一种基于宏基因组的人腺病毒分子分型和溯源方法及系统
文献发布时间:2023-06-19 10:40:10
技术领域
本发明涉及生物信息技术领域,尤其涉及一种基于宏基因组的人腺病毒分子分型和溯源方法及系统。
背景技术
人腺病毒(Human adenovirus,HAdV)属于腺病毒科(Adenoviridae)哺乳动物腺病毒属(Mastadenovirus),基因组全长约34.7kb,属于dsDNA,可感染多种黏膜组织,如胃肠道、呼吸道、泌尿生殖道及眼角结膜等,导致自限性的黏膜感染甚至严重的致死性感染。HAdV表面有3种主要的囊膜蛋白是构成囊膜的重要成分,也是用于诊断的重要抗原。在基因学上,编码这三类蛋白的区域是病毒基因组中变异最大的区域,是人腺病毒基因研究的热点区域。根据血凝、纤维基因长度、基因组GC含量等免疫学、生物学和生物化学特征,人腺病毒分为7个亚型/种:HAdV-A~G,又分为100种以上基因型/血清型。特定的HAdV亚型和基因型与特定的疾病、流行病学环境和人口风险组群相关。HAdV-1~7、11、14、21、35、55均与呼吸道疾病相关,且HAdV-7、35、55致病性较强。HAdV-7可分为多种基因型(如HAdV-7a等),其中7d基因型最常造成严重的感染。HAdV-40、41、52与胃肠炎相关,HAdV-4、8、9、19、37、53、54、56、64与流行性角膜结膜炎相关。对人腺病毒进行分类单元(亚型或基因型)的分型鉴定不仅在临床诊断、治疗和预后,也在监测人腺病毒流行和迁移等公卫领域有着重要的意义。
目前人腺病毒鉴定和分型方法包括:病毒分离鉴定、免疫学方法、核酸检测以及基于二代测序的扩增子测序、全基因组测序,但这些方法存在病毒培养、对样本的先验认知等局限性,或难以全面覆盖在临床上有重要意义的基因型。近年开始逐渐发展的宏基因组测序技术(Metagenomic sequencing),以特定生境中的整个微生物群落作为研究对象,直接提取临床样本的全部微生物组的核酸进行测序注释和比对分析。该技术弥补了以上方法的不足,无需培养,无需样本的先验知识,较全面的覆盖人腺病毒的各个亚型和基因型,但是目前主要应用于科学研究领域,其策略为测序reads(读序)直接比对参考基因组,基于比对质量(一致性位点百分数和比对reads数)进行分型,但当测序reads较短时,存在假阳性率较高的问题;或者将reads组装为全基因组后比对/进化分析以分型,但需要以样本有较高病毒丰度为前提,不适用于微量/痕量样本的鉴定分型。我国需要更灵敏、更全面的临床重要人腺病毒宏基因组分型技术,对微量/痕量的病毒样本进行亚型和基因型分型鉴定,满足临床人腺病毒诊断、治疗和流行病学病毒溯源的需求。
发明内容
本发明提出了一种基于宏基因组的人腺病毒分子分型和溯源方法及系统,用以解决背景技术中提到的目前人腺病毒的宏基因组测序分型需要测序reads较长、样本中病毒含量较高,通过组装、比对策略对病毒进行种水平的注释,且不能提供基因型水平的分型鉴定,故而无法对病毒含量较低的临床样本进行较全面的人腺病毒分型鉴定的问题。
一种基于宏基因组的人腺病毒分子分型和溯源方法,包括以下步骤:
构建包含分型层级分类系统的人腺病毒分型数据库,获取人腺病毒的分类单元和完整基因组/标记基因序列,确定人腺病毒每个分类单元的完整基因组/标记基因序列;
获取临床样本的宏基因组测序原始数据,对其进行预处理获得目标宏基因组数据;
利用预设宏基因组测序数据双重比对注释系统分析所述目标宏基因组数据,以所述人腺病毒分型数据库为基础,确定所述临床样本是否有目标人腺病毒分类单元;
当确定所述临床样本中有目标人腺病毒分类单元时,利用预设临床报告系统确定所述目标人腺病毒分类单元对应的目标关联特征,生成人腺病毒分型鉴定报告。
优选的,所述构建包含分型层级分类系统的人腺病毒分型数据库,获取人腺病毒的分类单元和完整基因组/标记基因序列,确定人腺病毒每个分类单元的完整基因组/标记基因序列,包括:
从NCBI Taxonomy数据库中提取人腺病毒亚型及基因型名称,构建第一人腺病毒分类单元列表;
按HAdV分型格式标准化分类单元命名,合并不规范的分类单元,建立分型层级分类系统;
从当前公共数据库中(NCBI GenBank、RefSeq)下载人腺病毒的完整基因序列及注释信息,获得第一基因序列集;
应用正则公式提取所述完整基因序列的注释信息中分类单元关键字进行序列分类单元注释,并基于分类单元合法性、序列质量合法性进行过滤,获得第二基因序列集;
将所述第二基因序列集中的序列进行聚类,过滤异常序列,获得第三基因序列集;
基于所述第三基因序列集中序列的分类单元,获得第一人腺病毒分类单元列表,去除所述第一人腺病毒分类单元列表中缺乏有效参考基因序列的第一分类单元,并补充未纳入的具有有效参考基因序列的第二分类单元,获得第二人腺病毒分类单元列表;
将所述第三基因序列集的分类单元注释与所述第二人腺病毒分类单元列表进行校对与标准化,获得人腺病毒分型层级中每个层级结构中的分类单元及其基因序列;
将所述每个层级结构中的人腺病毒分类单元以及基因序列进行存储,并建立人腺病毒分类单元基因参考序列索引,获得目标人腺病毒分型数据库。
优选的,所述基因序列包括:基因组和标记基因序列。
优选的,所述利用预设宏基因组测序数据双重比对注释系统分析所述目标宏基因组数据,以所述人腺病毒分型数据库为基础,确定所述临床样本是否有目标人腺病毒分类单元,包括:
将所述目标人腺病毒分型数据库中的基因序列作为参考序列。
构建将宏基因组数据与参考序列进行比对,确定匹配的参考序列的算法流程;
确定根据所述匹配的参考序列,基于目标人腺病毒分型数据库中的参考序列索引,确定目标人腺病毒分类单元的匹配流程;
将所述算法流程和匹配流程构建为宏基因组测序数据双重比对注释系统;
将将所述目标宏基因组数据输入到所述预设宏基因组测序数据双重比对注释系统,确定所述临床样本是否有目标人腺病毒分类单元。
优选的,所述算法流程包括:以基因组序列为参考序列的WhScore算法和UniScore算法流程,和以标记基因序列为参考序列的UniScore算法流程。
优选的,所述匹配流程包括:应用匹配的基因组参考序列的分类单元确定分型,和应用匹配的标记参考基因序列的分类单元确定分型。
优选的,所述获取临床样本的宏基因组测序原始数据,对其进行预处理获得目标宏基因组数据,包括:
过滤所述宏基因组原始测序数据中质量值低于2,碱基数占整个read的40%的第一reads;
切除所述宏基因组原始测序数据中特定滑窗内平均质量小于20的碱基;
过滤所述宏基因组原始测序数据中平均质量小于20的第二reads、含N数量大于5的第三reads以及长度小于50的第四reads。
优选的,所述当确定所述临床样本中有目标人腺病毒分类单元时,利用预设临床报告系统确定所述目标人腺病毒分类单元对应的目标关联特征,生成人腺病毒分型鉴定报告,包括:
通过文献收集和临床大样本的挖掘,构建不同人腺病毒分类单元和相关特征的关联表,利用所述不同人腺病毒分类单元和相关特征的关联表构建所述预设临床报告系统;
在所述关联表中搜索所述目标人腺病毒分类单元,确定所述目标人腺病毒分类单元对应的目标关联特征;
自动生成分型结果,包含注释的目标分类单元(物种拉丁名、物种中文名、亚型/基因型、该亚型/基因型的相关特征、支持得分);
将数据库的客户信息导入报告模板,将所述分型结果导入所述报告模板的相应表格;
以PDF格式生成最终的人腺病毒分型鉴定报告。
优选的,确定所述临床样本是否有目标人腺病毒分类单元的步骤包括:
应用已知样本数据中正常标签数据及其相应的病毒分子分型类别的结果,确定病毒分型模型的主要参数组及其阈值,应用该模型对未确定样本中的正常标签数据进行预测,获得预测的分型结果;
正常标签数据分型模型相关参数组的似然函数定义:
其中,L(θ)为似然函数,
其中,Ad
θ为分布模型参数组:
θ={φ
其中,φ
转换式(1)、(2)为下面形式:
其中,
估算第k次的参数组并统计来源于分型对象Ad
假设潜在变量
上式转化为:
优化似然函数为:
其中,F为第k次的似然函数,
设定Y
循环迭代,每次最大化第k轮的
其中,
ρ表示标签数据来源测序平台的错误率,δ表示标签数据错误来源比对算法的错误率,j为read标签数据;
重复迭代(默认20循环,初始参数为随机产生),直至
一种基于宏基因组的人腺病毒分子分型和溯源系统,该系统包括:
构建模块,用于构建人腺病毒分型数据库,对人腺病毒进行分类,确定每个类别中对应人腺病毒的分类单元以及每个分类单元的全基因组/标记基因序列;
解析模块,用于获取目标临床感染样本,解析出所述目标临床感染样本的宏基因组原始序列;
第一确定模块,用于将所述宏基因组原始序列进行预处理,并输入到所述人腺病毒分型数据库进行检索计算以确定目标临床感染样本中目标人腺病毒分类单元;
第二确定模块,用于当确认目标临床感染样本中有目标人腺病毒分类单元时,确定目标人腺病毒分类单元对应的目标临床特征;
生成模块,用于根据目标人腺病毒分类单元和目标临床特征生成人腺病毒分型鉴定报告。
本发明的其它特征和优点将在随后的说明书中阐述,并且,部分地从说明书中变得显而易见,或者通过实施本发明而了解。本发明的目的和其他优点可通过在所写的说明书以及附图中所特别指出的结构来实现和获得。
下面通过附图和实施例,对本发明的技术方案做进一步的详细描述。
附图说明
附图用来提供对本发明的进一步理解,并且构成说明书的一部分,与本发明的实施例一起用于解释本发明,并不构成对本发明的限制。
图1为本发明所提供的一种基于宏基因组的人腺病毒分子分型和溯源方法的工作流程图;
图2为本发明所提供的一种基于宏基因组的人腺病毒分子分型和溯源方法的另一工作流程图;
图3为本发明所提供的一种基于宏基因组的人腺病毒分子分型和溯源系统的结构示意图。
具体实施方式
这里将详细地对示例性实施例进行说明,其示例表示在附图中。下面的描述涉及附图时,除非另有表示,不同附图中的相同数字表示相同或相似的要素。以下示例性实施例中所描述的实施方式并不代表与本公开相一致的所有实施方式。相反,它们仅是与如所附权利要求书中所详述的、本公开的一些方面相一致的装置和方法的例子。
人腺病毒(Human adenovirus,HAdV)属于腺病毒科(Adenoviridae)哺乳动物腺病毒属(Mastadenovirus),基因组全长约34.7kb,属于dsDNA,可感染多种黏膜组织,如胃肠道、呼吸道、泌尿生殖道及眼角结膜等,导致自限性的黏膜感染甚至严重的致死性感染。HAdV表面有3种主要的囊膜蛋白是构成囊膜的重要成分,也是用于诊断的重要抗原。在基因学上,编码这三类蛋白的区域是病毒基因组中变异最大的区域,是人腺病毒基因研究的热点区域。根据血凝、纤维基因长度、基因组GC含量等免疫学、生物学和生物化学特征,人腺病毒分为7个亚型/种:HAdV-A~G,又分为100种以上基因型/血清型。特定的HAdV亚型和基因型与特定的疾病、流行病学环境和人口风险组群相关。HAdV-1~7、11、14、21、35、55均与呼吸道疾病相关,且HAdV-7、35、55致病性较强。HAdV-7可分为多种基因型(如HAdV-7a等),其中7d基因型最常造成严重的感染。HAdV-40、41、52与胃肠炎相关,HAdV-4、8、9、19、37、53、54、56、64与流行性角膜结膜炎相关。对人腺病毒进行分型不仅在临床诊断、治疗和预后,也在监测人腺病毒流行和迁移等公卫领域有着重要的意义。
目前人腺病毒鉴定和分型方法包括:病毒分离鉴定、免疫学方法、核酸检测以及基于二代测序的扩增子测序、全基因组测序,但这些方法存在病毒培养、对样本的先验认知等局限性,或难以全面覆盖在临床上有重要意义的基因型。近年开始逐渐发展的宏基因组测序技术(Metagenomic sequencing),以特定生境中的整个微生物群落作为研究对象,直接提取临床样本的全部微生物组的核酸进行测序注释和比对分析。该技术弥补了以上方法的不足,无需培养,无需样本的先验知识,较全面的覆盖人腺病毒的各个亚型和基因型,但是目前主要应用于科学研究领域,其策略为测序reads(读序)直接比对参考基因组,基于比对质量(一致性位点百分数和比对reads数)进行分型,但当测序reads较短时,存在假阳性率较高的问题;或者将reads组装为全基因组后比对/进化分析以分型,但需要以样本有较高病毒丰度为前提,不适用于微量/痕量样本的鉴定分型。我国需要更灵敏、更全面的临床重要人腺病毒宏基因组分型技术,对微量/痕量的病毒样本进行亚型和基因型分型鉴定,满足临床人腺病毒诊断、治疗和流行病学病毒溯源的需求。为了解决上述问题,本实施例公开了一种基于宏基因组的人腺病毒分子分型和溯源方法。
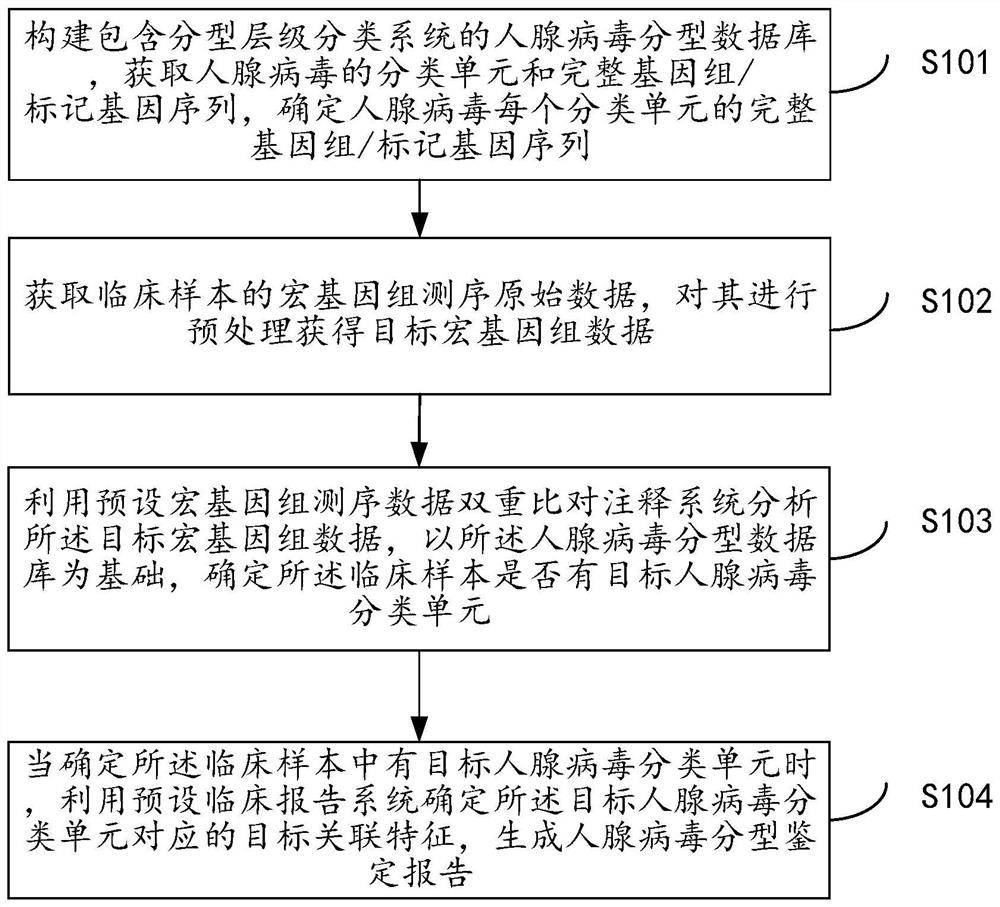
一种基于宏基因组的人腺病毒分子分型和溯源方法,如图1所示,包括以下步骤:
步骤S101、构建包含分型层级分类系统的人腺病毒分型数据库,获取人腺病毒的分类单元和完整基因组/标记基因序列,确定人腺病毒每个分类单元的完整基因组/标记基因序列;
步骤S102、获取临床样本的宏基因组测序原始数据,对其进行预处理获得目标宏基因组数据;
步骤S103、利用预设宏基因组测序数据双重比对注释系统分析所述目标宏基因组数据,以所述人腺病毒分型数据库为基础,确定所述临床样本是否有目标人腺病毒分类单元;
步骤S104、当确定所述临床样本中有目标人腺病毒分类单元时,利用预设临床报告系统确定所述目标人腺病毒分类单元对应的目标关联特征,生成人腺病毒分型鉴定报告;
在本实施例中,上述临床样本包括:人类脑脊液、血液、胸水、腹水、肺泡灌洗液等人腺病毒感染或疑似人腺病毒感染的临床样本。
上述技术方案的工作原理为:构建包含分型层级分类系统的人腺病毒分型数据库,获取人腺病毒的分类单元和完整基因组/标记基因序列,确定人腺病毒每个分类单元的完整基因组/标记基因序列,获取临床样本的宏基因组测序原始数据,对其进行预处理获得目标宏基因组数据,利用预设宏基因组测序数据双重比对注释系统分析所述目标宏基因组数据,以所述人腺病毒分型数据库为基础,确定所述临床样本是否有目标人腺病毒分类单元,当确定所述临床样本中有目标人腺病毒分类单元时,利用预设临床报告系统确定所述目标人腺病毒分类单元对应的目标关联特征,生成人腺病毒分型鉴定报告。
上述技术方案的有益效果为:通过全面的包含分型层级分类系统的人腺病毒分型数据库和基于宏基因组测序数据的双重比对注释系统,对病毒含量较低的目标临床样本进行人腺病毒的分型鉴定,克服了目前现有技术与方法的多方面局限性,能够对病毒含量偏低的临床样本进行亚型和基因型分型鉴定,可一次性对人腺病毒的全部临床重要分型进行鉴定,减少额外筛查时间,快速地生成人腺病毒分型鉴定报告,可以帮助医生及时进行诊断、治疗和预后,解决了现有技术中病毒培养、对样本的先验认知等局限性,或难以全面覆盖在临床上有重要意义的基因型的问题,以及目前人腺病毒的宏基因组测序分型需要测序reads较长、样本中病毒含量较高,通过组装、比对策略对病毒进行种水平的注释,且不能提供基因型水平的分型鉴定,故而无法对病毒含量较低的临床样本进行较全面的人腺病毒分型鉴定的问题。
在一个实施例中,所述构建包含分型层级分类系统的人腺病毒分型数据库,获取人腺病毒的分类单元和完整基因组/标记基因序列,确定人腺病毒每个分类单元的完整基因组/标记基因序列,包括:
从NCBI Taxonomy数据库中提取人腺病毒亚型及基因型名称,建立第一人腺病毒分类单元列表;
按HAdV分型格式标准化分类单元命名,合并不规范的分类单元,构建分型层级分类系统;
从当前公共数据库中(NCBI GenBank、RefSeq)下载人腺病毒的完整基因序列及注释信息,获得第一基因序列集;
应用正则公式提取所述完整基因序列的注释信息中分类单元关键字进行序列分类单元注释,并基于分类单元合法性、序列质量合法性进行过滤,获得第二基因序列集;
将所述第二基因序列集中的序列进行聚类,过滤异常序列,获得第三基因序列集;
基于所述第三基因序列集中序列的分类单元,获得第一人腺病毒分类单元列表,去除所述第一人腺病毒分类单元列表中缺乏有效参考基因序列的第一分类单元,并补充未纳入的具有有效参考基因序列的第二分类单元,获得第二人腺病毒分类单元列表;
将所述第三基因序列集的分类单元注释与所述第二人腺病毒分类单元列表进行校对与标准化,获得人腺病毒分型层级中每个层级结构中的分类单元及其基因序列;
将所述每个层级结构中的人腺病毒分类单元以及基因序列进行存储,并建立人腺病毒分类单元基因参考序列索引,获得目标人腺病毒分型数据库;
在本实施例中,上述HAdV分型命名规则如下:
第1级为大类(Human mastadenovirus A、B、C、D、E、F、G);
第1.5级为次级大类(Human adenovirus B1、B2、D10等);
第2级为基因型(可选1位字母+1-3位数字);
第3级为下级基因型(基因型+1位字母)
第4级为下下级基因型(下级基因型+1位数字)。
在本实施例中,上述包含分型层级分类系统的人腺病毒分型数据库中,包括但不限于95个人腺病毒分类单元(7个亚型+88个基因型);
上述技术方案的有益效果为:通过构建全面、经过校对的人腺病毒分型层级数据库,包含了目前临床重要的人腺病毒亚型和基因型的基因组和标记基因序列,以及校对后的分型注释信息,同时,通过在所述目标人腺病毒分型数据库中建立人腺病毒各个层级结构中分类单元的参考基因序列索引可以将人腺病毒的各个分类单元与基因组/标记基因序列关联,相比于NCBI Taxonomy,使分类单元的特征信息更为全面和准确的分类结构,也覆盖了Taxonomy尚未纳入的分类单元,在人腺病毒分型层级数据库中标准化了人腺病毒的分型命名规则,更方便查找和定位各个基因型。
在一个实施例中,所述基因序列包括:基因组和标记基因序列。
在一个实施例中,所述获取临床样本的宏基因组测序原始数据,对其进行预处理获得目标宏基因组数据,包括:
过滤所述宏基因组原始测序数据中质量值低于2,碱基数占整个read的40%的第一reads;
切除所述宏基因组原始测序数据中特定滑窗内平均质量小于20的碱基;
过滤所述宏基因组原始测序数据中平均质量小于20的第二reads、含N数量大于5的第三reads以及长度小于50的第四reads。
上述技术方案的有益效果为:通过对宏基因组测序原始数据进行数据质控可以去除掉宏基因组原始数据中的无用干扰数据,进而后续可以更加准确地根据高质量的目标宏基因组数据确定目标临床感染样本的人腺病毒分类单元,提高了输入数据的准确性和分型鉴定的可靠性。
在一个实施例中,如图2所示,所述利用预设宏基因组测序数据双重比对注释系统分析所述目标宏基因组数据,以所述人腺病毒分型数据库为基础,确定所述临床样本是否有目标人腺病毒分类单元,包括:
步骤S201、将所述目标人腺病毒分型数据库中的基因序列作为参考序列;
步骤S202、构建将宏基因组数据与参考序列进行比对,确定匹配的参考序列的算法流程;
步骤S203、确定根据所述匹配的参考序列,基于目标人腺病毒分型数据库中的参考序列索引,确定目标人腺病毒分类单元的匹配流程;
步骤S204、将所述算法流程和匹配流程构建为宏基因组测序数据双重比对注释系统。
步骤S205、将所述目标宏基因组数据输入到所述预设宏基因组测序数据双重比对注释系统,确定临床样本是否有目标人腺病毒分类单元。
在本实施例中,上述算法流程包括以基因组序列为参考序列的WhScore算法和UniScore算法流程,和以标记基因序列为参考序列的UniScore算法流程,其中WhScore算法公式如下:
WhScore=max(∑ASscore(1),…,∑ASscore(i))
其中:ASscore=∑(identities,mismatches)-∑(gap penalties);
i=参考基因组或基因序列的总数;
UniScore算法公式如下:
UniScore=∑ASscore(unique reads)/(Coverage of genome)
其中:unique reads(特异性读序)定义为对齐分数最高的单一类别参考序列(基因组/基因)有且仅有一个的读序。
在本实施例中,所述匹配流程包括:应用匹配的基因组参考序列的分类单元确定分型,和应用匹配的标记参考基因序列的分类单元确定分型。
上述技术方案的有益效果为:基于WhScore算法和WhScore算法,以及完整基因组参考序列和标记基因参考序列的双重比对策略,提高分型结果的敏感性,克服了现有相关技术分型鉴定的局限性,尤其是样本中病毒含量较低时可实现分型鉴定,基于大样本分析后的比对得分加权算法,提高比对的准确性,可以综合地评估出目标临床样本所感染的具体的人腺病毒的亚型/基因型。
在一个实施例中,所述当确定所述临床样本中有目标人腺病毒分类单元时,利用预设临床报告系统确定所述目标人腺病毒分类单元对应的目标关联特征,生成人腺病毒分型鉴定报告,包括:
通过文献收集和临床大样本的挖掘,构建不同人腺病毒分类单元和相关特征的关联表,利用所述不同人腺病毒分类单元和相关特征的关联表构建所述预设临床报告系统;
在所述关联表中搜索所述目标人腺病毒分类单元,确定所述目标人腺病毒分类单元对应的目标关联特征;
自动生成分型结果,包含注释的目标分类单元(物种拉丁名、物种中文名、亚型/基因型名称、该亚型/基因型的相关特征、支持得分);
将数据库的客户信息导入报告模板,将所述分型结果导入所述报告模板的相应表格;
以PDF格式生成最终的人腺病毒分型鉴定报告。
上述技术方案的有益效果为:通过构建不同人腺病毒分类单元和相关特征的关联表,尤其是临床相关的特征,可以直接从关联表中获得目标人腺病毒分类单元对应的目标特征,以便为医生评估临床诊断、治疗与预后提供参考,有着较高可靠性和实用性。自动化报告系统快速生成报告,包括目标临床样本所感染的人腺病毒的亚型/基因型,以及该分类单元的相关特征,支持该分类单元匹配的得分等,帮助医生及时进行诊断、治疗和预后。
在一个实施例中,确定所述临床样本是否有目标人腺病毒分类单元的步骤包括:
应用已知样本数据中正常标签数据及其相应的病毒分子分型类别的结果,确定病毒分型模型的主要参数组及其阈值,应用该模型对未确定样本中的正常标签数据进行预测,获得预测的分型结果;
正常标签数据分型模型相关参数组的似然函数定义:
其中,L(θ)为似然函数,
其中,Ad
θ为分布模型参数组:
θ={φ
其中,φ
转换式(1)、(2)为下面形式:
其中,
估算第k次的参数组并统计来源于分型对象Ad
假设潜在变量
上式转化为:
优化似然函数为:
其中,F为第k次的似然函数,
设定Y
循环迭代,每次最大化第k轮的
其中,
ρ表示标签数据来源测序平台的错误率,δ表示标签数据错误来源比对算法的错误率,j为read标签数据;
重复迭代(默认20循环,初始参数为随机产生),直至
上述技术方案的有益效果为:通过对获取的目标临床感染样本的宏基因组原始序列,以EM算法(Expectation-maximization)为基础为基础,构建正常标签数据分型模型,应用已知样本数据中正常标签数据及其相应的病毒分子分型类别的结果,确定病毒分型模型的主要参数组及其阈值,计算序列匹配到人腺病毒分型数据库中分型对象基因组/标记基因上的碱基比率、错配碱基比率、漏配概率等,并进行循环迭代,确定并预测该原始序列所属人腺病毒分类单元基因组/标记基因,提高分型结果的敏感性和准确性,克服了现有相关技术分型鉴定的局限性,尤其是样本中病毒含量较低时可实现人腺病毒的亚型/基因型鉴定。
本实施例还公开了一种种基于宏基因组的人腺病毒分子分型和溯源系统,如图3所示,该系统包括:
构建模块301,用于构建人腺病毒分型数据库,对人腺病毒进行分类,确定每个类别中对应人腺病毒的分类单元以及每个分类单元的全基因组/标记基因序列;
解析模块302,用于获取目标临床感染样本,解析出所述目标临床感染样本的宏基因组原始序列;
第一确定模块303,用于将所述宏基因组原始序列进行预处理,并输入到所述人腺病毒分型数据库进行检索计算以确定目标临床感染样本中目标人腺病毒分类单元;
第二确定模块304,用于当确认目标临床感染样本中有目标人腺病毒分类单元时,确定目标人腺病毒分类单元对应的目标临床特征;
生成模块305,用于根据目标人腺病毒分类单元和目标临床特征生成人腺病毒分型鉴定报告。
上述技术方案的工作原理及有益效果在方法权利要求中已经说明,此处不再赘述。
本领域技术用户员在考虑说明书及实践这里公开的公开后,将容易想到本公开的其它实施方案。本申请旨在涵盖本公开的任何变型、用途或者适应性变化,这些变型、用途或者适应性变化遵循本公开的一般性原理并包括本公开未公开的本技术领域中的公知常识或惯用技术手段。说明书和实施例仅被视为示例性的,本公开的真正范围和精神由下面的权利要求指出。
应当理解的是,本公开并不局限于上面已经描述并在附图中示出的精确结构,并且可以在不脱离其范围进行各种修改和改变。本公开的范围仅由所附的权利要求来限制。
- 一种基于宏基因组的人腺病毒分子分型和溯源方法及系统
- 一种基于宏基因组的人腺病毒分子分型和溯源方法及系统