有机发光材料的应用,利用所述有机发光材料的有机光电性能制备OLEDs器件的发光层材料;所述OLEDs器件包括玻璃和玻璃上依次设置的导电玻璃衬底层、空穴注入层、空穴传输层、发光层、电子传输层及阴极层,所述的发光层由主体材料和掺杂材料组成,发光层的主体材料或掺杂材料为所述的热致延迟荧光性能的有机发光材料。
本发明的有益效果:
(1)本发明所制备的热致延迟荧光性能的有机发光材料,为含有环芴酮二噻吩单元的D-A型结构(Donor-Acceptor型),具有良好的空穴/电子传输性能,高的荧光量子产率,可广泛应用于有机电致发光器件领域。
(2)本发明所制备的热致延迟荧光性能的有机发光材料,可应用于防伪、化学生物检测、生物成像等领域。
(3)本发明所制备的环芴酮二噻吩中间体在有机相中溶解性良好,可用于蒸镀器件,也适用于旋涂器件。
(4)本发明所述的热致延迟荧光性能的有机发光材料、环芴酮二噻吩中间体的制备方法,合成简单高效,产物易纯化,可规模化生产及应用。
附图说明:
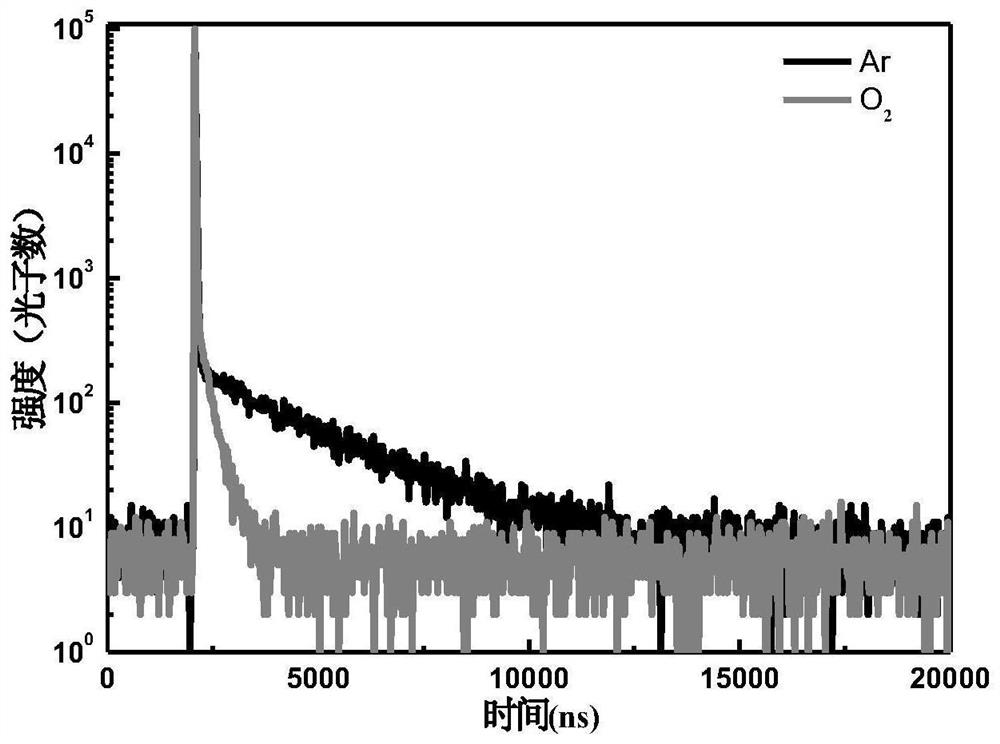
图1为本发明实施例5制得的热致延迟荧光性能的有机发光材料在有氧和无氧中的瞬态寿命情况。
图2为本发明实施例6制得的热致延迟荧光性能的有机发光材料的热失重分析图。
图3为本发明的有机电致荧光发光器件结构示意图;其中,1为导电玻璃衬底层,2为空穴注入层,3为空穴传输层,4为发光层,5为电子传输层,6为阴极层。
具体实施方式
通过以下实例对本发明作进一步的说明,其目的在于帮助更好的理解本发明的内容,但这些具体实施方案不以任何方式限制本发明的保护范围。本发明中所用的原料:3,3-二溴-2,2-联噻吩、咔唑、3,6-二叔丁基咔唑、二苯胺、4,4′-二甲基二苯胺、吩噁嗪、吩噻嗪、9,9′-二甲基吖啶、9,9′-二苯基吖啶、亚氨基芪、对甲基二苯酚、吡啶硼酸等均可在市场上购得。
实施例1
(1)中间体2,6-二溴-环芴酮二噻吩的合成,合成路线如下所示:
3,3-二溴-2,2-联噻吩(2.55g,7.87mmol)加入200ml西兰克瓶,抽通三次,加入乙醚50ml,放入已预冷至-78℃的低温箱,缓慢加入正丁基锂(8.04ml,18.89mmol),反应2小时后加入二甲氨基甲酰氯100ml,搅拌过夜至室温,冰水淬灭,乙醚萃取,无水硫酸钠干燥,硅胶柱层析分离,二氯甲烷/石油醚(v:v)=1:1作为淋洗剂,产品为深紫色粉末,产率:68%,元素分析理论值C
将摩尔比为1:2的上述得到的深紫色粉末环芴酮二噻吩和N-溴代琥珀酰亚胺加入含有乙酸的有机溶剂中,混合溶液于室温下反应过夜,淬灭反应,分离纯化,得到双边带溴中间体B,即2,6-二溴-环芴酮二噻吩,产品为深紫色针状,产率:96%,元素分析理论值C
(2)中间体2,6-二碘-环芴酮二噻吩的合成,合成路线如下所示:
采用实施例1-(1)中同样的方法制备环芴酮二噻吩,选用环芴酮二噻吩(1.00g,5.20mmol),N-碘代丁二酰亚胺(2.57g,11.44mmol),三氟乙酸(0.20ml),乙氰15ml,加入150ml圆底烧瓶,室温下反应过夜,10ml饱和氯化钠水溶液淬灭,二氯甲烷萃取,无水硫酸钠干燥,硅胶柱层析分离,二氯甲烷/石油醚(v:v)=1:1作为淋洗剂,产品为紫色粉末1.84g。产率:88%,元素分析理论值C
(3)中间体2-氟-6-溴-环芴酮二噻吩的合成,合成路线如下所示:
采用实施例1-(1)中同样的方法制备2,6-二溴-环芴酮二噻吩,将2,6-二溴-环芴酮二噻吩,四甲基氟化胺以1:1的摩尔比,以二甲基亚砜为有机溶剂,于圆底烧瓶中反应,110℃下搅拌12小时,得到单边氟取代的产物D,即2-氟-6-溴-环芴酮二噻吩,产率:78%,元素分析理论值C
(4)中间体2-氯-6-溴-环芴酮二噻吩的合成,合成路线如下所示:
采用实施例1-(1)中同样的方法制备2,6-二溴-环芴酮二噻吩,将2,6-二溴-环芴酮二噻吩,氯化锂,三氯化铁以1:1:2的摩尔比,以乙腈为有机溶剂,于圆底烧瓶中反应,80℃下搅拌10小时,得到单边氯取代的产物E,即2-氯-6-溴-环芴酮二噻吩,产率:67%,元素分析理论值C
实施例2
选用2,6-二溴-环芴酮二噻吩(2.50g,7.14mmol),咔唑(1.19g,7.14mmol),碘化亚铜(0.10g,0.53mmol),碳酸钾(2.20g,15.9mmol),18-冠-6(0.14g,0.54mmol),邻二氯苯3ml,加入150ml圆底烧瓶,190℃下回流48小时,10ml饱和氯化钠水溶液淬灭,二氯甲烷萃取,无水硫酸钠干燥,硅胶柱层析分离,二氯甲烷/石油醚(v:v)=1:1作为淋洗剂,产率:68%,元素分析理论值C
实施例3
将2,6-二溴-环芴酮二噻吩与咔唑以1:2的摩尔比投入反应,可合成双边咔唑取代的环芴酮二噻吩,产率86%,元素分析理论值C
实施例4
将2-氟-6-溴-环芴酮二噻吩(1.00g,3.46mmol),吩噻嗪(0.76g,3.81mmol),叔丁醇钠(0.55g,5.71mmol),醋酸钯(0.025g,0.11mmol),三叔丁基膦四氟硼酸盐(0.095g,0.33mmol),甲苯50ml,加入100ml圆底烧瓶,110℃下回流48小时,10ml饱和氯化钠水溶液淬灭,二氯甲烷萃取,无水硫酸钠干燥,硅胶柱层析分离,二氯甲烷/石油醚(v:v)=1:1作为淋洗剂,产率:92%,元素分析理论值C
实施例5
将2,6-二溴-环芴酮二噻吩(1.00g,2.86mmol),9,9′-二甲基吖啶(1.31g,6.29mmol),叔丁醇钠(1.1g,11.42mmol),醋酸钯(0.025g,0.11mmol),三叔丁基膦四氟硼酸盐(0.19g,0.66mmol),甲苯50ml,加入100ml圆底烧瓶,110℃下回流48小时,10ml饱和氯化钠水溶液淬灭,二氯甲烷萃取,无水硫酸钠干燥,硅胶柱层析分离,二氯甲烷/石油醚(v:v)=1:1作为淋洗剂,产率:93%,元素分析理论值C
实施例6
将2-溴-6-咔唑基-环芴酮二噻吩(1.00g,2.29mmol),氰化亚铜(0.25g,2.75mmol),二甲基甲酰胺15ml,加入100ml圆底烧瓶,回流过夜,冷却至室温,倒入三氯化铁的盐酸溶液2ml,搅拌半小时,10ml饱和氯化钠水溶液淬灭,二氯甲烷萃取,无水硫酸钠干燥。硅胶柱层析分离,二氯甲烷作为淋洗剂,产率:87%,元素分析理论值C
实施例7
将2,6-二溴-环芴酮二噻吩,9,9′-二甲基吖啶为原料可高产率得到目标产物2-溴-6-(9,9′-二甲基)吖啶基-环芴酮二噻吩,将2-溴-6-(9,9′-二甲基)吖啶基-环芴酮二噻吩(1.00g,2.09mmol),吡啶-3-硼酸(0.31g,2.51mmol),四(三苯基膦)钯(0.32g,0.28mmol),碳酸钾(0.76g,5.54mmol),甲苯20ml,乙醇10ml,水5ml,加入100ml圆底烧瓶,110℃下回流48h,10ml饱和氯化钠水溶液淬灭,二氯甲烷萃取,无水硫酸钠干燥,硅胶柱层析分离,乙酸乙酯/二氯甲烷(v:v)=1:1作为淋洗剂。产率:82%。元素分析理论值C
实施例8
以2,6-二溴-环芴酮二噻吩,对溴苯硼酸为原料可高产率得到目标中间体2,6-(4,4′-二溴)二苯基-环芴酮二噻吩,产率:68%。元素分析理论值C
以2,6-(4,4′-二溴)二苯基-环芴酮二噻吩,9,9′-二甲基吖啶为原料,摩尔比为1:2可高产率得到目标产物,产率:90%,元素分析理论值C
实施例9
以2,6-(4,4′-二溴)二苯基-环芴酮二噻吩,9,9′-二甲基吖啶为原料,摩尔比为1:1可高产率得到单边取代的产物,将上述单边取代的产物为原料(1.00g,1.59mmol)加入200ml西兰克瓶,抽通三次,加入四氢呋喃50ml,放入已预冷至-78℃的低温箱,缓慢加入正丁基锂(1.26ml,2.95mmol),反应2小时后加入二苯基氯化膦(0.45g,1.90mmol),搅拌3小时后加入双氧水进行氧化得到目标产物,4ml稀盐酸淬灭,二氯甲烷萃取,无水硫酸钠干燥,硅胶柱层析分离,二氯甲烷/石油醚(v:v)=3:2作为淋洗剂,产率:78%,元素分析理论值C
通式中其他化合物的制备参考以上实例均可高效获得。
应用例:
电致发光蒸镀型器件的制备及结果
器件结构:ITO/MoO
如图3所示,本发明所制备的热致延迟荧光性能的有机发光材作为发光层客体的电致荧光器件,包括玻璃和导电玻璃(ITO)衬底层1,空穴注入层2(三氧化钼MoO
器件制备过程如下:电致发光器件按本领域已知方法制作,如按参考文献(Adv.Mater.2004,16,537.)公开的方法制作;具体方法为:在高真空条件下,在经过清洗的导电玻璃(ITO)衬底上依次蒸镀8nm的MoO
器件性能见下表:
由上表可以看到,使用本发明提供的热致延迟荧光材料制备的OLEDs器件效率,远高于传统荧光材料制备的有机电致发光器件效率。
综上所述,本发明为含有环芴酮二噻吩单元的D-A型的有机发光材料,具有良好的空穴/电子传输性能,高的荧光量子产率,在电致OLEDs器件中,引用此类材料,获得了优良的电致发光性能,其中包括低启动电压及较高的外量子效率等,有利于开发高效全彩显示器,可广泛应用于有机电致发光器件领域,本发明所制备的有机发光材料的发光性能适用于防伪、化学生物检测、生物成像等领域。
- 一种热致延迟荧光性能的有机发光材料及其制备方法和应用
- 一种深蓝色热延迟有机发光材料及其制备方法和应用