一种可见光响应的芳基偶氮吡唑聚合物及合成方法
文献发布时间:2023-06-19 11:05:16
技术领域
本发明属于光响应功能高分子领域,具体涉及一种可见光响应的芳基偶氮吡唑聚合物及合成方法。
背景技术
偶氮苯化合物是一种光致变色分子,通常它可以在紫外光照射下发生反式向顺式的异构化,而在绿光照射下发生顺式到反式的回复。基于可逆的光异构化,国内外众多科研人员报道了许多将偶氮苯接枝到聚合中以制备光响应功能高分子的研究,这些研究使其在光开关、光驱动、能量与信息储存等多种技术领域中崭露头角。然而,目前报道的偶氮苯衍生物通常局限于紫外光照射下完成反式向顺式的异构化,而且偶氮苯的异构化程度和半衰期均有待提高,这些不足极大的限制了光响应高分子功能更为广泛的应用。为了解决这一科学挑战,近年来合成了一种具有超常半衰期和高异构化程度的吡唑基偶氮类化合物。然而,这类化合物依然限于紫外光照射下完成反式到顺式的转化。因此,目前函需设计并合成能够在可见光波段内实现可逆的异构化同时还具有高异构化程度、长半衰期的偶氮类衍生物。鉴于此,本发明涉及一种可见光响应的芳基偶氮吡唑聚合物的合成方法,从而为光响应功能高分子在可见光照射下的应用提供了新策略。
发明内容
本发明的目的在于提供一种可见光响应的芳基偶氮吡唑聚合物的合成方法。本发明采用以下技术方案:
一种可见光响应的芳基偶氮吡唑聚合物,其特征是:将芳基偶氮吡唑衍生物通过迈克尔加成反应共价接枝在树枝状聚合物表面,得到的芳基偶氮吡唑聚合物的结构式如下:
如上所述一种可见光响应的芳基偶氮吡唑聚合物的合成方法,其步骤如下:
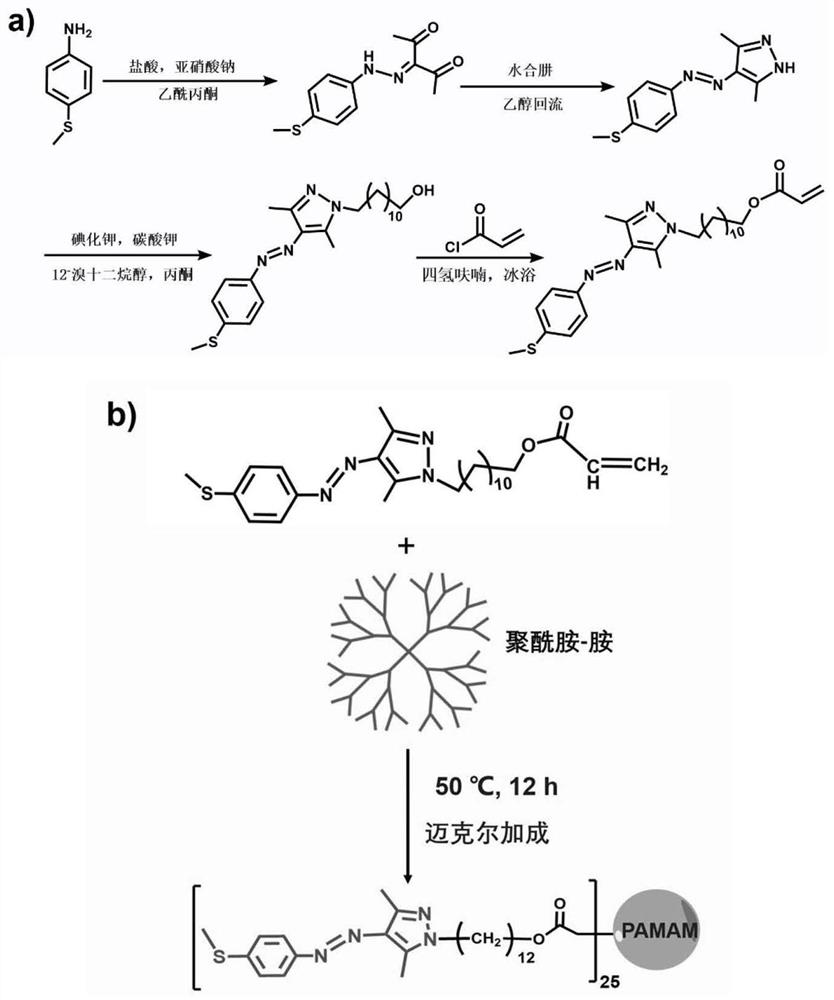
步骤一:芳基偶氮吡唑的制备:4-(甲硫基)苯胺溶解在浓盐酸与乙醇的混合溶液中并冷却到0-5℃。将亚硝酸钠水溶液缓慢滴加到上述溶液中并搅拌1-2h以制备重氮盐。将乙酰丙酮加到乙醇与水的混合溶液中随后加入乙酸钠并冷却到0-5℃。将上述重氮盐溶液缓慢滴加到该溶液中继续搅拌反应,将获得的黄色沉淀物过滤并用冷乙醇洗涤得到黄色的芳基偶氮乙酰丙酮粗产物。将芳基偶氮乙酰丙酮溶解在乙醇中然后向其中逐滴加入水合肼,混合液体在氮气氛围下搅拌加热到60-80℃回流12-48h。反应完成后冷却到室温,浓缩溶剂并用二氯甲烷溶解,再用饱和氯化钠溶液洗涤多次。将得到的有机层用无水硫酸镁干燥后浓缩得到芳基偶氮吡唑,最后通过柱层析提纯。
步骤二:芳基偶氮吡唑衍生物的制备:将芳基偶氮吡唑溶于乙腈中,然后依次加入12-溴十二烷基醇,碳酸钾和碘化钾,混合液在氮气氛围下加热到60-70℃回流反应24-48h。反应完成后,过滤除去无机盐并减压浓缩滤液至干燥,然后用二氯甲烷溶解萃取水相。有机层用无水硫酸钠干燥并浓缩得到中间产物,最后柱层析提纯得到期望的化合物。将得到的化合物溶于无水四氢呋喃中并冷却到0-5℃,随后加入三乙胺,然后加入丙烯酰氯和无水四氢呋喃的混合溶液并继续搅拌24-36h。反应完成后,过滤除去三乙胺盐,浓缩至干并用二氯甲烷溶解,饱和氯化钠溶液洗涤萃取多次,彻底除去三乙胺盐和丙烯酰氯。有机相用无水硫酸钠干燥后浓缩至干,最后通过柱层析提纯得到期望的芳基偶氮吡唑衍生物。
步骤三:芳基偶氮吡唑聚合物的制备:将芳基偶氮吡唑衍生物(AAP)在甲醇中充分溶解,然后缓慢滴加第三代聚酰胺-胺树枝状大分子(G3)的甲醇溶液。滴加完成后,在氮气氛围下将混合溶液置于40-50℃的油浴中搅拌反应24-36h。将反应后的溶液倒入大量冷乙醚中以沉淀出产物并除去过量的芳基偶氮吡唑衍生物。过滤后的固体在40-45℃下真空干燥30-36h得到干燥的偶氮聚合物。
进一步地,所述步骤一中4-(甲硫基)苯胺、亚硝酸钠、乙酰丙酮和乙酸钠的摩尔比为1:1:1:2~1:3:3:5;浓盐酸与乙醇的体积比为1:2~1:10;乙醇与水的体积比为1:1~4:1;芳基偶氮乙酰丙酮和水合肼的摩尔比为1:1~1:6;二氯甲烷与饱和氯化钠溶液的体积比为1:2~1:6。
进一步地,所述步骤一中盐水萃取次数为2~6次;粗产物柱层析提纯所用展开剂为乙酸乙酯和石油醚,其体积比为1:1~1:5。
进一步地,所述步骤二中芳基偶氮吡唑、碘化钾、碳酸钾和12-溴十二烷基醇的摩尔比为2:1:5:3~2:3:12:9;中间产物、三乙胺和丙烯酰氯的摩尔比为1:3:3~1:8:9。
进一步地,所述步骤二中二氯甲烷与饱和氯化钠溶液的体积比为1:2~1:8;饱和氯化钠溶液洗涤次数为2-5次;中间产物柱层析提纯所用展开剂为乙酸乙酯和正己烷,其体积比为1:1~3:2;芳基偶氮吡唑衍生物柱层析提纯所用展开剂为乙酸乙酯和石油醚,其体积比为1:3~2:1。
进一步地,所述步骤三中聚酰胺-胺树枝状大分子与芳基偶氮吡唑衍生物的摩尔比为1:30~1:60;所用甲醇与乙醚的体积比为1:20~1:40。
本发明技术关键点是:采用甲硫基取代的苯胺为原料通过标准的重氮化反应制备了可见光吸收的芳基偶氮吡唑。由于甲硫基的强供电子能力,芳基偶氮吡唑的吸收波长发生红移从而实现了可见光波段的显著吸收。相比于其他只能通过吸收紫外光异构化的偶氮杂环衍生物,这种新型芳基偶氮吡唑会有更为广泛的应用前景。此外,芳基偶氮吡唑在聚酰胺-胺树枝状聚合物表面的扩增还可以通过调控两者的摩尔比实现。
综上所述,本发明的有益效果是:
本发明设计合成的芳基偶氮吡唑聚合物能够在可见光照射下实现反式向顺式的高异构化程度和顺式到反式的快速回复,并且合成的芳基偶氮吡唑聚合物在室温黑暗条件下顺式异构体存储半衰期长达13天。此外,本发明设计合成的芳基偶氮吡唑聚合物合成工艺简单、产率高且易于量产。
附图说明
图1为本发明中合成流程图:
a)芳基偶氮吡唑衍生物的合成,
b)芳基偶氮吡唑聚合物的合成。
图2为实例中的核磁氢谱图(氘代氯仿):
a)芳基偶氮吡唑衍生物的核磁氢谱图,
b)基偶氮吡唑聚合物核磁氢谱图。
图3为实例中聚酰胺-胺、合成的芳基偶氮吡唑衍生物及聚合物的红外光谱图。
图4为实例中的紫外可见吸收光谱图:
a)紫色光(405nm,20mW/cm
b)绿色光(520nm,20mW/cm
具体实施方式
以下结合附图和具体实施例对本发明作进一步详细说明。
实施例1
1)4-(甲硫基)苯胺(2.78g,20mmol)溶解在浓盐酸(12M,0.5mL)与2mL乙醇的混合溶液中并冷却到0℃。将2mL亚硝酸钠(1.7g,24mmol)水溶液缓慢滴加到上述溶液中并搅拌1h以制备重氮盐。将乙酰丙酮(2.6g,26mmol)加到含50%乙醇的水溶液中随后加入乙酸钠(4.92g,60mmol)并冷却。将上述重氮盐溶液缓慢滴加到该溶液中并搅拌反应,将获得的黄色沉淀物过滤并用200mL含50%乙醇的水溶液洗涤,得到黄色的芳基偶氮乙酰丙酮粗产物。将芳基偶氮乙酰丙酮(2.5g,10mmol)溶解在50mL乙醇中然后向其中逐滴加入水合肼(0.6g,12mmol),混合液体在氮气氛围下搅拌加热到70℃回流24h。反应完成后冷却到室温,浓缩溶剂并用50mL二氯甲烷溶解,再用200mL饱和氯化钠溶液洗涤洗涤3次。将得到的有机层用10g无水硫酸镁干燥后浓缩得到芳基偶氮吡唑,最后通过柱层析提纯(乙酸乙酯:石油醚=1∶1)得到纯净的产物。
2)将芳基偶氮吡唑(1.5g,6mmol)溶于50mL乙腈中,然后依次加入12-溴十二烷基醇(2.05g,8mmol),碳酸钾(2.76g,20mmol)和碘化钾(1.32g,8mmol),混合液在氮气氛围下加热到68℃回流反应24h。反应完成后,过滤除去无机盐并减压浓缩滤液至干燥,然后用100mL二氯甲烷溶解并用300mL饱和氯化钠溶液洗涤洗涤。有机层用10g无水硫酸钠干燥并浓缩得到中间产物,最后柱层析提纯(乙酸乙酯:正己烷=3:1)得到期望的化合物。将得到的化合物(1.3g,3mmol)溶于50mL无水四氢呋喃中并冷却到0℃,随后加入三乙胺(0.91g,9mmol),然后加入丙烯酰氯(0.81g,9mmol)和10mL无水四氢呋喃的混合溶液并继续搅拌36h。反应完成后,过滤除去三乙胺盐,浓缩至干并用100mL二氯甲烷溶解,300mL饱和氯化钠溶液洗涤洗涤4次,彻底除去三乙胺盐和丙烯酰氯。有机相用10g无水硫酸钠干燥后浓缩至干,最后通过柱层析提纯(乙酸乙酯:石油醚=1:2)得到期望的芳基偶氮吡唑衍生物。
3)将芳基偶氮吡唑衍生物(0.72g,1.45mmol)在10mL甲醇中充分溶解,然后在冰浴中缓慢滴加聚酰胺-胺树枝状大分子(G3,200mg,0.029mmol)的甲醇溶液(5mL)。在N
实施例2
1)4-(甲硫基)苯胺(1.36g,10mmol)溶解在浓盐酸(12M,0.4mL)与3mL乙醇的混合溶液中并冷却到0℃。将1mL亚硝酸钠(1.2g,17mmol)水溶液缓慢滴加到上述溶液中并搅拌1.5h以制备重氮盐。将乙酰丙酮(2g,20mmol)加到含70%乙醇的水溶液中随后加入乙酸钠(4g,49mmol)并冷却。将上述重氮盐溶液缓慢滴加到该溶液中并搅拌反应,将获得的黄色沉淀物过滤并用200mL含70%乙醇的水溶液洗涤,得到黄色的芳基偶氮乙酰丙酮粗产物。将芳基偶氮乙酰丙酮(1g,4mmol)溶解在20mL乙醇中然后向其中逐滴加入水合肼(0.5g,10mmol),混合液体在氮气氛围下搅拌加热到80℃回流18h。反应完成后冷却到室温,浓缩溶剂并用30mL二氯甲烷溶解,再用100mL饱和氯化钠溶液洗涤萃取2次。将得到的有机层用10g无水硫酸镁干燥后浓缩得到芳基偶氮吡唑,最后通过柱层析提纯(乙酸乙酯:石油醚=1∶2)得到纯净的产物。
2)将芳基偶氮吡唑(0.8g,3.3mmol)溶于30mL乙腈中,然后依次加入12-溴十二烷基醇(2g,7.5mmol),碳酸钾(2.5g,18mmol)和碘化钾(1.0g,6mmol),混合液在氮气氛围下加热到80℃回流反应36h。反应完成后,过滤除去无机盐并减压浓缩滤液至干燥,然后用80mL二氯甲烷溶解并用200mL饱和氯化钠溶液洗涤洗涤。有机层用10g无水硫酸钠干燥并浓缩得到中间产物,最后柱层析提纯(乙酸乙酯:正己烷=3:2)得到期望的化合物。将得到的化合物(0.5g,1.2mmol)溶于20mL无水四氢呋喃中并冷却到0℃,随后加入三乙胺(0.8g,7.9mmol),然后加入丙烯酰氯(0.9g,10mmol)和20mL无水四氢呋喃的混合溶液并继续搅拌36h。反应完成后,过滤除去三乙胺盐,浓缩至干并用80mL二氯甲烷溶解,200mL饱和氯化钠溶液洗涤洗涤4次,彻底除去三乙胺盐和丙烯酰氯。有机相用10g无水硫酸钠干燥后浓缩至干,最后通过柱层析提纯(乙酸乙酯:石油醚=1:3)得到期望的芳基偶氮吡唑衍生物。
3)将芳基偶氮吡唑衍生物(1.4g,2.88mmol)在20mL甲醇中充分溶解,然后在冰浴中缓慢滴加聚酰胺-胺树枝状大分子(G3,500mg,0.072mmol)的甲醇溶液(20mL)。在N
实施例3
1)4-(甲硫基)苯胺(5g,36mmol)溶解在浓盐酸(12M,2mL)与10mL乙醇的混合溶液中并冷却到0℃。将5mL亚硝酸钠(6.9g,100mmol)水溶液缓慢滴加到上述溶液中并搅拌2h以制备重氮盐。将乙酰丙酮(10g,100mmol)加到含80%乙醇的水溶液中随后加入乙酸钠(10g,122mmol)并冷却。将上述重氮盐溶液缓慢滴加到该溶液中并搅拌反应,将获得的黄色沉淀物过滤并用300mL含80%乙醇的水溶液洗涤,得到黄色的芳基偶氮乙酰丙酮粗产物。将芳基偶氮乙酰丙酮(4g,16mmol)溶解在60mL乙醇中然后向其中逐滴加入水合肼(4g,80mmol),混合液体在氮气氛围下搅拌加热到80℃回流48h。反应完成后冷却到室温,浓缩溶剂并用80mL二氯甲烷溶解,再用400mL饱和氯化钠溶液洗涤萃取5次。将得到的有机层用10g无水硫酸镁干燥后浓缩得到芳基偶氮吡唑,最后通过柱层析提纯(乙酸乙酯:石油醚=1∶4)得到纯净的产物。
2)将芳基偶氮吡唑(3g,12mmol)溶于50mL乙腈中,然后依次加入12-溴十二烷基醇(10g,39mmol),碳酸钾(10g,72mmol)和碘化钾(2.0g,12mmol),混合液在氮气氛围下加热到75℃回流反应48h。反应完成后,过滤除去无机盐并减压浓缩滤液至干燥,然后用100mL二氯甲烷溶解并用300mL饱和氯化钠溶液洗涤洗涤。有机层用10g无水硫酸钠干燥并浓缩得到中间产物,最后柱层析提纯(乙酸乙酯:正己烷=1:1)得到期望的化合物。将得到的化合物(2g,4.8mmol)溶于60mL无水四氢呋喃中并冷却到0℃,随后加入三乙胺(2g,19.8mmol),然后加入丙烯酰氯(2g,22mmol)和30mL无水四氢呋喃的混合溶液并继续搅拌48h。反应完成后,过滤除去三乙胺盐,浓缩至干并用30mL二氯甲烷溶解,200mL饱和氯化钠溶液洗涤洗涤5次,彻底除去三乙胺盐和丙烯酰氯。有机相用10g无水硫酸钠干燥后浓缩至干,最后通过柱层析提纯(乙酸乙酯:石油醚=3:2)得到期望的芳基偶氮吡唑衍生物。
3)将芳基偶氮吡唑衍生物(0.74g,1.52mmol)在15mL甲醇中充分溶解,然后在冰浴中缓慢滴加聚酰胺-胺树枝状大分子(G3,300mg,0.043mmol)的甲醇溶液(15mL)。在N
综上所述,本发明提供了一种可见光响应的芳基偶氮吡唑聚合物的合成方法。本发明实施例1中芳基偶氮吡唑衍生物及聚合物通过核磁氢谱(图2)得到证明。本发明实施例中芳基偶氮吡唑聚合物的化学结构通过红外光谱(图3)得到证明。本发明实施例中芳基偶氮吡唑聚合物异构化和回复通过紫外可见吸收光谱(图4)得到证明。
- 一种可见光响应的芳基偶氮吡唑聚合物及合成方法
- 一种具有光响应性的甲基丙烯酸甲酯类偶氮聚合物及合成方法