用于制备3-5-氨基-4-(3-氰基苯甲酰基)-吡唑化合物的合成方法
文献发布时间:2023-06-19 11:05:16
发明领域
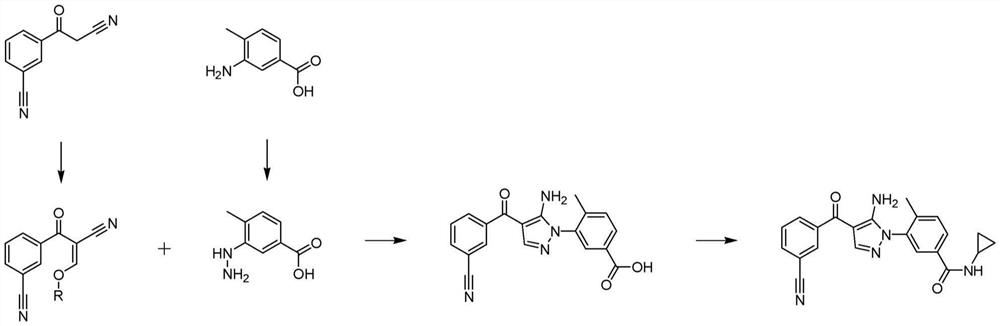
本发明公开了新化合物3-[5-氨基-4-(3-氰基苯甲酰基)-1H-吡唑-1-基]-4-甲基苯甲酸及其盐和溶剂化物、制备3-[5-氨基-4-(3-氰基苯甲酰基)-1H-吡唑-1-基]-4-甲基苯甲酸的方法及其在合成3-[5-氨基-4-(3-氰基苯甲酰基)-吡唑-1-基]-N-环丙基-4-甲基苯甲酰胺中的用途。
发明背景
化合物3-[5-氨基-4-(3-氰基苯甲酰基)-吡唑-1-基]-N-环丙基-4-甲基苯甲酰胺首先公开于国际专利申请WO2005/009973中,其中多种其它吡唑类和咪唑类化合物具有细胞因子抑制活性。WO2005/009973公开了这样的化合物可用于治疗与p38激酶,尤其是p38α激酶和p38β激酶相关的病况,包括慢性阻塞性肺病。WO2005/009973公开了3-[5-氨基-4-(3-氰基苯甲酰基)-吡唑-1-基]-N-环丙基-4-甲基苯甲酰胺作为一种这样的新的吡唑类p38激酶抑制剂。3-[5-氨基-4-(3-氰基苯甲酰基)-吡唑-1-基]-N-环丙基-4-甲基苯甲酰胺具有以下化学结构:
WO2005/009973描述了用于制备3-[5-氨基-4-(3-氰基苯甲酰基)-吡唑-1-基]-N-环丙基-4-甲基苯甲酰胺的方法。然而,上述方法没有考虑到商业规模的生产来设计。因此,先前公开的合成存在几个问题,这些问题使得所述合成不适于3-[5-氨基-4-(3-氰基苯甲酰基)-吡唑-1-基]-N-环丙基-4-甲基苯甲酰胺的商业规模生产。这类问题包括有害的或危险的副产物的产生,反应的不良控制,例如由于高度放热的反应热力学或过度的动力学,复杂的废物流管理,复杂的纯化工序,不可接受的不纯的产物和不良的产率。
迄今为止,没有适于商业规模的用于生产3-[5-氨基-4-(3-氰基苯甲酰基)-吡唑-1-基]-N-环丙基-4-甲基苯甲酰胺的简单方法。因此,尚未满足提供用于生产3-[5-氨基-4-(3-氰基苯甲酰基)-吡唑-1-基]-N-环丙基-4-甲基苯甲酰胺的有效方法和适用于商业规模的有用中间体化合物的需要。
发明概述
本发明提供了用于制备化合物的方法,所述方法包括以下步骤:
a)使式A化合物
与化合物3反应
以提供化合物5或其盐或其溶剂化物;
b)任选地分离所述化合物5或其盐或其溶剂化物;以及
c)任选地使所述化合物5或其盐或其溶剂化物与环丙胺反应以提供化合物6或其盐或其溶剂化物;
其中R是直链或支链的C1-C5烷基。
本发明还提供了通过上述方法获得的产物。
本发明还提供所述化合物5或其盐或其溶剂化物。
本发明还提供了通过上述方法获得的产物或化合物5作为中间体化合物在合成3-[5-氨基-4-(3-氰基苯甲酰基)-吡唑-1-基]-N-环丙基-4-甲基苯甲酰胺中的用途。
附图的简要说明
通过参考附图,可以更清楚地理解本文描述的实施方案的某些方面,这些附图旨在示例性说明本发明而不是限制本发明,并且其中:
图1是显示根据本发明将化合物1转化为式A化合物的示例性方法的方案。
图2是显示根据本发明将化合物4转化为化合物3的示例性方法的方案。首先将化合物4转化为重氮盐,然后转化为偕腙肼(hydrazyl)亚硫酸盐络合物,随后将其水解以提供化合物3。
图3是显示根据本发明由化合物3和式A化合物制备化合物5的示例性方法的方案。
图4是显示根据本发明将化合物5转化为3-[5-氨基-4-(3-氰基苯甲酰基)-吡唑-1-基]-N-环丙基-4-甲基苯甲酰胺的示例性方法的方案。
图5是显示根据本发明的3-[5-氨基-4-(3-氰基苯甲酰基)-吡唑-1-基]-N-环丙基-4-甲基苯甲酰胺的全合成的示例性方法的方案。
发明的详细说明
本发明提供了用于生产3-[5-氨基-4-(3-氰基苯甲酰基)-吡唑-1-基]-N-环丙基-4-甲基苯甲酰胺的方法和中间体化合物。应理解,所述方法可与本文未描述的其它步骤和方法组合以提供3-[5-氨基-4-(3-氰基苯甲酰基)-吡唑-1-基]-N-环丙基-4-甲基苯甲酰胺。还应理解,下文描述的方法可组合以提供3-[5-氨基-4-(3-氰基苯甲酰基)-吡唑-1-基]-N-环丙基-4-甲基苯甲酰胺的完整的合成路线。
应理解,当本文提及特定化合物如式A、1、2、3、4、5或6的化合物时,除非上下文另有要求,否则其盐及其溶剂化物旨在在本发明的范围内。
用于本发明的任何方面的起始材料可以是任何来源的。例如,起始材料可以是来自先前反应的粗产物,或起始材料可以是纯产物,其可商购获得或通过纯化来自先前反应的粗产物。
对于所述的所有合成步骤,在常规后处理之后,如果需要,可以通过常规纯化方法,例如色谱法、研磨、结晶或制备型HPLC来纯化粗产物。
本发明的方法优选用于材料的大规模生产(大于5kg)。
如本文所用,纯的或高纯的意指所述材料的纯度大于80wt%、或大于90wt%、优选大于95wt%,例如大于98wt%。本领域技术人员知晓用于测定纯度的方法。例如,纯度可以通过HPLC测定,例如含量测定(assay)(%w/w)或面积百分比(面积%),并且通常采用使用水基流动相和甲醇或乙腈的反相色谱法。
应当理解,当根据本发明的方法提及将一种组分添加到另一种组分的步骤时,除非上下文另有要求,否则将任一组分添加到另一种组分旨在包括在本发明的范围内。
根据本发明,式A化合物或其盐或其溶剂化物
可以通过包括以下步骤的方法制备,
a)使化合物1
与原甲酸三烷基酯反应以提供式A化合物或其盐或其溶剂化物,
其中R是所述原甲酸三烷基酯的烷基部分。
式A化合物可以从反应物料中分离,或者粗产物,例如在溶液中,可以用于随后的反应。例如,粗产物可用于式A化合物与化合物3之间的反应,如下文详细描述。
(3-肼基-4-甲基-苯甲酸)
根据本发明,式A的烷氧基亚甲基可以通过使化合物1与原甲酸三烷基酯反应来制备。根据式A的化合物是在制备3-[5-氨基-4-(3-氰基苯甲酰基)-吡唑-1-基]-N-环丙基-4-甲基苯甲酰胺中有用的中间体化合物。式A化合物与化合物3的环化产生关键的吡唑中间体化合物5和醇副产物。
(3-[5-氨基-4-(3-氰基苯甲酰基)-1H-吡唑-1-基]-4-甲基苯甲酸)
已发现提供与化合物5类似的吡唑的先前方法(例如WO2005/009973中所公开的那些方法)产生有害的副产物,例如苯胺。通过使用原甲酸三烷基酯产生式A的烷氧基亚甲基,随后式A化合物与化合物3的环化消除醇,其在安全性方面是优选的副产物。
提供与化合物5类似的吡唑的先前方法的另一个问题是环化反应的控制。已发现WO2005/009973中公开的产生吡唑的环化反应涉及急剧的放热反应曲线,这可能使得该反应不适于商业规模生产。通过提供具有较慢速率的反应,本发明解决了这个问题,该反应易于控制并避免了与急剧的放热反应曲线有关的安全问题。此外,较慢的反应速率是合乎需要的,因为它提供了增强的反应控制并减少了杂质的产生。
因此,本发明能够以商业规模安全地生产关键的中间体化合物5和3-[5-氨基-4-(3-氰基苯甲酰基)-吡唑-1-基]-N-环丙基-4-甲基苯甲酰胺。
使化合物1与原甲酸三烷基酯反应以提供式A化合物可包括一个或多个或所有的以下步骤:
a)将化合物1加入到非质子溶剂中;
b)任选地将有机酸酐加入到反应物料中;
c)蒸馏所述反应物料;
d)在蒸馏期间将原甲酸三烷基酯添加到反应物料中以提供式A化合物;
e)任选地将反应物料冷却,优选地冷却至小于约25℃;
f)任选地将反溶剂添加至所述反应物料中;
g)任选地将所述反应物料进一步冷却至约0℃至约5℃;
h)任选地过滤所述式A化合物,
i)任选地用所述反溶剂洗涤滤液;以及
j)任选地干燥所述滤液。
在一个实施方案中,分离式A化合物包括上述步骤(h)-(j)。
化合物1与原甲酸三烷基酯的反应可以产生醇副产物。醇副产物可以从反应中使用的原甲酸三烷基酯预测,例如原甲酸三乙酯可以产生乙醇作为副产物醇。优选在原甲酸三烷基酯与化合物1的反应期间去除醇副产物。去除醇副产物促进了反应向化合物2的转化。已经发现,去除醇副产物可以提高产率。在一个实施方案中,通过在非质子溶剂中蒸馏含有化合物1的反应物料并在蒸馏过程中将原甲酸三烷基酯加入到反应物料中来实现醇副产物的去除。在另一个实施方案中,通过向反应物料中加入有机酸酐如乙酸酐来实现醇副产物的去除。可以组合这些实施方案以进一步增强醇副产物的去除以允许进一步提高产率。
有机酸酐是由与同一氧原子键合的两个酰基组成的化合物,即酰基-O-酰基。本文考虑分别具有相同酰基和不同酰基的对称酸酐和混合酸酐。有机酸酐可以选自乙酸酐、丙酸酐、丁酸酐、乙酸丙酸酐和乙酸丁酸酐,优选为乙酸酐。
原甲酸三烷基酯可以是原甲酸直链或支链的C1-C5三烷基酯,在这种情况下,R可以是直链或支链的C1-C5烷基。原甲酸三烷基酯可以选自原甲酸三甲酯、原甲酸三乙酯、原甲酸三丙酯、原甲酸三丁酯和原甲酸三戊酯。
原甲酸三烷基酯可以是原甲酸三乙酯,在这种情况下,式A是化合物2。
(3-[2-氰基-2-(乙氧基亚甲基)乙酰基]苄腈)
原甲酸三烷基酯如原甲酸三乙酯与化合物1的反应可以产生醇如乙醇作为副产物。在该反应中,乙醇是优选的副产物,因为它是高度挥发性的并且与某些溶剂如甲苯和环己烷形成共沸混合物,这促进乙醇的消除和式A化合物的纯化。原甲酸三乙酯的使用导致与现有方法相比提高的产率。
此外,原甲酸三乙酯是优选的试剂,因为随后的化合物2与化合物3的环化产生关键的吡唑中间体化合物5和作为副产物的乙醇。乙醇是优选的副产物,因为它是高度挥发性的,这促进乙醇的消除和化合物5的纯化。化合物2的使用导致与现有方法相比提高的产率。在产物污染的情况下,乙醇也具有可接受的毒理学特性。
非质子溶剂是不为氢键供体的溶剂。非质子溶剂可选自甲苯、环己烷和二甲苯。原甲酸三烷基酯如原甲酸三乙酯与化合物1的反应可以产生醇如乙醇作为副产物。非质子溶剂优选与醇副产物形成共沸混合物。这促进醇副产物的消除和式A化合物的纯化。由于醇副产物可以从反应中使用的原甲酸三烷基酯预测,例如原甲酸三乙酯可以产生乙醇作为副产物醇,本领域技术人员将能够选择与副产物醇形成共沸混合物的合适的非质子溶剂。优选地,非质子溶剂是甲苯或环己烷,特别是当使用原甲酸三乙酯时。已知甲苯和环己烷与乙醇形成共沸混合物。发明人已经发现,形成共沸混合物促进乙醇的消除和化合物2的纯化。此外,醇副产物的去除促进了反应向化合物2的转化。优选地,非质子溶剂是甲苯。
蒸馏反应物料的步骤可以包括在约60℃至约130℃下在真空下(例如在约500mBar至约1Bar下)蒸馏反应物料。优选地,反应物料在约105℃和约800mBar至约900mBar下蒸馏。应当理解,本领域技术人员将能够选择用于蒸馏的合适条件,并且本发明中的蒸馏包括回流。
如本文所用,反溶剂是式A化合物不溶或难溶于其中的溶剂,例如小于5wt%或小于1wt%或甚至小于0.1wt%可溶。应当理解,本领域技术人员将能够选择用于给定的式A化合物的合适的反溶剂。所述反溶剂可以选自戊烷、己烷、环己烷、庚烷及它们的蒸馏馏分,并且除非上下文另有要求,否则其所有异构体都旨在包括在本发明的范围内。优选地,反溶剂是正庚烷。
根据本发明,可以通过上述方法获得产物。
根据本发明,提供了化合物2、3-[2-氰基-2-(乙氧基亚甲基)乙酰基]苄腈或其盐或其溶剂化物。
本发明还提供了通过上述方法获得的产物或化合物2作为中间体化合物在合成3-[5-氨基-4-(3-氰基苯甲酰基)-吡唑-1-基]-N-环丙基-4-甲基苯甲酰胺中的用途。
根据本发明,化合物3或其盐或其溶剂化物
可以通过包括以下步骤的方法制备
a)将化合物4与无机酸混合;
b)向反应物料中加入亚硝酸盐或有机亚硝酸酯衍生物以形成重氮盐;
c)向所述反应物料中加入含硫的还原剂以形成偕腙肼硫络合物,例如偕腙肼亚硫酸盐络合物;
d)水解偕腙肼硫络合物以提供化合物3;以及
e)任选地分离化合物3或其盐或其溶剂化物。
化合物3可以从反应物料中分离。例如,可以纯化粗产物以产生纯形式的化合物3,其可用于式A化合物与化合物3之间的反应,如下文详细描述。
在一个实施方案中,分离化合物3包括过滤化合物3,用水洗涤滤液和任选地干燥滤液。
过滤方法是本领域技术人员已知的。示例性方法包括过滤器干燥装置、离心过滤和膜过滤。
根据本发明,使用亚硝酸盐或有机亚硝酸酯衍生物以形成重氮盐,随后通过含硫的还原剂将重氮盐还原以形成偕腙肼硫络合物,例如偕腙肼亚硫酸盐络合物,将化合物4转化成化合物3。偕腙肼硫络合物的水解提供化合物3。化合物3是合成3-[5-氨基-4-(3-氰基苯甲酰基)-吡唑-1-基]-N-环丙基-4-甲基苯甲酰胺的关键中间体化合物。已发现先前可获得的制备化合物3的方法太复杂且昂贵以至于不适于放大。因此,尚未满足提供适于商业规模的制备化合物3的方法的需要。
亚硝酸盐如亚硝酸钠和有机亚硝酸酯衍生物如亚硝酸烷基酯适合用于商业规模。亚硝酸盐是容易获得的试剂,并且在最终产物污染的情况下具有可接受的毒理学特性。
一些先前的将苯胺衍生物转化成肼衍生物的方法,例如那些使用三苯基膦的方法,由于在处理那些方法的废物流方面的复杂性而不适于商业规模。例如,含磷废物的负面影响,例如富营养化,已被充分证明。本发明通过提供不需要使用含磷试剂来提供化合物3的方法来解决该问题。
本发明人还发现先前的将苯胺衍生物转化成肼衍生物的方法需要复杂的分离和纯化步骤以提供可接受的纯产物。据信这些低效率降低了所需产物的产率。使用含硫的还原剂(例如亚硫酸钠)和亚硝酸盐或有机亚硝酸酯衍生物(例如亚硝酸钠)提供了化合物3的简便合成法,其产生在商业规模上可管理的废物流。此外,发现根据本发明的方法产生被有效去除的副产物,得到高纯度产物。
在化合物3-[5-氨基-4-(3-氰基苯甲酰基)-吡唑-1-基]-N-环丙基-4-甲基苯甲酰胺的先前合成中,使用氯化亚锡将化合物4转化为化合物3。然而,发现使用氯化亚锡会导致含锡杂质的污染,该杂质必须被去除以提供可接受的产物。含锡杂质的去除可以通过数种方法进行,包括在硅胶上过滤。然而,发现额外的纯化步骤导致产物产率的降低。本发明要求保护的方法允许从3-[5-氨基-4-(3-氰基苯甲酰基)-吡唑-1-基]-N-环丙基-4-甲基苯甲酰胺的总合成中消除氯化亚锡,从而消除含锡杂质污染最终产物的风险。除了在减少污染和提高产品安全性方面的益处之外,本发明要求保护的方法还允许通过减少产品损失(例如归因于在先前合成中使用的额外的纯化步骤)来提高产率。
化合物3或其盐或其溶剂化物可以通过包括一个或多个或所有的以下步骤的方法制备
a)将化合物4与无机酸混合;
b)任选地将反应物料冷却,优选冷却至小于约10℃;
c)将亚硝酸盐或有机亚硝酸酯衍生物加入到反应物料中以形成重氮盐,优选在小于约10℃的温度下;
d)将含硫的还原剂加入到反应物料中,优选在小于约10℃的温度下;
e)任选地加热反应物料,优选加热至大于约50℃,更优选加热至约60℃;
f)任选地进一步加热反应物料至大于约60℃,优选加热至约80℃;
g)水解偕腙肼硫络合物以提供化合物3,优选地通过向反应物料中加入无机酸来实现水解;
h)任选地将反应物料冷却,优选冷却至小于约30℃,优选冷却至约20℃至约30℃,更优选冷却至约25℃;
i)任选地将反应物料的pH调节至约5至约7,优选约5.6至约5.8;
j)任选地过滤化合物3;
k)任选地用水洗涤滤液;
l)任选地干燥滤液;
m)任选地将干燥的滤液在水中重新浆化(混合);
n)任选地分离化合物3;
o)任选地用水洗涤化合物3;以及
p)任选地干燥化合物3。
无机酸可以选自HCl、H
在反应中,化合物4与亚硝酸盐源反应以形成重氮盐。亚硝酸盐源为硝酸盐或有机亚硝酸酯衍生物。
亚硝酸盐可以选自碱金属亚硝酸盐、碱土金属亚硝酸盐和亚硝酸银。示例性的碱金属亚硝酸盐包括亚硝酸锂、亚硝酸钠和亚硝酸钾。示例性的碱土金属亚硝酸盐包括亚硝酸镁和亚硝酸钙。
有机亚硝酸酯衍生物是具有式R-ONO的有机化合物。有机亚硝酸酯衍生物可以是亚硝酸烷基酯,例如直链或支链C1-C5亚硝酸酯。有机亚硝酸酯衍生物可以选自亚硝酸乙酯、亚硝酸丙酯、亚硝酸丁酯和亚硝酸戊酯。
优选地,亚硝酸盐源是亚硝酸钠。
含硫的还原剂可以选自亚硫酸盐、亚硫酸氢盐和连二亚硫酸盐。示例性的亚硫酸盐包括碱金属亚硫酸盐,例如亚硫酸锂、亚硫酸钠和亚硫酸钾;碱土金属亚硫酸盐,例如亚硫酸镁和亚硫酸钙;和亚硫酸银。示例性的亚硫酸氢盐包括碱金属亚硫酸氢盐,例如亚硫酸氢钠和亚硫酸氢钾;和碱土金属亚硫酸氢盐,例如亚硫酸氢钙。示例性的连二亚硫酸盐是连二亚硫酸钠。
优选地,含硫的还原剂是亚硫酸钠。
在一个实施方案中,在水解偕腙肼硫络合物以提供化合物3之后,将反应物料的pH调节至约5至约7,优选约5.6至约5.8。不希望受理论束缚,据信该步骤消除了某些不希望的盐副产物的产生,导致较高纯度的产物。
在一个实施方案中,在将含硫的还原剂添加至反应物料之后,将反应物料加热,优选加热至大于约50℃,更优选加热至约60℃。随后可将反应物料进一步加热至大于约60℃,优选加热至约80℃。随后可将反应物料冷却,优选冷却至小于约30℃,优选冷却至约20℃至约30℃,更优选冷却至约25℃。发现在分离化合物3之前,例如在调节反应物料的pH之前,将反应物料冷却至约20℃至约30℃,导致改善的产物纯度。不希望受理论束缚,据信该步骤消除了某些不希望的盐副产物的产生,导致较高纯度的产物。
在一个实施方案中,将粗滤液在水中重新浆化(混合),然后分离化合物3,用水洗涤并任选地干燥。在该实施方案中,分离化合物3可以包括过滤化合物3。本发明人已经发现,该额外的处理步骤显著改善了杂质如盐副产物的去除,导致较高纯度的产物。
根据本发明,可以通过上述方法获得产物。
本发明还提供了通过上述方法获得的产物作为中间体化合物在合成3-[5-氨基-4-(3-氰基苯甲酰基)-吡唑-1-基]-N-环丙基-4-甲基苯甲酰胺中的用途。
根据本发明,化合物可以通过包括以下步骤的方法制备
a)使式A化合物
与化合物3反应
以提供化合物5或其盐或其溶剂化物,
其中R是直链或支链的C1-C5烷基。
所述方法还可以包括以下步骤中的一个或两个,
b)分离化合物5或其盐或其溶剂化物;以及
c)使化合物5与环丙胺反应以提供化合物6或其盐或其溶剂化物。
(3-[5-氨基-4-(3-氰基苯甲酰基)-吡唑-1-基]-N-环丙基-4-甲基苯甲酰胺)
在一个实施方案中,分离化合物5包括过滤化合物5,用水洗涤滤液并干燥滤液。分离化合物5可以还包括在用水洗涤滤液后用甲醇洗涤滤液。
式A化合物可以来自任何来源。例如,所述材料可以是来自先前反应的粗产物,或者所述材料可以是纯产物,其可商购获得或通过纯化来自先前反应的粗产物。还考虑了粗产物和纯产物的混合物。例如,式A的来源可以是如上所述的来自化合物1与原甲酸三烷基酯的反应的粗产物,或者该来源可以是纯形式的式A。应当理解,技术人员将知道用于获得先前反应的粗产物的适合的方式,用于进一步反应,例如用于一锅合成或伸缩合成。
化合物3的来源可以是纯形式的化合物3。在一个实施方案中,化合物3的来源具有通过Karl Fischer滴定测定的小于1%的含水量。化合物3的来源可商购获得,或通过纯化来自先前反应的粗产物,例如如上所述的从化合物4向化合物3的转化而获得。应理解的是,技术人员将知道用于纯化粗产物的适合的方式以适合用于进一步的反应。
根据本发明,通过使式A化合物与化合物3反应来制备化合物5。化合物5是制备3-[5-氨基-4-(3-氰基苯甲酰基)-吡唑-1-基]-N-环丙基-4-甲基苯甲酰胺的关键中间体化合物。
式A化合物与化合物3的环化产生关键的吡唑中间体化合物5和醇副产物。已发现提供与化合物5类似的吡唑的先前方法(例如WO2005/009973中所公开的那些方法)产生有害副产物,例如苯胺。式A化合物与化合物3的反应消除醇,其在安全性方面是优选的副产物。
提供与化合物5类似的吡唑的先前方法的另一个问题是环化反应的控制。已经发现WO2005/009973中公开的用于产生吡唑的环化反应涉及急剧的放热反应曲线,这可能使得该反应不适于商业规模的生产。本发明通过提供具有较慢速率的反应来解决这个问题,该反应易于控制并避免了与急剧的放热反应曲线有关的安全问题。此外,较慢的反应速率是合乎需要的,因为它提供了增强的反应控制并减少了杂质的产生。
因此,本发明要求保护的方法能够以商业规模安全生产关键的中间体化合物5和3-[5-氨基-4-(3-氰基苯甲酰基)-吡唑-1-基]-N-环丙基-4-甲基苯甲酰胺。
在一个实施方案中,R可以是C
化合物2是优选的试剂,因为化合物2与化合物3的环化产生关键的吡唑中间体化合物5和作为副产物的乙醇。乙醇是优选的副产物,因为它是高度挥发性的,这有利于其消除和中间体化合物5的纯化。不希望受理论束缚,据信化合物2的使用导致比现有方法改善的产率。在产物污染的情况下,乙醇也具有可接受的毒理学特性。
可以通过包括一个或多个或所有的以下步骤的方法制备化合物5,
a)在极性非质子溶剂中,任选地在无水条件下,任选地在大于约90℃的温度下,优选在约105℃下,将式A化合物加入到化合物3中,以提供化合物5或其盐或其溶剂化物;
b)任选地将反应物料冷却,优选冷却至小于约70℃,更优选冷却至约60℃;
c)任选地将水加入到所述反应物料中;
d)任选地进一步冷却反应物料,优选冷却至小于约30℃,更优选冷却至约20℃;
e)任选地过滤化合物5;
f)任选地用水洗涤滤液;
g)任选地用甲醇洗涤滤液;以及
h)任选地干燥滤液,
其中R是直链或支链的C1-C5烷基。
所述方法可以还包括使化合物5与环丙胺反应以提供化合物6的步骤。使化合物5与环丙胺反应以提供化合物6可以包括一个或多个或所有的以下步骤
a)将化合物5加入到极性非质子溶剂中;
b)将上述反应物料加热,优选加热至大于约30℃的温度,更优选加热至约40℃的温度;
c)任选地在极性非质子溶剂中加入偶联试剂;
d)任选地吹扫反应容器,优选以去除CO
e)向反应物料中加入环丙胺;
f)保持反应物料以提供化合物6;
g)任选地冷却,优选冷却至小于约30℃,更优选冷却至约25℃;
h)任选地向反应物料中添加水;
i)任选地过滤化合物6;
j)任选地用水洗涤滤液;以及
k)任选地干燥滤液。
在分离化合物6(其在一个实施方案中包括上述步骤(i)-(k))之后,化合物6可以随后用极性非质子溶剂/水重结晶进行重结晶。本发明人已经发现该重结晶步骤为最终产物提供了更大的产物质量一致性。
在式A与化合物3的反应和随后的化合物5向化合物6的转化中,极性非质子溶剂可以选自二甲亚砜、二甲基乙酰胺、二甲基甲酰胺和N-甲基-2-吡咯烷酮。优选地,极性非质子溶剂是二甲亚砜。二甲亚砜是容易获得的并且特别适合用于商业规模。
在一个实施方案中,可以使用的偶联剂选自CDI、HOBt和HATU,优选CDI。使用CDI的反应的原子经济性显著大于使用不同偶联剂的类似反应。此外,CDI容易获得并且特别适合用于商业规模。
根据本发明,可以通过上述方法获得产物。
根据本发明,提供了化合物5、3-[5-氨基-4-(3-氰基苯甲酰基)-1H-吡唑-1-基]-4-甲基苯甲酸,或其盐或其溶剂化物。
本发明还提供了通过上述方法获得的产物或化合物5作为中间体化合物在合成3-[5-氨基-4-(3-氰基苯甲酰基)-吡唑-1-基]-N-环丙基-4-甲基苯甲酰胺中的用途。
现在将参考以下实施例进一步描述本发明,这些实施例旨在示例性说明,而不限制所附权利要求的范围。
实施例
实施例1
相对于化合物1的引入量(1.0eq)来描述所述方法。
将乙酸酐(2.0eq)加入到3-(氰基乙酰基)苄腈(化合物1)(1.0eq)在甲苯(4.5vol)中的混合物中并加热至105℃。将压力缓慢降低至800-900mbar直至获得轻微回流。在蒸馏期间加入原甲酸三乙酯(1.5eq)。将反应随后搅拌1h。将反应冷却至25℃并将产物结晶。在0.5h内加入正庚烷(5vol)。将反应进一步冷却至小于5℃并搅拌1h。将产物滤出,用正庚烷(2×2vol)洗涤并在真空下干燥。获得3-[2-氰基-2-(乙氧基亚甲基)乙酰基]苄腈(化合物2),为红色固体,产率90%,纯度98%(HPLC,面积%)。
实施例2
相对于化合物4的引入量(1.0eq)描述所述方法。
将盐酸(30%,3.7eq)加入到3-氨基-4-甲基苯甲酸(化合物4)(1.0eq)在水(1.5vol)中的悬浮液中。将悬浮液冷却至小于10℃并搅拌0.5小时。缓慢加入亚硝酸钠(1.1eq)的水(0.85vol)溶液,同时保持温度小于10℃。将反应随后搅拌0.5小时。在保持温度小于15℃的情况下,将亚硫酸钠(4.9eq)在水(10vol)中的冷却(<5℃)的悬浮液加入到反应混合物中。将反应随后搅拌1小时。将所得混合物加热至60℃,持续1h。加入盐酸(30%,7.2eq),并将混合物在80℃下搅拌2小时。将混合物冷却至25℃。加入氢氧化钠水溶液(33%)(最终pH:5.6-5.8)。将混合物随后搅拌1小时,过滤,用水(2×4vol)洗涤并真空干燥,得到3-肼基-4-甲基-苯甲酸(化合物3),产率63%,纯度87%(HPLC,w/w%)。
将滤饼在25℃下在水(6vol)中重新浆化1h。将混合物过滤,用水(2×4vol)洗涤并在真空下干燥,得到3-肼基-4-甲基-苯甲酸(化合物3),产率61%,纯度96%(HPLC,面积%)。
实施例3
相对于化合物2的引入量(1.0eq)描述所述方法。
将3-[2-氰基-2-(乙氧基亚甲基)乙酰基]苄腈(化合物2)(1.0eq)的二甲亚砜(4vol)溶液在105℃下在1.5小时内加入到3-肼基-4-甲基-苯甲酸(化合物3)(1.2eq)的二甲亚砜(20vol)溶液中。将反应随后搅拌1h。在60℃下在1小时内添加水(24vol)并随后将混合物冷却至20℃。将混合物随后搅拌1小时,过滤,用水(2vol)洗涤,用冷甲醇(<5℃,2×4vol)洗涤并在真空下干燥。获得3-[5-氨基-4-(3-氰基苯甲酰基)-1H-吡唑-1-基]-4-甲基苯甲酸(化合物5),为淡黄色固体,产率52%,纯度98%(HPLC,面积%)。
实施例4
相对于化合物2的引入量(1.0eq)描述所述方法。
在105±5℃下,在1.25±0.25h内,将3-[2-氰基-2-(乙氧基亚甲基)乙酰基]苄腈(化合物2)(1.0eq)的二甲亚砜(4vol)溶液加入到3-肼基-4-甲基-苯甲酸(化合物3)(1.2eq)的二甲亚砜(20vol)溶液中。将反应随后搅拌约1小时。将混合物的温度调节至60±5℃。在60±5℃下在1±0.25h的时间内向混合物中加入水(24vol)。将混合物的温度调节至20±5℃。将混合物随后搅拌1.5±0.5h,然后过滤。在20±5℃下将滤液与水(2vol)混合,然后过滤并在真空下干燥。获得3-[5-氨基-4-(3-氰基苯甲酰基)-1H-吡唑-1-基]-4-甲基苯甲酸(化合物5)。
实施例5
相对于化合物5的引入量(1.0eq)描述所述方法。
将DMSO(5vol)中的1,1’-羰基二咪唑(CDI)(1.3eq)在40℃下加入到3-[5-氨基-4-(3-氰基苯甲酰基)-1H-吡唑-1-基]-4-甲基苯甲酸(化合物5)(1.0eq)在DMSO(5vol)中的悬浮液中。将反应随后搅拌1小时。通过真空除去所形成的CO
实施例6
相对于化合物5的引入量(1.0eq)描述所述方法。
将3-[5-氨基-4-(3-氰基苯甲酰基)-1H-吡唑-1-基]-4-甲基苯甲酸(化合物5)(1.0eq)在DMSO(6.5vol)中的悬浮液在40±5℃下在约0.5h的时间段内加入到1,1'-羰基二咪唑(CDI)(1.3eq)的DMSO(5vol)溶液中。将反应随后搅拌1.25±0.25h。通过真空除去所形成的CO
- 用于制备3-5-氨基-4-(3-氰基苯甲酰基)-吡唑化合物的合成方法
- 制备3-5-氨基-4-(3-氰基苯甲酰基)-吡唑-1-基-N-环丙基-4-甲基苯甲酰胺的多晶型的方法