杂环激酶抑制剂及其用途
文献发布时间:2023-06-19 11:21:00
说明
本发明涉及激酶抑制剂,特别是蛋白激酶的抑制剂,所述蛋白激酶包括蛋白酪氨酸激酶LCK、ABL、SRC、KIT、SIK家族和/或其突变体。尽管在结构上与达沙替尼(dasatinib)相似,但是本发明的激酶抑制剂可以显示出与达沙替尼不同的一种或多种某些特性。同样,本发明涉及包含所述激酶抑制剂中的一种或多种的药物组合物。本发明的激酶抑制剂或药物组合物可用于治疗疾病或病症,例如增殖性疾病,例如白血病或实体瘤。激酶抑制剂或药物组合物可以用于与达沙替尼用于相应疾病的治疗方案相对应、相似或不同的治疗方案,特别是可以与一种或多种其他治疗剂(例如免疫检查点抑制剂)一起用于联合治疗方案。
激酶抑制剂是一种阻断激酶作用的酶抑制剂。此类激酶的部分非限制性列举包括ABL、AKT、BCR-ABL、BLK、BRK、c-KIT、c-MET、CDK1、CDK2、CDK3、CDK4、CDK5、CDK6、CDK7、CDK8、CDK9、CDK10、cRAF1、CSK、EGFR、ERBB2、ERBB3、ERBB4、ERK、PAK、FES、FGFR1、FGFR2、FGFR3、FGFR4、FGFR5、FGR、FIT-1、FPS、FRK、FYN、HCK、IGF-1R、INS-R、JAK、KDR、LCK、LYN、MEK、p38、PDGFR、PIK、PKC、PYK2、ROS、SIK1、SIK2、SIK3、SRC、TIE、TIE2、TRK和ZAP70。激酶是在蛋白质或其他有机分子上添加磷酸基团的酶,已被证明是大多数细胞功能的关键调节剂,包括细胞信号传导、增殖、分化、新陈代谢、存活、凋亡、运动、DNA损伤修复等。磷酸化,特别是由于蛋白质磷酸化控制缺陷引起的信号转导失调,涉及多种疾病。例如与激酶的异常活性(例如,活性增加)有关的疾病。此类疾病包括但不限于增生性疾病(例如,癌症、良性肿瘤、病理性血管生成、炎性疾病和自身免疫性疾病)以及过敏和CNS疾病。
蛋白质酪氨酸激酶(PTK)是与ATP结合作为底物的磷酸化肽和蛋白质中酪氨酸残基的酶。PTK尤其包括受体蛋白酪氨酸激酶(RPTK),包括表皮生长因子激酶家族的成员(例如HER1和HER2)、血小板衍生生长因子(PDGF)和在血管生成中起作用的激酶(例如TIE2和KDR);此外,还包括非受体蛋白酪氨酸激酶,包括SYK、JAK和SRC激酶家族的成员(例如SRC、FYN、LYN、LCK和BLK激酶)。蛋白质丝氨酸/苏氨酸激酶(STK)是使肽和蛋白质中丝氨酸或苏氨酸侧链的氧原子磷酸化的酶。STK尤其包括AKT1、Aurora激酶、BRAF、MAP激酶、PLK1、SIK1、SIK2和SIK3。
抑制蛋白激酶以及因此抑制底物肽或蛋白的磷酸化已显示可用于治疗许多疾病。例如,阿法替尼(afatinib)是一种ERBB抑制剂,可用于治疗非小细胞肺癌;阿西替尼(axitinib)是一种VEGFR、PDGFR和c-KIT抑制剂,可用于治疗肾细胞癌;博舒替尼(bosutinib)是一种ABL/BCR-ABL抑制剂,可用于治疗慢性粒细胞性白血病;卡博替尼(cabozantinib)是一种c-MET和VEGFR2抑制剂,可用于治疗甲状腺癌;克唑替尼(crizotinib)是一种ALK、HGFR和c-MET抑制剂,可用于治疗非小细胞肺癌;达沙替尼(dasatinib)是一种ABL/BCR-ABL、SRC和c-KIT抑制剂,可用于治疗慢性粒细胞性白血病;埃洛替尼(erlotinib)是一种EGFR抑制剂,可用于治疗非小细胞肺癌和胰腺癌。吉非替尼(gefitinib)是一种EGFR抑制剂,可用于治疗非小细胞肺癌;伊马替尼(imatinib)是一种ABL/BCR-ABL抑制剂,可用于治疗慢性粒细胞性白血病;拉帕替尼(lapatinib)是一种HER2抑制剂,可用于治疗乳腺癌;尼洛替尼(nilotinib)是一种ABL/BCR-ABL抑制剂,可用于治疗慢性粒细胞性白血病;帕唑帕尼(pazopanib)是一种VEGFR、PDGFR和c-KIT抑制剂,可用于治疗肾细胞癌和软组织肉瘤;帕博西尼(palbociclib)是一种CDK4和CDK6抑制剂,可用于治疗ER阳性和HER2阴性的乳腺癌;帕纳替尼(ponatinib)是一种ABL/BCR-ABL、BEGFR、PDGFR、FGFR、EPH、SRC、c-KIT、RET、TIE2和FLT3抑制剂,可用于治疗慢性粒细胞性白血病和急性淋巴细胞性白血病;瑞格非尼(regorafenib)是一种RET、VEGFR和PDGFR抑制剂,可用于治疗大肠癌和胃肠道间质瘤;瑞博西尼(ribociclib)是一种细胞周期蛋白D1/CDK4和CDK6抑制剂,可用于治疗HR阳性、HER2阴性的晚期或转移性乳腺癌;鲁索替尼(ruxolitinib)是一种JAK抑制剂,可用于治疗骨髓纤维化;索拉非尼(sorafenib)是一种VEGFR、PDGFR、BRAF和c-KIT抑制剂,可用于治疗肾细胞癌和肝细胞癌;舒尼替尼(sunitinib)是一种VEGFR和PDGFR抑制剂,可用于治疗肾细胞癌、胃肠道间质瘤和胰腺神经内分泌肿瘤;托法替尼(tofacitinib)是一种JAK抑制剂,可用于治疗类风湿关节炎;凡德他尼(vandetanib)是一种VEGFR、EGFR、RET和BRK抑制剂,可用于治疗甲状腺癌;以及维拉非尼(vemurafenib)是一种BRAF抑制剂,可用于治疗恶性黑色素瘤。
鉴于激酶和相关疾病的数量众多,一直存在对各种激酶具有选择性的新抑制剂的需求,这些抑制剂可能对治疗相关疾病有用;特别是,仍然需要新的激酶抑制剂、药物组合物/制剂及其在治疗与一种或多种激酶的异常活性有关的疾病中的用途;特别地,仍然需要替代现有激酶抑制剂(例如达沙替尼)的新抑制剂。
一种特定的激酶抑制剂是达沙替尼(N-(2-氯-6-甲基苯基)-2-[[6-[4-(2-羟乙基)-1-哌嗪基]-2-甲基-4-嘧啶基]氨基]-5-噻唑羧酰胺,一水合物;图1A),由Bristol-Myers Squibb以“SPRYCEL”出售,适用于治疗患有以下疾病的成年患者:(i)处于慢性期的新近诊断出的费城染色体阳性(Ph+)慢性粒细胞性白血病(CML);(ii)对先前治疗(包括伊马替尼)有耐药性或不耐受性的慢性、加速或(髓样或淋巴样)母细胞期(Ph+)CML;以及(iii)对先前治疗有耐药性或不耐受性的费城染色体阳性的急性淋巴细胞白血病(Ph+ALL)。在欧盟,达沙替尼还适用于治疗对先前治疗(包括伊马替尼)有耐药性或不耐受性的患有新近诊断出的慢性期Ph+CML(Ph+CML-CP)或Ph+CML-CP的儿科患者,在美国,它也可用于慢性期Ph+CML的儿科患者。
值得注意的是,尽管已对达沙替尼进行了大量试验,但除CML或Ph+ALL以外,在美国或欧洲没有针对其他癌症的治疗指征;特别是,截至2018年9月,达沙替尼尚未明确用于任何实体瘤。确实,许多使用达沙替尼研究其可能用于治疗实体瘤的临床试验已提前终止(例如由于毒性问题),或者未能报告有力甚至令人鼓舞的结果。例如,根据clincialtrials.gov上的信息(2018年9月9日),达沙替尼仅进行了一次3期实体瘤测试:在一项针对去势抵抗性前列腺癌联合多西紫杉醇的单一研究(“READY”试验(NCT00744497))中,但在此试验中达沙替尼无法比单用多西他赛提高整体生存率(Araujo et al.2013,Lancet Oncol.14:13017),尽管在早期试验中有人暗示其对未接受过化学治疗的去势抵抗性前列腺的活性(例如Araujo et al.2012,Cancer 118:63)。尽管已经针对其他癌症(例如乳腺癌、皮肤癌、胰腺癌、脑癌或肺癌)进行了多项试验,但达沙替尼仍未显示出令人满意的疗效或耐受性,并且尚未进行针对任何这些癌症的3期试验。特别是,在针对局部晚期无法切除的胰腺患者的双盲2期试验中,与单用吉西他滨相比,达沙替尼与单用吉西他滨合用时并未显示出整体生存率的提高(Evens et al.2017,Annal.Onc.28:354)。但是,最近,一些旨在针对具有表达特定药物靶标的特定癌症(包括实体瘤)的患者选择“靶向”疗法的专业试验,可能会根据患者的靶标情况对达沙替尼进行测试。例如(i)“TAPUR”试验(“TheTargeted Agent and Profiling Utilization Registry”,https://www.tapur.org,NCT02693535)将达沙替尼纳入一个或多个基于以下一个或多个目标的治疗方案中:BCR-ABL、SRC、KIT、PDGFRB、EPHA2、FYN、LCK、YES1;以及(ii)澳大利亚黑色素瘤研究所的一项临床试验(NCT02645149),其中涉及患有BRAF和NRAS野生型不可切除的III期或IV期转移性黑色素瘤患者,其已经进展或无法接受标准疗法(通常是免疫疗法),包括达沙替尼作为一种可能的治疗方法,取决于患者癌症中发现的KIT突变。达沙替尼也是BMS“FRACTION-Lung”2期试验(NCT02750514)的一个可能分支,在该试验中,它可能与免疫肿瘤药物纳武单抗(nivolumab)联合用于晚期非小细胞肺癌患者。该试验的其他部分将纳武单抗与其他免疫肿瘤药物联合使用。
因此,特别需要新型激酶抑制剂,其可用于治疗未指明用达沙替尼治疗的癌症(尤其是实体瘤)、和/或达沙替尼尚未显示出有希望的结果的癌症。特别地,需要用于治疗一种或多种癌症的新型激酶抑制剂,该癌症例如是乳腺癌、肺癌(例如非小细胞癌)、胰腺癌或前列腺癌(例如去势或激素抵抗)以及黑色素瘤。还需要新的激酶抑制剂用于治疗髓样或淋巴母细胞癌,例如白血病,优选用于治疗一种或多种Ph+白血病,例如CML和/或ALL。
达沙替尼被描述为以下激酶的纳摩尔浓度下的抑制剂:BCR-ABL,SRC家族(SRC、LCK、YES、FYN),c-KIT,EPHA2和PDGFR-β;与达沙替尼对Ph+白血病的适应症特别相关的地方是它对杂合蛋白激酶BCR-ABL的抑制作用。
BCL-ABL激酶与白血病癌细胞(特别是CML细胞)的22号染色体(被称为“费城染色体”(或费城易位))中特定遗传异常的存在直接相关。遗传物质在9号染色体和22号染色体之间的相互易位,将9号染色体的ABL1基因并置在22号染色体的BCR基因上,从而形成了称为“BCR-ABL”的杂合蛋白的编码序列:一种“永远在线”、导致细胞分裂失控的蛋白酪氨酸激酶。绝大多数CML病例和20-30%的ALL病例为Ph+。第一种选择性BRC-ABL抑制剂伊马替尼(STI571),被诺华公司以“GLEEVEC/GLIVEC”出售,被认为是治疗Ph+白血病的突破。然而,尽管总生存期有所增加,但伊马替尼治疗期间产生的耐药性使科学家发现,大多数此类耐药性是由于BCR-ABL突变的出现,特别是ABL衍生的激酶结构域内的氨基酸取代而产生的(对于综述,请参见Rossari&Orciuolo.2018,J.Hemat.Oncol.11:84,通过引用整体并入本文)。
对伊马替尼治疗的患者的BCR-ABL突变状态和存活可能性的分析表明,BCR-ABL激酶ABL位置的磷酸酶环(P环)内的突变是最常见的,但P环外的(更罕见)突变(特别是激酶域内的突变)与伊马替尼治疗的CML患者的总体生存期降低有关(Jabbour et al.2006,Leukemia 20:1767)。此后已经鉴定并描述了许多新兴的BCR-ABL突变(参见Manley etal.2005,Biochem.Biophys.Acta 1754:3的表1、以及Rossari&Orciuolo 2018的表1,其还描述了其他突变达沙替尼的激酶靶标;这两个均通过引用特别地并入本文)。特别是,在BCR-ABL的ATP结合区域(针对野生型ABL蛋白标出的位置)中发现以下突变:V299L、F311L、T315I、T315A、F317L和F317V。的确,达沙替尼最初是作为用于CML二线治疗的“第二代”BCR-ABL抑制剂而开发的,该疾病已对伊马替尼产生抗药性,推测是由于这些突变中的一种或另一种的出现而引起的。根据建模研究,预计达沙替尼将与ABL激酶的多个构象结合,这被认为可以解释为什么达沙替尼可抑制ABL的几种构象改变突变,而伊马替尼则不抑制。确实,一项回顾性分析比较了使用达沙替尼或伊马替尼进行一线治疗期间的突变发展,发现与伊马替尼治疗(12个不同的位点)相比,使用达沙替尼治疗(4个不同的位点)出现的不同突变位点更少(Hughes et al.2015,Leukemia 29:1832,特别是其图1)。然而,重要的是:(i)发生任何类型突变的患者总比例大致相同(17/259名达沙替尼患者和18/260名伊马替尼患者);(ii)达沙替尼治疗后出现的大多数突变位点都在ATP结合区(3/4个突变位点);以及(iii)在达沙替尼治疗期间(11/17)出现的最常见的突变是所谓的“守门员”残基处的T315I突变,该突变仍赋予对达沙替尼抑制BCR-ABL激酶的抗性。ProQinase ABL1激酶“Wildtypeand Mutant Panel”提供了一组可针对激酶抑制剂进行测试的BCR-ABL突变体,包括ABL1野生型蛋白(氨基酸P118-S525)和代表BCR-ABL最常见的伊马替尼耐药突变体形式的突变体形式:G250E、Q252H、Y253F、E255K、T315I、F317I、M351T和H396P(www.proqinase.com)。
T315I突变是最频繁出现的BCR-ABL突变之一:发生于2%至20%的CML病例中(Nicolini et al.2009,Blood 114:5271)。这种突变对达沙替尼的抑制有抗性,这是达沙替尼作为激酶抑制剂的一个潜在缺点,它刺激了被称为“帕纳替尼”(由Incyte&Takeda以ICLUSIG出售)的“第三代”BCR-ABL抑制剂的发展。但是,尽管帕纳替尼确实确实能强烈抑制BCR-ABL激酶的T315I突变(2.0nM的体外IC50),但它是比达沙替尼更为混杂的激酶抑制剂,并且还抑制了许多其他激酶,包括至少VEGFR、PDGFR、FGFR、EPH受体和SRC激酶家族成员以及KIT、RET、TIE2和FLT3的体外IC50浓度在0.1至20nM之间。此外,由于“存在威胁生命的血栓和严重狭窄的血管的危险”,帕纳替尼在美国的销售于2013年10月被暂时中止。在2013年12月,部分解除了该暂停,并向帕纳替尼发布了修订的处方信息,新的“黑匣子警告”和“风险评估和缓解策略”,以更好地评估使用该药物的风险和益处。此外,帕纳替尼在美国的价格(每年可能花费138,000美元)也受到批评。因此,帕纳替尼显示出实质性的缺点,因此仍然需要新的激酶抑制剂,特别是那些有可能更有效、安全、容易和/或廉价地治疗Ph+白血病(或其他癌症)的激酶抑制剂;和/或与其他激酶抑制剂(例如达沙替尼或帕纳替尼)相比,对SRC、ABL/BCR-ABL和/或LCK更具选择性的抑制剂。
然而,与伊马替尼相比,达沙替尼对BCR-ABL并不是特别特异,并且与大量其他激酶结合和/或抑制大量其他激酶(请参阅:Bantscheff et al.2007,Nat.Biotech.25:1035的图3;Anastassiadis et al.2012,Nat.Biotech.29:1039的补充图2)。特别地,与伊马替尼相比,达沙替尼被描述为更有效地结合和/或抑制许多其他激酶,包括:BTK、CSK、EPHB2、EPHB4、FYN、GAK、KIT、LYN、QIK、QSK、RIPK2、SRC、TEC、TESK2、YES和ZAK。更具体地说,显示达沙替尼是盐诱导性激酶的重要抑制剂,三个家族成员SIK1、SIK2和SIK3的IC50值分别<3nM、<3nM和18nM(Ozanne et al.2015,Biochem.J.465:271;也描述于同时待审的PCT/EP2018/060172)。的确,鉴于达沙替尼是选择性较低的激酶抑制剂(这是另一个潜在的缺点),该选择性的降低可能与治疗达沙替尼所面临的非微不足道的毒性挑战有因果关系,尤其是血小板减少症的发生率增加(Wei et al.2010,J.Hemat.Oncol.3:47)。
如上所述,达沙替尼是一种有效的KIT抑制物,该酪氨酸激酶受体正成为治疗某些癌症的越来越感兴趣的靶标(Babei et al.2016,Drug Des.Dev.Thera.,10:2443),尤其是因为已在白血病、卵巢癌和黑色素瘤等癌症中检测到KIT基因的突变。还已知达沙替尼还可以至少抑制黑素瘤中最常见的KIT突变(Woodman et al.2009,J.Clin.Onc.27:9019)。但是,抑制KIT,尤其是抑制某些酪氨酸激酶抑制剂对抗FLT3和KIT的相对活性,已与骨髓抑制和其他副作用(例如脱发)有关(Galanis and Levis 2015:Haematologica 100:e89)。确实,达沙替尼治疗与严重的骨髓抑制有关(见下文)。
盐诱导性激酶(salt-inducible kinase,SIK)构成丝氨酸酪氨酸激酶亚家族,属于单磷酸腺苷激活激酶(AMPK)家族。到目前为止,已经确定了三个成员(SIK1、2和3)。在激酶结构域中,SIK1与SIK2和SIK3的氨基酸同源性分别为78%和68%。在高盐饮食喂养的大鼠的肾上腺中大量表达的SIK1(也称为SIK和SNF1LK)的克隆,导致随后的主要在脂肪组织中表达的SIK2(也称为QIK、KIAA0781和SNF1LK2)的克隆,以及相当普遍的SIK3(也称为QSK、KIAA0999或L19)(Katoh et al.2004,Mol.Cell.Endocrinol.217:109)。这三种SIK具有相似的结构,具有一个N端激酶结构域(催化结构域)、一个中间泛素相关结构域(被认为对LKB1的磷酸化很重要)和一个长C末端序列(被认为是进一步由PKA磷酸化的位点)。但是,各种SIK涉及许多不同的角色。例如,各种SIK已牵涉到生物学过程中,如骨细胞对甲状旁腺激素的反应(Wein et al.2016,Nature Commun.7:13176),以响应胃泌素诱导SIK1并抑制胃腺癌细胞迁移(Selvik et al.2014,PLoS ONE 9:e112485)。盐诱导的激酶(特别是SIK3)的其他潜在作用在同时待审的PCT/EP2018/060172中进一步描述,SIK3是涉及肿瘤细胞对细胞介导的免疫反应,特别是肿瘤细胞对TNF的抵抗的基因。
因此,仍然需要新的激酶抑制剂,尤其是那些与达沙替尼抑制的激酶表现出不同激酶特性的激酶抑制剂。例如,新的激酶抑制剂,其:(i)相对于其他激酶而言对关键疾病相关激酶(例如ABL/BCR-ABL、SRC、LCK和/或EPHA2、EPHA4、CSF-R1、HCK、ACK1和/或KIT)更具有特异性,比达沙替尼对一种或多种此类其他激酶所显示的特异性更大;(ii)以与达沙替尼不同的方式抑制关键疾病或副作用相关激酶(例如对KIT和/或FLT3的抑制);和/或(iii)抑制疾病相关激酶的一个或多个突变体,特别是对一种或其他激酶抑制剂具有抗性的突变体,例如ABL/BCR-ABL或KIT的突变体。
此外,尽管达沙替尼主要在人体中被细胞色素P450酶3A4(CYP3A4)代谢,但它也是CYP3A4的时间依赖性抑制剂。实际上,如果患者同时服用强效CYP3A4抑制剂(例如酮康唑(ketoconazole)、伊曲康唑(itraconazole)、克拉霉素(clarithromycin)、阿扎那韦(atazanavir)、茚地那韦(indinavir)、奈法唑酮(nefazodone)、奈非那韦(nelfinavir)、利托那韦(ritonavir)、沙奎那韦(saquinavir)、泰利霉素(telithromycin)和伏立康唑(voriconazole)),则必须显著降低达沙替尼的剂量(例如,从每天100mg减至每天20mg),因为它们可能会使达沙替尼的血浆浓度增加到潜在的不安全水平。葡萄柚汁也可能会增加达沙替尼的血浆浓度,也应避免。因此,仍然需要表现出不同于达沙替尼的细胞色素P450抑制模式(例如,对CYP3A4的抑制)的新型激酶抑制剂。
重要的是,发生骨髓抑制后,应停止(或减少)达沙替尼的剂量和给药。确实,骨髓抑制在美国达沙替尼处方信息中仅描述为“警告和注意事项”,因为使用达沙替尼的治疗与严重的(NCI CTC 3或4级)血小板减少、中性粒细胞减少和贫血有关。在达沙替尼的所有临床研究中,除了引起人类血小板减少症外,(i)1%的患者发生了严重的中枢神经系统(CNS)出血(包括死亡);(ii)4%的患者发生了严重的胃肠道出血,包括死亡事故,通常需要中断治疗和输血;以及(iii)其他严重出血病例发生在2%的患者中。
达沙替尼的其他“警告和注意事项”包括:(x)它与体液潴留留有关,在临床试验中有高达10%的患者报告严重体液潴留;(y)它有可能延长心室复极化(QT间期),并且在临床试验中多达1%的CML患者经历了QT延长;以及(z)258名服用达沙替尼的患者中有5.8%发生了心脏不良反应,其中包括1.6%的心肌病、心力衰竭充血、舒张功能障碍、致命性心肌梗塞和左心室功能不全。实际上,已知达沙替尼是hERG的抑制剂(NDA 21-986的药理/毒性研究与评价,第31页)。hERG(人类“以太相关基因”)是一个离子通道,可促进心脏的电活动并协调心脏的跳动。当该通道在细胞膜上传导电流的能力受到抑制或损害时(例如,通过给药),可能会导致“长期QT综合征”,这可能是致命的。因此,仍然需要显示出与达沙替尼不同的对hERG的抑制作用的新型激酶抑制剂。例如,提供对hERG表现出比达沙替尼更大的IC50的新激酶抑制剂将是有利的。
与其他BCR-ABL抑制剂相比,达沙替尼具有极短的半衰期:总体平均终末半衰期仅为3-5小时(完整处方信息的第12.3节“药代动力学”)。与之形成鲜明对比的是:伊马替尼的消除半衰期约为18小时;博舒替尼的终末期消除半衰期平均半衰期为22.5小时;尼洛替尼的表观消除半衰期约为17小时;帕纳替尼的几何平均终末消除半衰期约为24小时。不受理论的束缚,达沙替尼的半衰期短——指示每天一次剂量——可能解释了当天晚些时候体内药物浓度较低和/或与给药后不久体内药物浓度峰值/较高剂量相关的副作用所导致的活动受限。因此,仍然需要表现出更长的半衰期特性(例如,比达沙替尼显示的半衰期更长)的新的激酶抑制剂。例如,有利的激酶抑制剂可以是比达沙替尼更稳定的激酶抑制剂,例如通过在血浆和/或肝微粒体稳定性测定中表现出更长的半衰期。
如可以从EMA和FDA的相应网站上分别找到的,可以在完整处方信息的各个产品特征摘要(SmPC)中找到达沙替尼的其他预防措施、不良事件和其他处方信息(分别显示在下面,访问日期为2018年8月20日,并且每个内容的全部内容通过引用合并于此):(i)http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000709/WC500056998.pdf,和(ii)https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/021986s7s8lbl.pdf。
已经合成了达沙替尼的许多变体,并证明它们具有针对一种或多种激酶的体外生化抑制活性和/或对细胞的抗增殖作用。特别地,合成了这样的变体:(i)在达沙替尼的发现阶段,以了解和描述其结构-活性关系(SAR)(Lombardo et al 2004,J Med Chem 47:6658;Das et al 2006,J Med Chem 49:6819);(ii)提供替代的激酶抑制剂和/或候选药物(例如WO 2006/081172和WO 2008/033746)。其中描述的达沙替尼的变体在羧酰胺上带有苯基部分。这些公开证明了其他位置以及在其上可以被取代的取代基的实质范围,其提供了作为激酶抑制剂和/或具有细胞抗增殖活性的化合物(图8)。
WO 2018/193084(本申请人,并且于2018年10月25日公开)公开了一种带有吡啶基部分的达沙替尼变体。Beutner等人((2018,Org Lett 20:4218)描述了一种形成具有挑战性的酰胺键的方法,其中包括某些吡啶的吡嗪和嘧啶的酰胺键。Pennington等人(2017,JMed Chem 60:3552)描述了在芳族和杂芳族环系统中用N原子取代CH基团可能对分子和生理特性产生影响。但是,从经验上证明,进行这种取代可提高统计学上的效力,而不仅仅是偶然性:Abbott内部数据的匹配分子对分析(MMPA)(Hajduk&Sauer 2008,J Med Chem 51:553)发现与大多数取代基取代一样,通过交换CH基团和N原子来增加或减少效力的可能性大致相同。的确,该分析进一步揭示了用这种替代物实现效力提高10倍的可能性小于十分之一,而实现100倍的可能性小于100之一。与研究此类替代物改善结合亲和力的效果时观察到的概率相似(Hu et al 2014,F1000Research 3:36;de la Vega de Leon et al2014,MedChemComm5:64)。
因此,本发明的一个目的是提供一种或多种激酶抑制剂,其具有一种或多种针对这些或其他问题中的一种或多种的特性(例如,通过体外和/或体内测定法显示的那些)。在其他目的中,本发明提供了达沙替尼的替代和/或改进的激酶抑制剂(或一种或其他激酶抑制剂,例如本文所述的那些)。例如,一种激酶抑制剂,其可以展现出与达沙替尼(或一种或其他激酶抑制剂,如此处所描述的那些)不同的和/或改进的一种或多种功能(例如,激酶选择性)和/或ADMET特性,将是特别有利的。通过如本文中任何地方公开或定义的主题,例如通过所附权利要求的主题,解决了本发明的基础的目的。
发明概述
大体上,通过简要描述,可以将本发明的主要方面概括如下:
在第一方面,本发明提供了一种化合物,其选自下式的激酶抑制剂:
以及其溶剂化物、盐、N-氧化物、络合物、多晶型物、晶体形式、外消旋混合物、非对映异构体、对映异构体、互变异构体、构象异构体、同位素标记形式、前药及其组合;其中R
在第二方面,本申请提供了一种药物组合物,其包含第一方面的化合物和任选的药学上可接受的载体。
在第三方面,本申请提供了第一方面的化合物或第二方面的药物组合物,其用于治疗。
在第四方面,本申请提供第一方面的化合物或第二方面的药物组合物,其用于治疗个体(特别是人类患者)的疾病、病症或病状的方法,其中所述疾病或病状与激酶有关。
本文公开了本发明的其他方面。
附图说明
附图示出了:
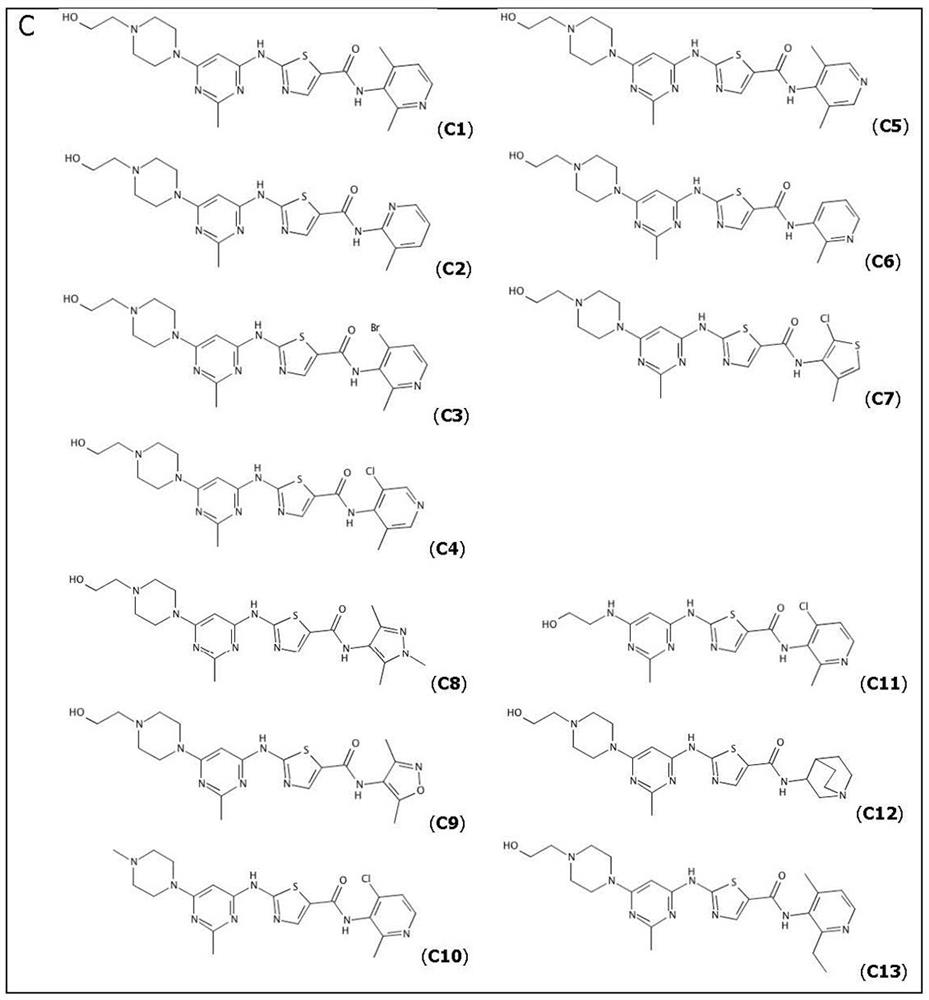
图1:描述了以下化合物的化学结构:(A)达沙替尼(化合物A8),N-(2-氯-6-甲基苯基)-2-((6-(4-(2-羟甲基)哌嗪-1-基)-2-甲基嘧啶-4-基)氨基)噻唑-5-羧酰胺;(B)激酶抑制剂B3,N-(4-氯-2-甲基吡啶-3-基)-2-((6-(4-(2-羟乙基)哌嗪-1-基)-2-甲基嘧啶-4-基)氨基)噻唑-5-羧酰胺;(C)某些其他式(I)C1至C13的激酶抑制剂;以及(D)某些其他式(I)D1至D10的激酶抑制剂。
图2:(在A至E中)描述了与达沙替尼(A8,右栏)相比,式(I)的激酶抑制剂(B3,左栏)对激酶(A)ABL1;(B)SRC;(C)SIK1;(D)SIK2;和(E)SIK3的抑制活性;并且描述了(F至J中)式(I)的其他激酶抑制剂(C3,左栏;C12右栏)对激酶(F)ABL1;(G)SRC;(H)SIK1;(I)SIK2;和(J)SIK3的抑制活性。X轴化合物浓度(M)和Y轴激酶活性(%)。
图3:描绘了B3(式(I)的激酶抑制剂)、达沙替尼(A8)和C7(式(I)的另一种激酶抑制剂)的按残留活性百分比计(在1uM化合物处)对激酶抑制的选择性:****<25%残留活性;***25%至<50%残留活性;**50%至<75%残留活性;*>75%残留活性。蛋白激酶家族的分类(Manning et al.Science 6December 2002:Vol.298no.5600pp.1912-1934):AGC:包含PKA、PKG和PKC家族;CAMK:钙/钙调蛋白依赖性蛋白激酶;CK1:酪蛋白激酶样;CMGC:包含CDK、MAPK,GSK3和CLK系列;TK:酪氨酸激酶;TKL:酪氨酸激酶样;STE:Yeast Sterile 7、Sterile 11、Sterile 20激酶的同系物。##组成型活性激酶。
图4:描述了与达沙替尼(A8;Y轴)相比,式(I)的激酶抑制剂B3(X轴)抑制激酶的选择性(按1uM化合物的残留活性百分比计):(A)轴显示残留活性的整个范围;并且(B)轴表示0至50%残留活性的范围。
图5:描述了与达沙替尼(A8,右栏)相比,式(I)的激酶抑制剂(B3,左栏)对激酶(A)FLT3;(B)SYK;(C)KIT;和(D)LCK的抑制活性。X轴化合物浓度(M)和Y轴激酶活性(%)。
图6:描绘了(A)激酶抑制剂B3和(B)A8(达沙替尼)对肿瘤细胞对体外TNF攻击的敏感化;圆圈:化合物(浓度如图所示)加rHuTNF(10ng/mL);正方形:没有rHuTNF的单独化合物(浓度如图所示)。
图7:描绘了使用实施例9中所述的M579-A2-luc以单独的各种浓度(正方形)或与10ng/mL TNF结合使用(圆圈)进行测定的情况下,PCT/EP2018/060172中所述的某些激酶抑制剂的相对肿瘤细胞存活率(通过细胞毒性/生存力归一化的RLU)。还显示了化合物对SIK家族成员和相关激酶ABL1和SRC的指示性抑制活性,通过表3所示的指示显示。(A)泛SIK和ABL1&SRC抑制剂,化合物B1;(B)ABL1&SRC抑制剂,化合物B8。(C)SIK1、SIK2和ABL1&SRC抑制剂,化合物B4。
图8:描绘了:(A)从Lombardo et al 2004(J Med Chem 47:6658)的表1中获得的达沙替尼变体的细胞抗增殖活性,显示了达沙替尼的各种衍生物针对所示细胞系的效力。a根据化合物暴露72h后的四唑鎓染料转化率确定抗增殖活性。IC50值报告为至少三个单独测定的平均值,或者在少于三个测量值的情况下报告为单个IC50值。除非括号中的SE值另有说明,否则平均值附近的可变性<50%;以及(B)来自Das et al 2006(J Med Chem 49:6819)的表4的达沙替尼变体的生化和细胞抗增殖活性。a n=3,单个值的变化,<20%。b n=3,单个值,<30%。
图9:描绘了与(A)达沙替尼(A8;X轴)和(B)另一种式(I)的激酶抑制剂B3(X轴)相比,式(I)的激酶抑制剂C7(Y轴)抑制激酶的选择性(按1uM化合物的残留活性百分比计)。虚线区域突出了在适用化合物之间基本上被不同地抑制的激酶组。
图10:描绘了与对照动物(灰色正方形)相比,每天一次(QD=A)和每天两次(BID=B)通过强饲法施用不同浓度33mg/kg(黑色正方形)和100mg/kg(灰色菱形)的C7的雌性C57Bl/6小鼠的体重。X轴:给药后天数;Y轴体重变化(%)。(C)通过LC-MS/MS测定的C7血浆水平。A=33mg/kg QD;B=100mg/kg QD;C=33mg/kg BID;D=100mg/kg BID。Y轴:C7的血浆浓度(nM)。
图11:描绘了(A)用媒介物(黑色正方形)、C7 100mg/kg QD(灰色六边形)、C7100mg/kg BID(灰色三角形)和A8(达沙替尼)30mg/kg QD A8(灰色圆圈)处理后,植入MC38细胞的小鼠的肿瘤生长动力学;Y轴=平均肿瘤体积(mm3)。误差条SEM。X轴:天数。统计显著性是通过双向方差分析进行计算的,其中包括Tukey的多重比较分析。***p<0.001;(B)(A)中小鼠的体重动力学。Y轴:平均体重变化(%)。
图12:描绘了式(I)的化合物C7对肿瘤微环境中存在的免疫细胞的免疫肿瘤作用。肿瘤内免疫浸润计算为肿瘤内CD45+细胞的百分比。统计显著性是通过单向方差分析进行计算的,其中包括Tukey的多重比较分析。(A)Y轴:CTL与Treg细胞的比例。(B)激活的CTL(CD25+CD69+);Y轴:CD45+细胞的百分比。(C)激活的CTL(粒酶B+);Y轴:CD45+细胞的百分比。(D)免疫抑制性M2样肿瘤相关巨噬细胞(TAM)(CD206+MHC-II+);Y轴:CD45+细胞的百分比;*p<0.05;**p<0.01;***p<0.00。
图13:描绘了化合物C7致敏的TNF杀伤细胞。(A)TNF诱导的PANC-1细胞凋亡。用370nM(“菱形”)、3333nM(“正方形”)的C7和仅DMSO(“星形”)处理PANC-1细胞,然后添加100ng/ml rHuTNF 120h(+rHuTNF=空心形状;-rHuTNF=实心形状;空心圆圈10ng/mLrHuTNF对照)。使用实时活细胞显微镜法评估细胞死亡,测量YOYO-1染料的核掺入(YOYO-1+细胞的面积/孔)。Y轴=肿瘤细胞死亡(um2/孔)。X轴=时间(h);(B)C7对TNF诱导(100ng/mlrMuTNF)的鼠MC38的细胞凋亡的影响(+rMuTNF=“菱形”;-rMuTNF=“圆圈”)。72小时后,使用CellTiter-Glo测定法测量细胞活力。将萤光素酶值标准化为使用不含抑制剂的rMuTNF处理的细胞(仅DMSO)。Y轴=生存力(%)。X轴=化合物浓度(nM)。
图14:描绘了:(A)化合物C7对NFKB活性的影响。用不同浓度的C7处理在NFKB启动子控制下表达荧光素酶的Reporter PANC-1细胞,然后添加10ng/ml rHuTNF 8小时(+rHuTNF=“菱形”;-rHuTNF=“圆圈”)。萤光素酶活性相对于无抑制剂的rHuTNF处理的PANC-1细胞(仅DMSO)标准化。Y轴=NFKB活性(%)。X轴=化合物浓度(nM);(B)化合物C7对HDAC4磷酸化的作用。用各种浓度的C7(在10ng/mL rHuTNF存在下)处理PANC-1细胞3小时。全细胞裂解液在Meso Scale Discovery(MSD)分析中使用抗HDAC4捕获和抗pHDAC4检测抗体进行分析。将HDAC4磷酸化标准化至未经处理的PANC-1细胞(仅DMSO)。Y轴=HDAC4磷酸化(%)。X轴=化合物浓度(nM)。
图15:描绘了:化合物C7对WSU-NHL(A)和DOHH-2(B)细胞系的生长抑制,分别显示约8nM和9nM的GI50。X轴:化合物浓度(M);Y轴:96小时时的百分比生长抑制(GI)。
本发明的详述
可以如下更详细地描述本发明及其特定的非限制性方面和/或实施例。
尽管可以进一步更详细地描述本发明,但是应当理解,本发明不限于本文描述的特定方法、方案和试剂,因为它们可以变化。还应理解,本文所用术语仅出于描述特定实施方案的目的,而无意限制本发明的范围,本发明的范围将仅受本文所述、本文所定义或以其他方式公开的内容所限制,特别是在任何分项实施例或所附权利要求中。
在此,更详细地描述本发明的某些元素。这些元素与特定实施例一起列出,但是,应当理解,它们可以以任何方式和任何数量组合以创建附加的实施例。各种描述的示例和优选实施例不应被解释为将本发明限制为仅明确描述的实施例。应该理解本申请的描述支持并包含将明确描述的实施例与任何数量的公开和/或优选元素相结合的实施例。此外,除非上下文另外指出,否则应当认为本申请的描述中公开了本申请中所有描述的元素的任何排列和组合。例如,如果在本发明化合物的一个实施方案中L为键,并且在本发明化合物的另一实施方案中R
除非另外定义,否则本文中使用的所有技术和科学术语具有与本领域普通技术人员通常理解的相同含义。
优选地,本文所用的术语如"A multilingual glossary of biotechnologicalterms:(IUPAC Recommendations)",H.G.W.Leuenberger,B.Nagel,和H.
除非另有说明,否则本发明的实施将采用化学、生物化学和重组DNA技术的常规方法,这些方法在本领域的文献中有解释(参见,例如,Molecular Cloning:A LaboratoryManual,2
在整个本说明书和随后的权利要求书中,除非上下文另有要求,否则词语“包括”以及诸如“含有”和“包含”之类的变体将被理解为暗示包括所述的成员、整数或步骤或成员、整数或步骤的组,但不排除任何其他成员、整数或步骤或成员、整数或步骤的组。术语“基本上由……组成”是指排除具有任何重要意义的其他成员、整数或步骤或具有任何重要意义的成员、整数或步骤的组。例如,基本上由本文所定义的成员/组分组成的药物组合物(例如本发明的任何方面所定义的化合物和任选的一种其他治疗剂)将排除其他治疗剂(除了本发明任何方面中定义的化合物和任选的一种其他治疗剂之外),但不排除痕量的污染物(例如,来自分离和纯化方法的污染物)(例如,相对于整个组合物,污染物的量(优选组合物所有存在的污染物的量)小于按重量计5%,例如小于按重量计4%、按重量计3%、按重量计2%、按重量计1%、按重量计0.5%、按重量计0.4%、按重量计0.3%、按重量计0.2%、按重量计0.1%、按重量计0.05%)和/或药学上可接受的赋形剂(例如载体,例如磷酸盐缓冲盐水,防腐剂等)。术语“由……组成”意指排除具有重要意义的所有其他成员、整数或步骤或具有重要意义的成员、整数或步骤的组。例如,由本文定义的成员/组分(例如本发明任何方面定义的化合物、一种赋形剂和任选的一种其他治疗剂)组成的药物组合物将排除相对于整个组合物来说其量超过按重量计2%的任何其他化合物(包括第二种试剂或进一步的赋形剂)(例如任何其他化合物的量大于按重量计1%、大于按重量计0.5%、大于按重量计0.4%、大于按重量计0.3%、大于按重量计0.2%、大于按重量计0.1%、大于按重量计0.09%、大于按重量计0.08%、大于按重量计0.07%、大于按重量计0.06%、大于按重量计0.05%、大于按重量计0.04%、大于按重量计0.03%、大于按重量计0.02%、大于按重量计0.01%)。术语“包括”涵盖术语“基本上由……组成”,而术语“基本上由……组成”又涵盖术语“由……组成”。因此,在本申请中的每次出现时,术语“包括”可以被术语“基本上由……组成”或“由……组成”代替。同样,在本申请中的每次出现时,术语“基本上由……组成”可以被术语“由……组成”代替。
在本文中使用时,“和/或”应被视为两个指定的特征或组件中具有或不具有另一个的特定公开。例如,“X和/或Y”将被视为(i)X、(ii)Y以及(iii)X和Y中的每一个的具体公开,就好像它们在本文中分别列出一样。
在本发明的上下文中,术语“大约”和“近似地”可互换使用,并且表示普通技术人员将理解以仍然确保所讨论的特征的技术效果的准确度区间。该术语通常表示与指示的数值相差±5%、±4%、±3%、±2%、±1%、±0.9%、±0.8%、±0.7%、±0.6%、±0.5%、±0.4%、±0.3%、±0.2%、±0.1%、±0.05%,例如±0.01%。如本领域技术人员将理解的,对于给定的技术效果,数值的这种特定偏差将取决于技术效果的性质。例如,与人为或工程技术效果相比,自然或生物技术效果通常可能具有更大的偏差。如本领域技术人员将理解的,对于给定的技术效果,数值的这种特定偏差将取决于技术效果的性质。例如,与人为或工程技术效果相比,自然或生物技术效果通常可能具有更大的偏差。
在描述本发明的上下文中(特别是在权利要求的上下文中)使用的术语“一”、“一个”和“该/所述”以及类似的附图标记应解释为涵盖单数和复数,除非在此另有说明或与上下文明显矛盾。
本文中数值范围的列举仅旨在用作分别指代该范围内的每个单独数值的速记方法。除非本文另外指出,否则每个单独的值都被并入说明书中,就好像它在本文中被单独引用一样。
除非本文另外指出或与上下文明显矛盾,否则本文描述的所有方法可以以任何合适的顺序执行。
本文提供的任何和所有示例或示例性语言(例如,“诸如/例如”)的使用仅旨在更好地说明本发明,并且不对以其他方式要求保护的本发明的范围构成限制。说明书中的任何语言都不应被解释为表示对实施本发明必不可少的任何未要求保护的要素。
在本说明书全文中引用了一些文档。无论是上文还是下文,本文引用的每个文件(包括所有专利、专利申请、科学出版物、制造商的说明书、说明书等)均通过引用全文并入本文。本文中的任何内容均不应解释为承认本发明无权凭借在先发明而早于这种公开。
如本文所使用,术语“[本]发明的”、“根据[本]发明的”、“依据[本]发明的”等意在指代本文描述和/或要求保护的发明的所有方面和实施方式。
应当理解,将本发明的教导应用于特定问题或环境,以及将本发明的变型或其附加特征(诸如另外的方面和实施例)包括在其中,将会是在根据本文所包含的教导的本领域技术人员的能力范围内的。
除非上下文另外指示,否则以上或以下列出的特征的描述和定义不限于本发明的任何特定方面或实施例,并且等同地适用于所描述的所有方面和实施例。
术语“烷基”是指饱和的直链或支链烃的单价基团。优选地,烷基包含1至12个(例如1至10个)碳原子,即1、2、3、4、5、6、7、8、9、10、11或12个碳原子(例如(例如1、2、3、4、5、6、7、8、9或10个碳原子),更优选1至8个碳原子,例如1至6或1至4个碳原子。示例性的烷基包括甲基、乙基、丙基、异丙基(也称为2-丙基或1-甲基乙基)、丁基、异丁基、叔丁基、正戊基、异戊基、仲戊基、新戊基、1,2-二甲基丙基、异戊基、正己基、异己基、仲己基、正庚基、异庚基、正辛基、2-乙基己基、正壬基、正癸基、正十一烷基、正十二烷基等。“取代的烷基”是指烷基的一个或多个(例如结合至烷基的氢原子数的1至最大,例如1、2、3、4、5、6、7、8、9或至多10个,例如1至5、1至4、或1至3、或1或2个)氢原子被氢以外的取代基取代(当取代一个以上的氢原子时,取代基可以是相同或不同)。优选地,除氢以外的取代基是本文所指定的第一级取代基、第二级取代基或第三级取代基,例如卤素、-OH、-NH
术语“亚烷基”是指饱和的直链或支链烃的双自由基。优选地,亚烷基包含1至12个(例如1至10个)碳原子,即1、2、3、4、5、6、7、8、9、10、11或12个碳原子(例如1、2、3、4、5、6、7、8、9或10个碳原子),更优选1至8个碳原子,例如1至6或1至4个碳原子。示例性的亚烷基包括亚甲基、亚乙基(即1,1-亚乙基、1,2-亚乙基)、亚丙基(即1,1-亚丙基、1,2-亚丙基(-CH(CH
术语“烯基”是指具有至少一个碳-碳双键的不饱和直链或支链烃的单价基团。通常,烯基中碳-碳双键的最大数目可以等于整数,该整数是通过如计算得到的:将烯基中的碳原子数除以2,而如果烯基中的碳原子数为奇数,则将除法结果向下舍入到下一个整数。例如,对于具有9个碳原子的烯基,碳-碳双键的最大数目为4。优选地,烯基具有1至6个(例如1至4个),即1、2、3、4、5或6个碳-碳双键。优选地,烯基包含2至12(例如2至10)个碳原子,即2、3、4、5、6、7、8、9、10、11或12个碳原子(例如2、3、4、5、6、7、8、9或10个碳原子),更优选2至8个碳原子,例如2至6个碳原子或2至4个碳原子。因此,在一个优选的实施方案中,烯基包含2至12个(例如2至10个)碳原子和1、2、3、4、5或6(例如1、2、3、4或5个)碳-碳双键,更优选它包含2至8个碳原子和1、2、3或4个碳-碳双键,例如2至6个碳原子和1、2、或3个碳-碳双键或2至4个碳原子和1或2个碳-碳双键。碳-碳双键可以呈顺式(Z)或反式(E)构型。示例性的烯基包括乙烯基、1-丙烯基、2-丙烯基(即烯丙基)、1-丁烯基、2-丁烯基、3-丁烯基、1-戊烯基、2-戊烯基、3-戊烯基、4-戊烯基、1-己烯基、2-己烯基、3-己烯基、4-己烯基、5-己烯基、1-庚烯基、2-庚烯基、3-庚烯基、4-庚烯基、5-庚烯基、6-庚烯基、1-辛烯基、2-辛烯基、3-辛烯基、4-辛烯基、5-辛烯基、6-辛烯基、7-辛烯基、1-壬烯基、2-壬烯基、3-壬烯基、4-壬烯基、5-壬烯基、6-壬烯基、7-壬烯基、8-壬烯基、1-癸烯基、2-癸烯基、3-癸烯基、4-癸烯基、5-癸烯基、6-癸烯基、7-癸烯基、8-癸烯基、9-癸烯基、1-十一碳烯基、2-十一碳烯基、3-十一碳烯基、4-十一碳烯基、5-十一碳烯基、6-十一碳烯基、7-十一碳烯基、8-十一碳烯基、9-十一碳烯基、10-十一碳烯基、1-十二碳烯基、2-十二碳烯基、3-十二碳烯基、4-十二碳烯基、5-十二碳烯基、6-十二碳烯基、7-十二碳烯基、8-十二碳烯基、9-十二碳烯基、10-十二碳烯基、11-十二碳烯基等。如果烯基连接至氮原子,则双键不能为氮原子的α。“取代的烯基”是指烯基中的一个或多个(例如与烯基结合的氢原子数的1至最大,例如1、2、3、4、5、6、7、8、9或至多10个,例如1至5、1至4、或1至3、或1或2个)的氢原子被氢以外的取代基取代(当取代一个以上的氢原子时,取代基可以是相同或不同)。优选地,除氢以外的取代基是本文所指定的第一级取代基、第二级取代基或第三级取代基,例如卤素或任选取代的芳基。取代的烯基的例子是苯乙烯基(即2-苯基乙烯基)。
术语“亚烯基”是指具有至少一个碳-碳双键的不饱和直链或支链烃的双自由基。通常,亚烯基中的碳-碳双键的最大数目可以等于整数,该整数是通过如计算得到的:将亚烯基中的碳原子数除以2,而如果亚烯基中的碳原子数为奇数,则将除法结果向下舍入到下一个整数。例如,对于具有9个碳原子的亚烯基,最大碳-碳双键数为4。优选地,亚烯基具有1至6(例如1至4),即1、2、3、4,5或6个碳-碳双键。优选地,亚烯基基团包含2至12(例如2至10)个碳原子,即2、3、4、5、6、7、8、9、10、11或12个碳原子(例如2、3、4、5、6、7、8、9或10个碳原子),更优选2至8个碳原子,例如2至6个碳原子或2至4个碳原子。因此,在优选的实施方案中,亚烯基包含2至12个(例如2至10个碳)原子和1、2、3、4、5或6(例如1、2、3、4或5个)碳-碳双键,更优选它包含2至8个碳原子和1、2、3或4个碳-碳双键,例如2至6个碳原子和1、2、或3碳-碳双键或2至4个碳原子和1或2个碳-碳双键。碳-碳双键可以呈顺式(Z)或反式(E)构型。示例性的亚烯基包括乙烯-1,2-二基、亚乙烯基(也称为亚乙烯基)、1-丙烯-1,2-二基、1-丙烯-1,3-二基、1-丙烯-2,3-二基、亚芳基、1-丁烯1,2-二基、1-丁烯-1,3-二基、1-丁烯-1,4-二基、1-丁烯-2,3-二基、1-丁烯-2,4-二基、1-丁烯-3,4-二基、2-丁烯1,2-二基、2-丁烯-1,3-二基、2-丁烯-1,4-二基、2-丁烯-2,3-二基、2-丁烯-2,4-二基、2-丁烯-3,4-二基等。如果亚烯基连接至氮原子,则双键不能为氮原子的α。“取代的亚烯基”是指亚烯基基团的一个或多个(例如与亚烯基结合的氢原子数的1至最大,例如1、2、3、4、5、6、7、8、9或至多10,例如1至5、1至4、或1至3或1或2)的氢原子被除氢以外的取代基取代(当取代一个以上的氢原子时,取代基可以是相同或不同)。优选地,除氢以外的取代基是本文所指定的第一级取代基、第二级取代基或第三级取代基,例如卤素或任选取代的芳基。取代的亚烯基的实例是1-苯基-乙烯-1,2-二基和2-苯基-乙烯-1,2-二基。
术语“炔基”是指具有至少一个碳-碳三键的不饱和直链或支链烃的单自由基。通常,炔基中碳-碳三键的最大数目可以等于整数,该整数是通过如计算得到的:将炔基中的碳原子数除以2,而如果炔基中的碳原子数为奇数,则将除法结果向下舍入到下一个整数。例如,对于具有9个碳原子的炔基,碳-碳三键的最大数目为4。优选地,炔基具有1至6个(例如1至4个),即1、2、3、4、5或6个,更优选1或2个碳-碳三键。优选地,炔基包含2至12个(例如2至10个)碳原子(例如1、2、3、4、5、6、7、8、9或10个碳原子),即2、3、4、5、6、7、8、9、10、11或12个碳原子,更优选2至8个碳原子,例如2至6个碳原子或2至4个碳原子。因此,在一个优选的实施方案中,炔基包含2至12个(例如2至10个)碳原子和1、2、3、4、5或6(例如1、2、3、4或5个(优选1、2或3))个碳-碳三键,更优选它包含2至8个碳原子和1、2、3或4个(优选1或2)个碳-碳三键,例如2至6个碳原子和1、2或3个碳-碳三键或2-4个碳原子和1或2个碳-碳三键。炔基的实例包括乙炔基、1-丙炔基、2-丙炔基、1-丁炔基、2-丁炔基、3-丁炔基、1-戊炔基、2-戊炔基、3-戊炔基、4-戊炔基、1-己炔基、2-己炔基、3-己炔基、4-己炔基、5-己炔基、1-庚炔基、2-庚炔基、3-庚炔基、4-庚炔基、5-庚炔基、6-庚炔基、1-辛炔基、2-辛炔基、3-辛炔基、4-辛炔基、5-辛炔基、6-辛炔基、7-辛炔基、1-壬基、2-壬炔基、3-壬炔基、4-壬炔基、5-壬炔基、6-壬炔基、7-壬炔基、8-壬炔基、1-癸炔基、2-癸炔基、3-癸炔基、4-癸炔基、5-癸炔基、6-癸炔基、7-癸炔基、8-癸炔基、9-癸炔基等。如果炔基连接至氮原子,则三键不能为氮原子的α。“取代的炔基”是指炔基的一个或多个(例如与炔基结合的氢原子数的1至最大,例如1、2、3、4、5、6、7、8、9或至多10,例如1-5、1-4、或1-3、或1或2)个氢原子被氢以外的取代基取代(当取代一个以上的氢原子时,取代基可以是相同或不同)。优选地,除氢以外的取代基是本文所指定的第一级取代基、第二级取代基或第三级取代基,例如卤素或任选取代的芳基。
术语“亚炔基”是指具有至少一个碳-碳三键的不饱和直链或支链烃的双自由基。通常,亚炔基中的碳-碳三键的最大数目可以等于整数,该整数是通过如计算得到的:将亚炔基中的碳原子数除以2,而如果亚炔基中的碳原子数为奇数,则将除法结果向下舍入到下一个整数。例如,对于具有9个碳原子的亚炔基,碳-碳三键的最大数目为4。优选地,亚炔基具有1至6(例如1至4),即1、2、3、4,5或6(例如1、2、3或4),更优选1或2个碳-碳三键。优选地,亚炔基包含2至12(例如2至10)个碳原子,即2、3、4、5、6、7、8、9、10、11或12个碳原子(例如2、3、4、5、6、7、8、9或10个碳原子,更优选2至8个碳原子,例如2至6个碳原子或2至4个碳原子。因此,在优选的实施方案中,亚炔基包含2至12个(例如2至10个)碳原子和1、2、3、4、5或6(例如1、2、3、4或5个(优选1、2或3))个碳-碳三键,更优选它包含2至8个碳原子和1、2、3或4个(优选1或2)个碳-碳三键,例如2至6个碳原子和1、2或3个碳-碳三键或2-4个碳原子和1或2个碳-碳三键。示例性的亚炔基包括乙炔基1,2-二基、1-丙炔基-1,3-二基、1-丙炔基-3,3-二基、1-丁炔基-1,3-二基、1-丁炔基-1,4-二基、1-丁基-3,4-二基、2-丁基-1,4-二基等。如果亚炔基连接至氮原子,则三键不能为氮原子的α。“取代的亚炔基”是指亚炔基的一个或多个(例如与亚炔基结合的氢原子数的1至最大,例如1、2、3、4、5、6、7、8、9或至多10,例如1至5、1至4,或1至3或1或2)氢原子被除氢以外的取代基取代(当取代一个以上的氢原子时,取代基可以是相同或不同)。优选地,除氢以外的取代基是本文所指定的第一级取代基、第二级取代基或第三级取代基,例如卤素或任选取代的芳基。
术语“芳基”或“芳族环”是指芳族环状烃的单自由基。优选地,芳基含有3至14个(例如5、6、7、8、9或10,例如5、6或10)个碳原子,其可以排列在一个环(例如苯基)或两个环中。或更多个稠环(例如萘基)。示例性的芳基包括环丙烯基、环戊二烯基、苯基、茚基、萘基、azulenyl基、芴基、蒽基和菲基。优选地,“芳基”是指含有6个碳原子的单环或含有10个碳原子的芳族双环系统。优选的实例是苯基和萘基。芳基不包括富勒烯。“取代的芳基”是指芳基中的一个或多个(例如与芳基结合的氢原子数的1至最大,例如1、2、3、4、5、6、7、8、9或至多10个,例如1-5、1-4、或1-3、或1或2个)氢原子被氢以外的取代基取代(当取代一个以上的氢原子时,取代基可以是相同或不同)。优选地,除氢以外的取代基是本文所指定的第一级取代基、第二级取代基或第三级取代基,例如卤素、-CN、硝基、OR
术语“杂芳基”或“杂芳族环”是指如上所定义的芳基,其中该芳基中的一个或多个碳原子被杂原子(例如O、S或N)取代。优选地,杂芳基是指五元或六元芳族单环,其中1、2或3个碳原子被O、N或S的相同或不同杂原子取代。可替代地,其表示芳族双环或三环系统,其中1、2、3、4或5个碳原子被O、N或S的相同或不同的杂原子取代。优选地,在杂芳基的每个环中,最大O原子数为1,最大S原子数为1,O和S原子的最大总数为2。例如,3至14元杂芳基包括单环杂芳基(例如5或6元)、双环杂芳基(例如9或10元)和三环杂芳基(例如13或14元)。示例性的杂芳基基团包括呋喃基、噻吩基、恶唑基、异恶唑基、恶二唑基(1,2,5-和1,2,3-)、吡咯基、咪唑基、吡唑基、三唑基(1,2,3-和1,2,4-)、四唑基、噻唑基、异噻唑基、噻二唑基(1,2,3-和1,2,5-)、吡啶基(也称为吡啶基)、嘧啶基、吡嗪基、三嗪基(1,2,3-、1,2,4-和1,3,5-)、苯并呋喃基(1-和2)、吲哚基、异吲哚基、苯并噻吩基(1-和2-)、1H-吲唑基、苯并咪唑基、苯并恶唑基、吲哚并嗪基、苯并异唑基、苯并噻唑基、苯并异噻唑基、喹啉基、异喹啉基、苯并二嗪基、喹喔啉基、喹唑啉基、苯并三嗪基(1,2,3-和1,2,4-苯并三嗪基)、哒嗪基、苯恶嗪基、噻唑并吡啶基、吡咯并噻唑基、苯并噻吩基、异噻吩并噻吩基、异苯并呋喃基、吲唑基、嘌呤基、喹啉基、邻苯二甲酰基、萘啶基(1,5-、1,6-、1,7-、1,8-和2,6-)、肉桂啉基、蝶啶基、咔唑基、菲啶基、,啶基、哌啶基、苯乙炔基邻苯二甲酰基(1,7-、1,8-、1,10-、3,8-和4,7-)、吩嗪基、恶唑并吡啶基、异恶唑并吡啶基、吡咯并恶唑基和吡咯并吡咯基。示例性的5元或6元杂芳基包括呋喃基、噻吩基、恶唑基、异恶唑基、恶二唑基(1,2,5-和1,2,3-)、吡咯基、咪唑基、吡唑基、三唑基(1,2,3-和1,2,4-)、噻唑基、异噻唑基、噻二唑基(1,2,3-和1,2,5)、吡啶基、嘧啶基、吡嗪基、三嗪基(1,2,3-、1,2,4-和1,3,5-)和哒嗪基。“取代的杂芳基”是指杂芳基中的一个或多个(例如与杂芳基键合的1至最大数目的氢原子,例如1、2、3、4、5、6、7、8、9或至多10个,例如1-5个、1-4个或1-3个或1个或2个)的氢原子被氢以外的取代基取代(当取代一个以上的氢原子时,取代基可以是相同或不同)。优选地,除氢以外的取代基是本文所指定的第一级取代基、第二级取代基或第三级取代基,例如卤素、-CN、硝基、OR
术语“环烷基”或“环脂族”表示具有优选3至14个碳原子(例如3至12或3至10个碳原子(即3、4、5、6、7、8、9、10、11、12、13或14个碳原子(例如3、4、5、6、7、8、9或10个碳原子),更优选3到7个碳原子)的环状非芳族形式的“烷基”和“烯基”。示例性的环烷基包括环丙基、环丙烯基、环丁基、环丁烯基、环戊基、环戊烯基、环己基、环己烯基、环庚基、环庚烯基、环辛基、环辛烯基、环壬基、环壬烯基、环癸基、环癸烯基和金刚烷基。术语“环烷基”还旨在包括其双环和三环形式。如果形成双环,则优选各个环在两个相邻的碳原子上彼此连接,但是,可替代地,两个环通过相同的碳原子连接,即,它们形成螺环系统或它们形成“桥连”环系统。环烷基的优选实例包括C
术语“杂环基”或“杂环”是指如上所定义的环烷基,其中环烷基中的1、2、3或4个环碳原子被杂原子取代(例如选自O、S、S(O)、S(O)
本文所用的不饱和化合物或基团的“部分氢化形式”是指通过将氢正式加成至最初的不饱和化合物或基团而不除去所有不饱和基团而除去了部分不饱和基团。术语“不饱和化合物或基团的完全氢化形式”在本文中与术语“全氢”可互换使用,并且是指通过将氢正式加成到最初的不饱和化合物或基团中,所有不饱和键都已被除去。例如,5元杂芳基的部分氢化形式(在环中含有2个双键,例如呋喃)包括所述5元杂芳基的二氢形式(例如2,3-二氢呋喃或2,5-二氢呋喃),而所述5元杂芳基的四氢形式(例如,四氢呋喃,即THF)是所述5元杂芳基的完全氢化(或全氢)形式。同样,对于在环上具有3个双键的6元杂芳基(例如吡啶基),部分氢化形式包括二氢和四氢形式(例如二氢和四氢吡啶基),而六氢形式(如杂芳基吡啶基,如哌啶基)是所述6-元杂芳基的完全氢化的(或全氢的)衍生物。因此,如果芳基或杂芳基的六氢形式仅被认为是部分氢化的形式,则该芳基或杂芳基包含至少四个由环原子之间的双键和三键组成的不饱和部分。
在烃的上下文中使用的术语“芳族”是指整个分子必须是芳族的。例如,如果单环芳基被氢化(部分或全部),则出于本发明的目的,所得的氢化环状结构被分类为环烷基。同样地,如果将双环或多环芳基(例如萘基)氢化,则出于本发明的目的,将所得的氢化双环或多环结构(例如1,2-二氢萘基)归类为环烷基(即使有一个环,例如1,2-二氢萘基,仍为芳香族)。在本申请中,在杂芳基和杂环基之间有相似的区别。例如,出于本发明的目的,吲哚基,即吲哚基的二氢变体,被分类为杂环基,因为双环结构中只有一个环是芳族的,而环原子中的一个是杂原子。
如本文所用,术语“多环”是指该结构具有两个或更多个(例如2、3、4、5、6、7、8、9或10),优选地为2、3、4、或5,更优选2、3或4个环。因此,根据本发明,术语“多环”不包括单环结构,其中该结构仅包含一个环。多环基团的例子是稠合结构(例如萘基或蒽基)、螺环化合物、通过单键或双键连接的环(例如联苯)和桥连结构(例如冰片基)。示例性多环结构是具有至少两个环的上述芳基、杂芳基、环烷基和杂环基。
术语“卤素”是指氟、氯、溴或碘。
术语“叠氮基”是指-N
术语“N-氧化物”是指胺氧化物或胺-N-氧化物,其为包含官能团(R
如本文其他地方所述,R
如本文所用,表述“一个R
其中
但排除以下结构:
如本文所用,术语“k元环”是指该环具有k个环原子。例如,对于吡唑基,k为5;因此,相对于吡唑基与化合物其余部分结合的环原子(基位置),邻位为位置2和5,位置k-1为位置4。此外,对于吡啶基为6元杂芳基的情况,相对于吡啶基与化合物的其余部分结合是环原子(基位置),邻位是位置2和6,位置k-1是位置5。
如本文所用,表述“
术语“任选(地)取代的”是指一个或多个(例如键合至基团的氢原子中1至最大数目,例如1、2、3、4、5、6、7、8、9,或最多10个,例如1至5、1至4或1至3或1或2个)氢原子可以被不同于氢的基团(即第一级取代基)取代,例如烷基(优选C
其中
R
R
X
典型的第一级取代基优选选自C
典型的第二级取代基优选选自C
典型的第三级取代基优选选自C
如本文所用,术语“任选的”或“任选地”是指随后描述的事件、情况或状况可能发生或可能不发生,并且该描述包括发生所述事件、情况或状况发生的情况以及不发生的情况。
“异构体”是具有相同分子式但结构(“结构异构体”)或官能团和/或原子的几何(空间)位置不同(“立体异构体”)的化合物。“对映异构体”是一对立体异构体,它们是彼此不可重叠的镜像。“外消旋混合物”或“外消旋物”包含等量的一对对映异构体,并用前缀(±)表示。“非对映异构体”是不可重叠的并且不是彼此的镜像的立体异构体。“互变异构体”是同一化学物质的结构异构体,其由于单个原子或原子团的迁移,可自发可逆地相互转化(即使是纯净的)。即,互变异构体彼此处于动态化学平衡。互变异构体的例子是酮-烯醇-互变异构体的异构体。“构象异构体”是仅通过围绕形式上单键的旋转就可以相互转化的立体异构体,并且尤其包括导致(杂环)环的3维形式不同的那些异构体,例如环己烷的椅子、半椅子、船形物和扭曲船形物的形式。
在本申请中所示的结构式可以被解释为包含一个以上的异构体的情况下,除非另外明确指出,否则所述结构式包括所有可能的异构体,因此包括每个单独的异构体。例如,其中R
在本文中,“多态性”是指固体材料(例如化合物)能够以一种以上的形式或晶体结构存在,即“多态性修饰”或“多态性形式”。术语“多晶型修饰”、“多晶型形式”和“多晶型”在本发明中可互换使用。根据本发明,这些“多晶型修饰”包括结晶形式、无定形形式、溶剂化物和水合物。主要是,存在不同的多晶型形式的原因在于在结晶过程中使用了不同的条件,例如以下:
·溶剂效应(极性和非极性溶剂的晶体堆积可能不同);
·某些杂质会抑制生长方式,并有利于亚稳多晶型物的生长;
·材料从中结晶出来的过饱和度(通常高于溶解度的浓度越高,形成亚稳态的可能性就越大);
·进行结晶的温度;
·共价键的几何形状(导致构象多态性的差异);
·改变搅拌条件。
多晶型物可能具有不同的化学、物理和/或药理特性,包括但不限于熔点、X射线晶体和衍射图、化学反应性、溶解度、溶解速率、蒸气压、密度、吸湿性、流动性、稳定性、致密性和生物利用度。在特定温度下,多晶型可以自发地从亚稳形式(不稳定形式)转变为稳定形式。根据奥斯特瓦尔德(Ostwald)的规则,通常首先结晶的不是最稳定的,而是最不稳定的多晶型物。因此,化合物(例如本发明的化合物)的质量、功效、安全性、可加工性和/或制造会受到多态性的影响。通常,由于转化成另一种多晶型物的可能性最小,因此选择化合物(例如本发明的化合物)的最稳定的多晶型物。然而,由于除了稳定性以外的原因,例如溶解度、溶解速度和/或生物利用度,可能会选择不是最稳定的多晶型形式的多晶型形式。
本文所用的术语材料的“晶体形式”是指所述材料的最小组分(即原子、分子或离子)形成晶体结构。这里所说的“晶体结构”是指结晶液体或固体中原子或分子的独特的三维排列,其特征在于图案、以特定方式排列的一组原子以及表现出远距离有序和对称的晶格。晶格是在三个维度上周期性重复的点的阵列,并且图案位于晶格的这些点上。晶格的亚单位是晶胞。晶格参数是晶胞边缘的长度以及它们之间的角度。晶体的对称性体现在其空间群中。为了描述晶体结构,需要以下参数:化学式、晶格参数、空间群、原子坐标和点位置的占据数。
本文所用的术语“材料的无定形形式”是指所述材料的最小组分(即原子、分子或离子)不是排列成晶格而是随机排列。因此,不同于其中存在短程有序(到下一个相邻原子的恒定距离)和长程有序(基本晶格的周期性重复)的晶体,仅短程有序以非晶形式存在。
如本文所用,术语“化合物的复合物”是指通过使该化合物与其他一个或多个其他分子缔合而生成的高级化合物。化合物的示例性复合物包括但不限于所述化合物的溶剂化物、簇和螯合物。
如本文所用,术语“溶剂化物”是指溶解的材料在溶剂(例如有机溶剂(例如,脂肪族醇(例如,甲醇、乙醇、正丙醇、异丙醇)、丙酮、乙腈、乙醚等)、水或其中两种或多种液体的混合物)中的加成配合物,其中加成配合物以晶体或混合晶体的形式存在。加成配合物中所含溶剂的量可以是化学计量的或非化学计量的。“水合物”是溶剂化物,其中溶剂是水。
在同位素标记的化合物中,一个或多个原子被质子数量相同但中子数量不同的相应原子取代。例如,氢原子可以被氘原子代替。可用于本发明化合物的示例性同位素包括氘、
术语“半衰期”是指消除分子的一半活性、数量或数目所需的时间。在本发明的上下文中,式(I)或(Ia)的化合物的半衰期指示所述化合物的稳定性。
术语“受试者/对象”、“患者”、“个体”或“动物”涉及多细胞动物,例如脊椎动物。例如,在本发明的上下文中,脊椎动物是哺乳动物、鸟类(例如家禽)、爬行动物、两栖动物、骨性鱼类和软骨鱼类,特别是前述任何一种的驯养动物以及圈养的动物(特别是脊椎动物),例如动物园的动物(特别是脊椎动物)。在本发明的上下文中,哺乳动物包括但不限于人类、非人类灵长类、驯养的哺乳动物(例如狗、猫、绵羊、牛、山羊、猪、马等)、实验室哺乳动物(例如小鼠、老鼠、兔子、豚鼠等)、以及圈养的哺乳动物(例如动物园的哺乳动物)。本文所用的术语“动物”也包括人类。鸟类的特定非限制性实例包括驯养的家禽,并且包括鸟类,例如鸡、火鸡、鸭、鹅、珍珠鸡、鸽子、野鸡等。而骨性或软骨性鱼类的特定非限制性实例包括适合通过水产养殖进行养殖的那些,并且包括骨性鱼类,例如鲑鱼、鳟鱼、鲈鱼、鲤鱼、鲶鱼等。
化合物达沙替尼(在本文中也称为化合物A8)具有以下结构:
在第一个方面,并且如本文可以进一步描述、定义、要求保护或以其他方式公开的,本发明提供了一种化合物,其选自下式的激酶抑制剂:
以及其溶剂化物、盐、N-氧化物、络合物、多晶型物、晶体形式、外消旋混合物、非对映异构体、对映异构体、互变异构体、构象异构体、同位素标记形式、前药及其组合;
其中:
R
R
R
R
R
R
L选自键、C
R
R
A选自S、O、NR
R
R
X独立地选自O、S和N(R
E是O或S;
B是N或CR
R
R
R
R
R
y是0到2的整数;
R
R
其中
R
R
X
在一个实施方案中,该激酶抑制剂具有式(II):
其中R
在式(II)的激酶抑制剂的一个实施方案中,R
在式(II)的激酶抑制剂的一个实施方案中,R
在式(II)的激酶抑制剂的一个实施方案中,R
在式(II)的激酶抑制剂的一个实施方案中,R
在式(II)的激酶抑制剂的另一个实施方案中,R
在一个实施方案中,该激酶抑制剂具有通式(III)
其中R
在式(III)的激酶抑制剂的一个实施方案中,R
在式(III)的激酶抑制剂的一个实施方案中,R
在一个相关方面,本发明提供了一种化合物,其选自式(Ia)的激酶抑制剂:
以及其溶剂化物、盐、N-氧化物、络合物、多晶型物、晶体形式、外消旋混合物、非对映异构体、对映异构体、互变异构体、构象异构体、同位素标记形式、前药及其组合;其中R
在式(Ia)的激酶抑制剂的一个实施方案中,R
其中上述a)、b)、c)、d)、e)、f)和g)中指定的每个基团任选地被一个、两个、三个、四个、五个或六个独立选择的R
在式(Ia)的激酶抑制剂的任何上述实施方案中,Hy可以被一个R
在式(Ia)激酶抑制剂的上述任何实施方案中,优选A为S和/或E为O。
在一个实施方案中,该激酶抑制剂具有通式(IV)
其中R
在式(IV)的激酶抑制剂的一个实施方案中,R
在式(IV)的激酶抑制剂的一个实施方案中,R
在式(IV)的激酶抑制剂的一个实施方案中,R
在一个实施方案中,该激酶抑制剂具有通式(V)
其中R
在式(V)的激酶抑制剂的一个实施方案中,R
在式(V)的激酶抑制剂的一个实施方案中,R
在式(V)的激酶抑制剂的一个实施方案中,R
在式(V)的激酶抑制剂的一个实施方案中,R
在式(V)的激酶抑制剂的一个实施方案中,R
在式(V)的激酶抑制剂的一个实施方案中,R
在式(V)的激酶抑制剂的任何上述实施方式(包括式(I)、(II)、(III)和(IV)的激酶抑制剂)中,R
在式(V)的激酶抑制剂(包括式(I)、(II)、(III)和(IV)的激酶抑制剂)的任何上述实施方案中,其中R
在式(V)的激酶抑制剂的一个实施方案中,R
其中
在式(V)的激酶抑制剂的一个实施方案中,R
其中
在式(V)的激酶抑制剂的一个实施方案中,R
其中
在式(V)的激酶抑制剂的一个实施方案中,R
其中
在式(V)的激酶抑制剂的任何上述实施方案中(包括式(I)、(II)、(III)和(IV)的激酶抑制剂),L可以选自键、C
在式(V)的激酶抑制剂的任何上述实施方案中(包括式(I)、(II)、(III)和(IV)的激酶抑制剂),其中R
在一个实施方案中,该激酶抑制剂具有通式(VI)
其中R
在一个实施方案中,该激酶抑制剂具有通式(VII)
其中R
在一个实施方案中,该激酶抑制剂具有通式(VIII)
其中R
(A)R
(B)R
(C)R
(D)R
(E)A选自S、O、NH、N(C
(F)B为N或CR
(G)E为O或S,优选为O。
在具有通式(VIII)的激酶抑制剂的一个优选实施方案中,R
在具有通式(VIII)的激酶抑制剂的进一步优选的实施方式中:
(A')R
(B')R
(C')R
(D')R
(E')A为S、O或N(CH
(F')B是N或CR
(G')E为O或S,优选为O。
在具有通式(VIII)的激酶抑制剂的一个优选实施方案中,R
在具有通式(VIII)的激酶抑制剂的进一步优选的实施方式中:
(A”)R
(B”)(a)R
(C”)R
(D”)R
(E”)A是S;
(F”)B为N;和/或
(G”)E是O。
在具有通式(VIII)的激酶抑制剂的一个优选实施方案中,R
在一个实施方案中,本发明的化合物选自表A和/或表B所示的化合物。
意图是本发明的化合物(特别是式(I)、(Ia)、(II)、(III)、(IV)、(V)、(VI)、(VII)和(VIII)(例如以下表A和/或表B中所描述的化合物)不仅涵盖所描述的化合物,还涵盖其溶剂化物(例如水合物)、盐(特别是药学上可接受的盐)、N-氧化物(尤其是R
选择的化合物,包括在本发明的范围内或在本发明的方法中使用的已合成和测试的化合物,和/或各自单独或以任何组合用于合成本发明的其他化合物的代表各种示例性或优选的R
表A:式(I)的激酶抑制剂。
*化合物B3包含在同时待审的PCT/EP2018/060172的表1中。
表B:其他的式(I)激酶抑制剂
在特定的实施方案中,本发明的化合物选自C7和C8和/或D1和/或D9,或在某些实施方案中,本发明的化合物为B3;以及它们的溶剂化物、盐、N-氧化物、络合物、多晶型物、晶体形式、互变异构体、构象异构体、同位素标记的形式、前药及其组合。
在另一个特定的实施方案中,本发明的化合物为C12或溶剂化物、盐、N-氧化物、络合物、多晶型物、结晶形式、外消旋混合物、非对映异构体、对映异构体、互变异构体、构象异构体、同位素标记形式、前药、或其组合。
在某些实施方案中,本发明可以涉及本发明的任何化合物的溶剂化物、盐、N-氧化物、络合物、外消旋混合物、非对映异构体、对映异构体、互变异构体、构象异构体、同位素标记形式、前药或其组合;例如这类化合物的溶剂化物、盐、络合物、外消旋混合物、非对映异构体、对映异构体、互变异构体、构象异构体、同位素标记形式或其组合。
在一个实施方案中,本发明的化合物不包括式(I)的以下群组(1)至(5)中的一个或多个的化合物(在群组(1)至(5)中,一个部分(例如甲基)是未取代的,除非明确指出所述部分是被取代的):
(1)当R
(2)当R
(3)当R
(i)R
(ii)R
(4)当E为O,B为CR
(5)当R
在一个实施方案中,本发明的化合物不包括具有式(Ia)的以下群组(6)至(8)的一个或多个化合物(在群组(6)至(8)中,一个部分(例如甲基)是未取代的,除非明确指出所述部分是被取代的):
(6)当Hy是
(i)R
(ii)当R
(7)当Hy为1-{(2E)-4-[(2-甲氧基乙基)氨基]-1-氧代-2-丁烯-1-基}哌啶-4-基,R
(8)当R
(i)R
(ii)Hy为1-(苯甲基)哌啶-4-基时,则R
(iii)Hy是1-(苯甲基)吡咯烷-3-基时,则R
在某些其他实施方案中,该化合物不是选自以下的一种:
·5-噻唑甲酰胺,2-[(6-氯-2-甲基-4-嘧啶基)氨基]-N-[2-[4-(2-羟乙基)-1-哌嗪基]-6-甲基苯基]-(CAS登记号:2048106-50-7),
·5-噻唑甲酰胺,2-[[7-[4-氰基-3-(三氟甲基)苯基]-5,6,7,8-四氢吡啶基[3,4-d]嘧啶-4-基]氨基]-N-[(1R)-1-(1,3,4-恶二唑-2-基)乙基]-4-(三氟甲基)-(CAS登记号:1831086-00-0),
·5-噻唑甲酰胺,2-[[6-甲氧基-7-[3-(4-吗啉基)丙氧基]-4-喹唑啉基]氨基]-N-(2-吡嗪基甲基)-(CAS登记号:385780-87-0),
·5-噻唑甲酰胺,2-[[6-甲氧基-7-[3-(4-吗啉基)丙氧基]-4-喹唑啉基]氨基]-N-[2-(3-吡啶基)乙基]-(CAS登记号:385780-82-5),
·5-噻唑甲酰胺,N-1H-吲哚-5-基-2-[[6-甲氧基-7-[3-(4-吗啉基)丙氧基]-4-喹唑啉基]氨基]-(CAS登记号:385780-79-0),
·5-噻唑甲酰胺,2-[[6-甲氧基-7-[3-(4-吗啉基)丙氧基]-4-喹唑啉基]氨基]-N-(2-噻吩甲基)-(CAS登记号:385780-69-8),
·5-噻唑甲酰胺,N-(2-呋喃基甲基)-2-[[6-甲氧基-7-[3-(4-吗啉基)丙氧基]-4-喹唑啉基]氨基]-(CAS登记号:385780-66-5),
·5-噻唑甲酰胺,2-[[6-甲氧基-7-[3-(4-吗啉基)丙氧基]-4-喹唑啉基]氨基]-N-2-吡啶基-(CAS登记号:385780-57-4),以及
·5-噻唑甲酰胺,2-[[6-甲氧基-7-[3-(4-吗啉基)丙氧基]-4-喹唑啉基]氨基]-N-4-吡啶基-(CAS登记号:385779-93-1)。
在其他某些其他实施方案中,该化合物不是选自以下的一种:
·咪唑并[4,5-d]吡咯并[2,3-b]吡啶-7-羧酰胺,N,N-二环丙基-6-乙基-1,6-二氢-1-甲基-4-[[4-甲基-5-[[(四氢-2H-吡喃-4-基)氨基]羰基]-2-噻唑基]氨基]-(CAS登记号:1271022-78-6),
·咪唑并[4,5-d]吡咯并[2,3-b]吡啶-7-羧酰胺,N,N-二环丙基-6-乙基-1,6-二氢-1-甲基-4-[[4-甲基-5-[[(四氢-1,1-二氧-3-噻吩基)氨基]羰基]-2-噻唑基]氨基]-(CAS登记号:1271022-57-1),
·咪唑并[4,5-d]吡咯并[2,3-b]吡啶-7-羧酰胺,N,N-二环丙基-6-乙基-1,6-二氢-1-甲基-4-[[4-甲基-5-[[[2-(4-吗啉基)乙基]氨基]羰基]-2-噻唑基]氨基]-(CAS登记号:1271022-45-7),以及
·咪唑并[4,5-d]吡咯并[2,3-b]吡啶-7-羧酰胺,N,N-二环丙基-6-乙基-1,6-二氢-1-甲基-4-[[5-[[甲基(四氢-1,1-二氧-3-噻吩基)氨基]羰基]-2-噻唑基]氨基]-(CAS登记号:1271021-43-2)
·2-[(6-{[3-(1H-咪唑-1-基)丙基]氨基}吡啶-2-基)氨基]-4-甲基-N-[1-(苯甲基)-1H-吲唑-5-基]-1,3-噻唑-5-羧酰胺(CAS登记号:302963-64-0)
·2-[(6-{[3-(1H-咪唑-1-基)丙基]氨基}吡啶-2-基)氨基]-N-[1-(苯甲基)-1H-吲唑-5-基]-1,3-噻唑-5-羧酰胺(CAS登记号:302963-55-9)。
在本文一个或多个方面的某些替代方案中,该化合物为N-(2-氯-6-甲基苯基)-2-((6-(4-(2-羟乙基)哌嗪-1-基)-2-甲基嘧啶-4-基)氨基)噻唑-5-羧酰胺(达沙替尼;A8)。
含有碱性官能团的本发明化合物可以与多种无机或有机酸形成盐。含有酸性官能团的本发明化合物可以与多种无机或有机碱形成盐。本发明化合物的示例性无机和有机酸/碱以及示例性酸/碱加成盐在以下“药物组合物”部分的“药学上可接受的盐”的定义中给出。包含碱性和酸性官能团的本发明化合物可以转化为碱或酸加成盐。发明化合物的中性形式可以通过使盐与碱或酸接触并以常规方式分离母体化合物来再生。
本发明的化合物可以是N-氧化物的形式,即它们可以包含官能团官能团≡N
本发明的化合物可以是前药形式。本发明化合物的前药是那些在给予个体后在生理条件下进行化学转化以提供本发明化合物的化合物。另外,可以在离体环境中通过化学或生化方法将前药转化为本发明的化合物。例如,当例如与合适的酶或化学试剂一起置于透皮贴剂储库中时,前药可以缓慢地转化为本发明的化合物。示例性前药是在体内可水解的酯(使用本发明的激酶抑制剂中包含的醇或羧基)或酰胺(使用本发明的激酶抑制剂中包含的氨基或羧基)。具体地,本发明的激酶抑制剂中包含的并且带有至少一个氢原子的任何氨基可以被转化为前药形式。典型的N前药形式包括氨基甲酸酯(1)、曼尼希(Mannich)碱(2)、烯胺(3)和烯胺酮(4)。
其中R
本发明化合物的特定前药形式是具有式(IXa)、(IXb)、(IXc)或(IXd)的那些(前药):
以及溶剂化物、盐、N-氧化物、络合物、多晶型物、晶体形式、外消旋混合物、非对映异构体、对映异构体、互变异构体、构象异构体、同位素标记形式及其组合,其中R
对于具有式(I)、(Ia)、(II)、(III)、(IV)、(V)、(VI)、(VII)、(VIII)、(IXa)、(IXb)、(IXc)和(IXd)中任一项并带有一个或多个羟基(即-OH)的本发明化合物,另一种特别的前药形式是其中两个或多个羟基中的至少一个被衍生化为选自-OP(O)(OR
在某些实施方案中,本发明可以涉及具有式(IXa)、(IXb)、(IXc)或(IXd)的前药的溶剂化物、盐、N-氧化物、络合物、外消旋混合物、非对映异构体、对映异构体、互变异构体、构象异构体、同位素标记形式或其组合;例如这种前药的溶剂化物、盐、络合物、外消旋混合物、非对映异构体、对映异构体、互变异构体、构象异构体、同位素标记形式或其组合。
在一个特定的实施方案中,本发明的化合物是标题“化合物”下指定的激酶抑制剂(例如,具有通式(I)、(Ia)、(II)、(III)、(IV)、(V)、(VI)、(VII)或(VIII)的激酶抑制剂)的水合物,合适地是一水合物或二水合物,或溶剂化物、盐(特别是药学上可接受的盐)、N-氧化物(尤其是R
在某些实施方案中,本发明的化合物(例如,如在标题“化合物”下指定的)可以是纯化的或(例如,基本上)纯的形式(例如,以其提供)。例如,化合物的纯度可以大于约50%,例如大于约60%、70%或80%,合适地大于约90%(特别是大于约95%,97%98%甚至99%)。也就是说,在某些这样的实施方案中,这样的化合物与仅有限量的杂质(例如,诸如在制造期间引入的那些杂质)一起存在,例如仅存在少量的杂质,包括其中化合物存在于基本上不存在杂质的地方的实施方案。化合物的纯度(例如,不存在、或杂质的存在程度)可以通过常规方法确定,例如通过HLPC。
在一个实施方案中,本发明提供了这样一种化合物,其HPLC面积小于约50%、40%、30%且合适地为10%或5%,优选HPLC面积小于约3%和2%,更优选HPLC的总杂质面积小于1%。如本文所用,术语“HPLC的%面积”是指HPLC色谱图中一个或多个峰的面积与HPLC色谱图中所有峰的总面积相比,以总面积的百分数表示。此外,化合物的纯度在本文中可以表示为“HPLC”纯度。这样,“HPLC纯度”是化合物峰下的面积除以HPLC色谱图中曲线下的总面积的计算值。合适地,通过HPLC,该化合物包含小于约10%的总杂质面积。更优选HPLC的总杂质面积小于约5%。
在相关的方面并且如将进一步描述、定义、请求保护或公开的,本发明提供了一个或多个容器,其中所述容器(每个独立地或全部共同地)包含第一个方面的激酶抑制剂(例如,具有通式(I)、(Ia)、(II)、(III)、(IV)、(V)、(VI)、(VII)或(VIII)的激酶抑制剂、或溶剂化物、盐(特别是药学上可接受的盐)、N-氧化物(尤其是R
另一方面,本发明提供了本发明的化合物(特别是以上关于式(I)、(Ia)、(II)、(III)、(IV)、(V)、(VI)、(VII)和(VIII))用作药物,例如用于治疗。在这个方面的一个实施方案中,本发明的化合物不包括具有式(I)并且属于如上所述的群组(1)、(2)、(3)、(4)和/或(5)的一个或多个的化合物(例如群组(1)(特别是当R
从实施例显而易见,发明人发现本发明的化合物以及其他结构相似的化合物抑制一种或多种选自ABL1/BCR-ABL、SRC、LCK、KIT、FLT3及其突变体和/或SIK1、SIK2和SIK3和/或PHA2、EPHA4、CSF-R1、HCK和ACK1;和/或NEK11、WEE1、WNK2、Aurora-A、Aurora-B和TBK1的蛋白酪氨酸激酶。在一个实施方案中,本发明的化合物表现出的药理学特性(选择性、生物利用度、毒性、副作用、剂量、患者依从性、相容性、稳定性、半衰期等)在至少一方面优于沙替尼所展现的药理学特性。
本发明中描述的化合物(特别是上面指定的化合物,例如式(I)、(Ia)、(II)、(III)、(IV)、(V)、(VI)、(VII))或(VIII)的化合物,特别是表A和/或表B的化合物)优选通过药物组合物施用于需要其的患者。因此,在第二方面,本发明提供了一种药物组合物,其包含如上文在标题“化合物”下指定的激酶抑制剂(例如,具有通式(I)、(Ia)、(II)、(III)、(IV)、(V)、(VI)、(VII)或(VIII)的激酶抑制剂)或溶剂化物、盐(特别是药学上可接受的盐)、N-氧化物(特别是R
因此,在一个实施方案中,药物组合物包含如上文在“化合物”标题下指定的激酶抑制剂和一种或多种药学上可接受的赋形剂。此外,药物组合物可以进一步包含一种或多种其他治疗剂。因此,在特定的实施方案中,药物组合物包含(i)如上文在“化合物”标题下指定的激酶抑制剂和一种或多种其他治疗剂;或(ii)如上文在“化合物”标题下指定的激酶抑制剂、一种或多种其他治疗剂和一种或多种药学上可接受的赋形剂。
术语“药学上可接受的”是指不与药物组合物的活性成分(例如本发明的激酶抑制剂,单独或与一种或多种其他治疗剂组合)的(例如治疗)作用相互作用的材料的无毒性物。
可以通过任何途径,例如肠内或肠胃外,将药物组合物施用至个体。
如本文所用,表述“肠内施用”和“肠内地施用”是指所施用的药物被胃和/或肠吸收。肠内施用的例子包括口服和直肠施用。如本文所用,表述“肠胃外施用”和“肠胃外地施用”是指除肠内施用以外的施用方式,通常通过注射或局部施用,包括但不限于静脉内、肌肉内、动脉内、鞘内、囊内、骨内、眶内、心内、皮内、腹膜内、经气管、皮下、表皮下、关节内、包膜下、脑内、脑室内、蛛网膜下、脊柱内、硬膜外和胸骨内给药(例如通过注射和/或输注)以及局部给药(例如表皮、吸入、或通过粘膜(如颊、舌下或阴道))。
本发明的化合物通常以“药学上可接受的量”和“药学上可接受的制剂”的形式施用。这样的组合物可以包含盐、缓冲剂、防腐剂、载体和任选的其他治疗剂。“药学上可接受的盐”包括例如酸加成盐,其可以例如通过将化合物的溶液与药学上可接受的酸(例如盐酸、硫酸、富马酸、马来酸、琥珀酸、乙酸、苯甲酸、柠檬酸、酒石酸、碳酸或磷酸)的溶液混合而形成。此外,在化合物带有酸性部分的情况下,其合适的药学上可接受的盐可包括碱金属盐(例如钠或钾盐);碱土金属盐(例如钙盐或镁盐);以及与合适的有机配体形成的盐(例如铵盐、季铵盐和胺阳离子,这些阳离子是使用抗衡阴离子,例如卤化物、氢氧根、羧酸根、硫酸根、磷酸根、硝酸根、烷基磺酸根和芳基磺酸根形成的)。药学上可接受的盐的说明性实例包括但不限于乙酸盐、己二酸盐、藻酸盐、精氨酸盐、抗坏血酸盐、天冬氨酸盐、苯磺酸盐、苯甲酸盐、碳酸氢盐、硫酸氢盐、酒石酸氢盐、硼酸盐、溴化物、丁酸盐、依地酸钙、樟脑酸盐、樟脑磺酸盐、右旋樟脑磺酸、碳酸盐、氯化物、柠檬酸盐、克拉维酸盐、环戊烷丙酸盐、二葡萄糖酸盐、二盐酸盐、十二烷基硫酸盐、依地酸盐(edetate)、乙二磺酸盐(edisylate)、依托酸盐(estolate)、乙磺酸盐(esylate)、乙磺酸酯(ethanesulfonate)、甲酸盐、富马酸盐、半乳糖酸盐(galactate)、半乳糖醛酸盐(galacturonate)、葡庚糖酸盐(gluceptate)、葡庚糖酸盐(glucoheptonate)、葡糖酸盐(gluconate)、谷氨酸盐(glutamate)、甘油磷酸盐(glycerophosphate)、对α-羟乙酰氨基苯胂酸盐(glycolylarsanilate)、半硫酸盐(hemisulfate)、庚酸盐、己酸盐、己基间苯二酸酯、氢溴酸胺、氢溴酸盐、盐酸盐、氢碘化物、2-羟基-乙磺酸盐、羟基萘甲酸酯、碘化物、异丁酸酯、异硫代酸酯、乳酸酯、乳糖酸乙酯、月桂酸酯、月桂烷硫酸酯、苹果酸酯、马来酸酯、丙二酸酯、扁桃酸盐、甲磺酸盐、甲基磺酸盐、硫酸二甲酯、粘酸盐、2-萘磺酸盐、萘甲酸盐、烟酸盐、硝酸盐、N-甲基葡糖胺铵盐、油酸盐、草酸盐、巴莫酸盐(内含酸盐)、棕榈酸酯、泛酸盐、果胶酸盐、过硫酸盐、3-苯基丙酸酯、磷酸盐/二磷酸盐、邻苯二甲酸盐、苦味酸盐、新戊酸酯、聚半乳糖醛酸盐、丙酸盐、水杨酸盐、硬脂酸盐、硫酸盐、辛二酸盐、琥珀酸盐、单宁酸盐、酒石酸盐、茶氯酸盐(teoclate)、甲苯磺酸盐、三乙基碘、十一烷酸酯、戊酸酯等(参见,例如,Berge et al.,"PharmaceuticalSalts",J.Pharm.Sci.,66,pp.1-19(1977))。
当在本文中使用时,术语“赋形剂”旨在表示药物组合物中不是活性成分的所有物质(例如,在所用的量/浓度上不表现出任何治疗作用的治疗上惰性的成分),例如载体、粘合剂、润滑剂、增稠剂、表面活性剂、防腐剂、稳定剂、乳化剂、缓冲剂、调味剂、着色剂或抗氧化剂。
本发明中描述的组合物可以包含药学上可接受的载体。如本文所用,“药学上可接受的载体”包括生理相容的任何和所有溶剂、分散介质、包衣、等渗和吸收延迟剂等。“药学上可接受的载体”可以是固体、半固体、液体或其组合的形式。优选地,载体适合于肠内(例如口服)或肠胃外给药(例如静脉内、肌内、皮下、脊髓或表皮给药(例如,通过注射或输注))。根据给药途径,可以将活性化合物(例如本发明的化合物)单独或与一种或多种其他治疗剂组合包被在一种材料中以保护一种或多种活性化合物免受可能会使活性化合物失活的酸和其他自然条件的作用。
可用于根据本发明的药物组合物中的合适的水性和非水性载体的实例包括水(例如注射用水)、乙醇、多元醇(例如甘油、丙二醇、聚乙二醇等)、盐、碳水化合物、糖醇或氨基酸的水溶液(例如盐水溶液或氨基酸水溶液)及其合适的混合物和/或缓冲形式、植物油(例如橄榄油)、以及可注射的有机酯(例如油酸乙酯)。例如,通过使用包衣材料,例如卵磷脂,在分散液的情况下通过保持所需的粒径,以及通过使用表面活性剂,可以保持适当的流动性。
药学上可接受的载体包括无菌水溶液或分散液和用于临时制备无菌注射溶液或分散液的无菌粉末。这种介质和试剂用于药物活性化合物的用途是本领域已知的。除非任何常规介质或试剂与活性化合物不相容,否则可考虑将其用于根据本发明的药物组合物中。
另外的治疗剂可以与本发明的化合物(特别是以上指定的那些,例如式(I)、(Ia)、(II)、(III)、(IV))、(V)、(VI)、(VII)或(VIII))一起、在其之前或之后给药或掺入组合物中。在一个实施方案中,本文所述的药物组合物包含如上所述的本发明的激酶抑制剂(例如具有通式(I)、(Ia)、(II)、(III)、(IV)、(V)、(VI)、(VII)或(VIII)或溶剂化物、盐(特别是药学上可接受的盐)、N-氧化物(尤其是R
“另外的治疗剂”(在一个实施方案中不是本文所指定的式(I)或(Ia)的激酶抑制剂,或在另一实施方案中可以是式(I)或(Ia))可选自可用于治疗作为增生性疾病(例如,癌症,例如本文其他地方所描述、定义或公开的疾病)和/或由以下造成或与以下有关的病症、疾病或病状的任何化合物:(i)激酶(例如SRC、ABL/BCR-ABL、LCK、SIK1、SIK2、SIK3、FLT3和/或KIT;和/或PHA2、EPHA4、CSF-R1、HCK和ACK1;和/或NEK11、WEE1、WNK2、Aurora-A、Aurora-B和TBK1)的(例如错误的)表达和/或活性;和/或(ii)对(例如细胞介导的)免疫应答的细胞抗性。合适的其他治疗剂的实例在本文其他地方定义或公开,包括:EGFR抑制剂,吉西他滨,多西他赛和免疫检查点抑制剂(例如PD1、PDL1、CTLA-4、LAG3或IDO1的抑制剂,尤其是选自nivolumab、relatlimab、ipilimumab和BMS-986205的免疫检查点抑制剂),TNF或TNFR1-或TNFR2信号激动剂,过继细胞疗法,包括针对肿瘤抗原的CAR T细胞,疫苗,包括基于树突状细胞的疫苗,或向对象施用时能够诱导或诱导与增殖性疾病有关的细胞暴露于TNF或TNFR1信号传导激动剂的试剂。所述另外的治疗剂可以诱导加性或协同治疗作用。
本文所述的药物组合物除本发明的激酶抑制剂外还可包含至少一种,例如1、2、3、4、5、6、7或8种其他治疗剂。根据本教导,可以将至少一种另外的治疗剂与本发明的激酶抑制剂一起配制在单一药物组合物中。或者,可以将药物组合物构造成成套试剂盒,其中本发明的激酶抑制剂以第一制剂的形式提供,并且至少一种其他治疗剂以第二制剂(即第二药物组合物)的形式提供。可以在使用之前将第一药物组合物和第二药物组合物合并。换句话说,在施用药物组合物之前,可以将包含另外的治疗剂的制剂添加到包含本发明的激酶抑制剂的第一药物组合物中。或者,本教导设想施用配制在第一药物组合物中的本发明的激酶抑制剂和施用配制在第二药物组合物中的至少一种其他治疗剂。药物组合物可以同时或连续施用。例如,可以在第一时间点施用第一药物组合物,并且可以在第二时间点施用第二药物组合物,其中时间点可以分开例如0或至多1、2、3、4、5或10分钟,至多1、2、3、4、5或10小时,至多1、2、3、4、5或10天,至多1、2、3、4、5或10周,至多1、2、3、4、5或10个月,或至多1、2、3、4、5或10年。
组合物还可包含佐剂,例如防腐剂、稳定剂、湿润剂、乳化剂、pH缓冲剂和分散剂。可以通过灭菌程序和/或通过包含各种抗菌和抗真菌剂(例如对羟基苯甲酸酯、三氯叔丁醇、苯酚山梨酸等)来确保防止微生物的存在。还可能需要在组合物中包括等渗剂,例如糖、氯化钠等。另外,可通过包含延迟吸收的试剂例如单硬脂酸铝和明胶来延长可注射药物形式的吸收。
不管选择的给药途径如何,都可以通过合适的水合形式使用的活性化合物和/或根据本发明的药物组合物通过本领域技术人员已知的常规方法配制成药学上可接受的剂型(参见例如Remington,"The Science and Practice of Pharmacy"edited by Allen,Loyd V.,Jr.,22nd edition,Pharmaceutical Sciences,September 2012;Ansel etal.,"Pharmaceutical Dosage Forms and Drug Delivery Systems",7th edition,Lippincott Williams&Wilkins Publishers,1999)。
可以通过本领域已知的多种方法来施用药物组合物。如本领域技术人员将理解的,施用途径和/或方式将根据所需结果而变化。包含一种或多种活性化合物的药物组合物可以与载体一起制备,所述载体将保护一种或多种活性化合物免于快速释放,例如控释制剂,包括植入物、透皮贴剂和微囊递送系统。可以使用可生物降解的生物相容性聚合物,例如乙烯乙酸乙烯酯、聚酸酐、聚乙醇酸、胶原蛋白、聚原酸酯和聚乳酸。制备此类组合物的方法是本领域技术人员通常已知的。参见,例如,Sustained and Controlled Release DrugDelivery Systems,J.R.Robinson,ed.,Marcel Dekker,Inc.,New York,1978。
为了通过某些施用途径施用本发明的化合物,可能有必要用防止其失活的材料包被该化合物或与该化合物共同施用。例如,可以将化合物以适当的载体(例如脂质体)或稀释剂的形式给予个体。药学上可接受的稀释剂包括盐水和缓冲水溶液。脂质体包括水包油包水型CGF乳剂以及常规脂质体(Strejan et al.,J.Neuroimmunol.7:27(1984))。
药物组合物通常在生产和储存条件下是无菌的和稳定的。可以将组合物配制为溶液、微乳剂、脂质体或其他适合高药物浓度的有序结构。载体可以是溶剂或分散介质,其包含例如水、乙醇、多元醇(例如甘油、丙二醇和液体聚乙二醇等)及其合适的混合物。可以例如通过使用诸如卵磷脂的包衣,在分散的情况下通过维持所需的粒径以及通过使用表面活性剂来维持适当的流动性。在许多情况下,优选在组合物中包括等渗剂,例如糖、多元醇,如甘露醇、山梨糖醇或氯化钠。通过在组合物中包括延迟吸收的试剂,例如单硬脂酸盐和明胶,可以实现可注射组合物的延长吸收。
注射用组合物应是无菌的,并且其流动性应达到可以通过注射器输送的程度。除水外,载体可以是等渗缓冲盐溶液、乙醇、多元醇(例如甘油、丙二醇和液态聚乙二醇等)及其合适的混合物。可以通过将所需量的活性化合物与所需的一种或上述成分的组合掺入适当的溶剂中,然后进行灭菌微滤,来制备无菌注射溶液。
通常,通过将活性化合物掺入无菌载体中来制备分散体,所述无菌载体包含基本分散介质和上述所需的其他成分。对于用于制备无菌注射溶液的无菌粉末,优选的制备方法是真空干燥和冷冻干燥(冻干),其从其先前无菌过滤的溶液中产生活性成分和任何其他所需成分的粉末。
调整剂量方案以提供最佳的所需反应(例如治疗反应)。例如,可以施用单次推注,可以随时间施用数个分开的剂量,或者可以根据治疗情况的紧急程度按比例减少或增加剂量。以剂量单位形式配制肠胃外组合物特别有利,以易于给药和剂量均匀。如本文所用,剂量单位形式是指适合作为待治疗个体的单位剂量的物理上离散的单位;每个单元包含预定量的活性化合物,该活性化合物经计算可与所需的药物载体一起产生所需的治疗效果。根据本发明使用的剂量单位形式的规范由以下决定并直接取决于以下因素:(a)活性化合物的独特特性和要实现的特定治疗效果,以及(b)复合此类活性化合物以治疗个体敏感性的技术领域固有的局限性。
药学上可接受的抗氧化剂的实例包括:(1)水溶性抗氧化剂,例如抗坏血酸、盐酸半胱氨酸、硫酸氢钠、偏亚硫酸氢钠、亚硫酸钠等;(2)油溶性抗氧化剂,例如棕榈酸抗坏血酸酯、丁基羟基茴香醚(BHA)、丁基羟基甲苯(BHT)、卵磷脂、没食子酸丙酯、α-生育酚等;和(3)金属螯合剂,例如柠檬酸、乙二胺四乙酸(EDTA)、山梨糖醇、酒石酸、磷酸等。
对于治疗/药物制剂,根据本发明的组合物包括适合于肠内给药(例如口服或直肠)或肠胃外给药(例如鼻、局部(包括阴道、颊和舌下))的组分。所述组合物可以方便地以单位剂型存在,并且可以通过药学领域中已知的任何方法来制备。可以与载体材料组合以产生药物组合物(例如单一剂型)的活性成分的量(特别是本发明化合物的量)将根据所治疗的个体和以及特定的施用方式而有所不同。可与载体材料组合以产生单一剂型的活性成分的量通常将是产生治疗效果的组合物的量。
通常,在100%(对于药物制剂/组合物)中,活性成分的量(特别是本发明化合物的量,任选地与其他治疗活性剂(如果存在于药物制剂/组合物中)一起)的范围为约0.01%至约99%,优选为约0.1%至约70%,最优选为约1%至约30%,其中剩余部分优选由一种或多种药学上可接受的赋形剂组成。
以单位剂型和/或当施用给个体或用于治疗时的活性成分(例如本发明的化合物)的量可以在每单位、给药或治疗约0.1mg至约1000mg的范围内(例如约1mg至约500mg,例如约10mg至约200mg)。在某些实施方案中,可以使用个体的质量或体表面积来计算此类活性成分的合适量,包括约1mg/kg至10mg/kg(例如约2mg/kg至5mg/kg),或约1mg/m
根据本发明的药物组合物中的活性成分的实际剂量水平可以变化,以获得对于特定患者、组合物和方式有效达到期望的治疗反应而对患者无毒的活性成分的量。所选剂量水平将取决于多种药代动力学因素,包括所用特定组合物的活性、给药途径、给药时间、所用特定化合物的排泄速率、治疗持续时间、与所使用的特定组合物结合使用的其他药物、化合物和/或材料、所治疗患者的年龄、性别、体重、状况、一般健康状况以及既往病史、以及医学领域众所周知的类似因素。
具有本领域普通技术的医师或兽医可以容易地确定并开出(例如,治疗上)有效量的所需药物组合物。例如,医师或兽医可以以低于获得所需治疗效果所需水平的本发明化合物的剂量开始,并逐渐增加剂量直至获得所需效果。通常,根据本发明的组合物的合适的日剂量将是有效产生治疗效果的最低剂量的化合物的量。这样的有效剂量通常将取决于上述因素。优选以口服、静脉内、肌内、腹膜内或皮下给药,优选在靶部位附近给药。如果需要,则药物组合物的(例如,治疗上)有效日剂量可以整天以适当间隔分别以两个、三个、四个、五个、六个或更多个亚剂量施用,任选地以单位剂型施用。尽管可以单独施用根据本发明的化合物,但是优选以药物制剂/组合物的形式施用该化合物。
对于口服给药根据本发明的药物组合物可以采取例如通过常规方法与药学上可接受的赋形剂制备的片剂或胶囊剂的形式,所述赋形剂例如是粘合剂(例如预糊化的玉米淀粉、聚乙烯吡咯烷酮、羟丙基甲基纤维素)、填充剂(例如乳糖、微晶纤维素、磷酸氢钙)、润滑剂(例如硬脂酸镁、滑石粉、二氧化硅)、崩解剂(例如马铃薯淀粉、淀粉羟乙酸钠)或润湿剂(例如十二烷基硫酸钠)。用于口服的液体制剂可以是例如溶液剂、糖浆剂或混悬剂的形式,或者可以以干燥产品的形式存在,以在使用前用水或其他合适的赋形剂来配制。这样的液体制剂可以通过常规方法与药学上可接受的添加剂一起制备,所述添加剂例如是悬浮剂(例如山梨糖醇、糖浆、纤维素衍生物、氢化食用脂肪)、乳化剂(例如卵磷脂、阿拉伯胶)、非水媒介物(例如杏仁油、油性酯、乙醇、分馏植物油)、防腐剂(例如对羟基碳酸甲酯或对羟基苯甲酸丙酯、山梨酸)。所述制剂还可以视需要包含缓冲盐、调味剂、着色剂和甜味剂。可以适当地配制口服给药的制剂以控制释放本发明的药物组合物。
在一个实施方案中,化合物以例如最多100mg/kg体重的浓度口服施用(例如最多50mg/kg体重、至多40mg/kg体重、最多30mg/kg体重、最多20mg/kg体重、最多10mg/kg体重、最多5mg/kg体重、最多4mg/kg体重、最多3mg/kg体重、最多2mg/kg体重、最多1mg/kg体重)。
在一个实施方案中,化合物以例如最多10mg/kg体重的浓度经肠胃外施用(例如静脉内、肌肉内或皮下)(例如最多5mg/kg体重、最多4mg/kg体重、最多3mg/kg体重、最多2mg/kg体重、最多1mg/kg体重、最多0.5mg/kg体重、最多0.4mg/kg体重、最多0.3mg/kg体重、最多0.2mg/kg体重、最多0.1mg/kg体重)。
所述药物组合物可以与传统的粘合剂和载体(例如甘油三酸酯)一起配制成栓剂。口服制剂可包括标准载体,例如药用级的甘露醇、乳糖、淀粉、硬脂酸镁、糖精钠、纤维素、碳酸镁等。
为了通过吸入给药,使用合适的推进剂(例如二氯二氟甲烷、三氯氟甲烷、二氯四氟乙烷、二氧化碳、氮气或其他合适的气体))从加压包装或喷雾器以气雾剂形式方便地递送根据本发明的药物组合物。在使用加压气雾剂的情况下,可以通过提供一个阀门来确定计量单位,从而确定剂量单位。可以配制用于吸入器或吹入器的例如明胶的胶囊和药筒,其包含根据本发明的药物组合物的粉末混合物和合适的粉末基质(例如乳糖或淀粉)。
根据本发明的药物组合物可以配制成用于通过注射,例如通过推注或连续输注进行肠胃外给药。在一个实施方案中,本发明的化合物或组合物可以通过长时间(例如超过24小时)的缓慢连续输注给药,以减少毒性副作用。给药也可以通过持续输注2至24小时(例如2至12小时)来进行。该方案可以根据需要重复一次或多次,例如在6个月或12个月后。
在另一个实施方案中,根据本发明的化合物或组合物通过维持疗法给药,例如每周一次,持续6个月或更长时间。
注射剂可以单位剂型存在(例如,在玻璃瓶中、在多剂量容器中),并带有添加的防腐剂。根据本发明的药物组合物可以采取诸如在油性或水性运载体中的悬浮液、溶液或乳剂的形式,并且可以包含配制剂,例如悬浮剂、稳定剂或分散剂。或者,该试剂可以是粉末形式,以便在使用前与合适的媒介物(例如无菌无热原水)一起配制。通常,用于静脉内施用的组合物是在无菌等渗水性缓冲液中的溶液。必要时,组合物还可包含增溶剂和局部麻醉剂,例如利多卡因,以减轻注射部位的疼痛。通常,成分以单位剂型单独提供或混合在一起提供,例如,作为干燥的冻干粉末或无水浓缩物,放在指示活性剂含量的密闭容器中,例如安瓿或小药囊中。当组合物要通过输注给药时,可以用装有无菌药物级水或盐水的输液瓶分配。在通过注射施用组合物的情况下,可以提供安瓿瓶的无菌注射用水或盐水,以便可以在施用之前将成分混合。
适用于阴道给药的根据本发明的组合物还包括含有本领域已知合适的载体的子宫托、棉塞、乳膏、凝胶、糊剂、泡沫或喷雾制剂。用于根据本发明的组合物的局部或透皮给药的剂型包括散剂、喷雾剂、软膏剂、糊剂、乳膏剂、洗剂、凝胶剂、溶液剂、贴剂和吸入剂。可以在无菌条件下将活性化合物与药学上可接受的载体以及可能需要的任何防腐剂、缓冲剂或推进剂混合。
治疗/药物组合物可以与本领域已知的医疗装置一起施用。例如,在一个优选的实施方案中,根据本发明的治疗/药物组合物可以与无针皮下注射装置一起施用,例如US 5,399,163、US 5,383,851、US 5,312,335、US 5,064,413、US 4,941,880、US 4,790,824或US4,596,556中公开的装置。可用于本发明的众所周知的植入物和模块的例子包括在以下文献中描述的:US 4,487,603,其公开了一种用于以受控的速率分配药物的可植入的微输液泵;US 4,486,194,其公开了一种用于通过皮肤施用药物的治疗装置;US 4,447,233,其公开了一种用于以精确的输注速率输送药物的药物输注泵;US 4,447,224,其公开了一种用于连续药物输送的可变流量植入式输注设备;US 4,439,196,其公开了具有多室隔室的渗透性药物递送系统;以及US 4,475,916,其公开了一种渗透药物输送系统。
许多其他这样的植入物、递送系统和模块是本领域技术人员已知的。在某些实施方案中,可以配制根据本发明的化合物以确保在体内的适当分布。例如,血脑屏障(BBB)排除了许多高度亲水的化合物。为了确保本发明的化合物穿过BBB(如果需要的话),可以将它们配制成例如脂质体。对于制造脂质体的方法,参见,例如US 4,522,811、US 5,374,548和US 5,399,331。脂质体可包含一个或多个部分,所述部分被选择性地运输到特定细胞或器官中,并因此增强靶向药物递送(参见,例如,V.V.Ranade(1989)J.Clin.Pharmacol.29:685)。示例性的靶向部分包括叶酸或生物素(参见,例如,Low等人的US 5,416,016);甘露糖苷(Umezawa et al.,(1988)Biochem.Biophys.Res.Commun.153:1038);抗体(P.G.Bloemanet al.(1995)FEBS Lett.357:140;M.Owais et al.(1995)Antimicrob.AgentsChemother.39:180)以及表面活性剂蛋白A受体(Briscoe et al.(1995)Am.J.Physiol.1233:134)。
在一个实施方案中,根据本发明的化合物被配制成脂质体。在一个更优选的实施方案中,脂质体包括靶向部分。在最优选的实施方案中,通过推注将脂质体中的化合物递送至接近所需区域的部位。这样的基于脂质体的组合物应具有易于注射的程度的流动性,在制造和储存条件下应是稳定的,并且应被保存以抵抗诸如细菌和真菌之类的微生物的污染作用。
用于治疗/疗法的“治疗有效剂量”可以通过客观反应来衡量,该反应可以是完全的也可以是部分的。完全缓解(CR)的定义是没有任何有关疾病、病症或疾病的临床、放射学或其他证据。疾病减少超过50%会导致部分反应(PR)。进展的中位时间是表征客观肿瘤反应的持久性的量度。
治疗/疗法的“治疗有效剂量”也可以通过其稳定疾病、病症或疾病进展的能力来衡量。化合物抑制一种或多种蛋白激酶或降低与增生性疾病相关的细胞(例如癌细胞)的活力的能力可以通过使用熟练的从业者已知的合适的体外测定来评估,例如本文所述的那些(尤其是在以下“实施例”中)。或者,可以通过在本领域技术人员已知的合适的动物模型系统(例如本文所述的那些)中检查化合物的能力来评估本发明中描述的化合物的性能(特别是在以下“实施例”中)。治疗有效量的根据本发明的化合物可以治愈、疗愈、减轻、减缓、改变、补救、改善、改进或影响病状、病症或疾病或病状、病症或疾病的症状或易感性个体的病状、病症或疾病。本领域普通技术人员将能够基于诸如个体的大小、个体的症状的严重程度、以及所选择的特定的组成或给药途径之类的因素来确定这些量。
如果需要,则根据本发明的药物组合物也可以包装或分配器的形式提供,其可以含有一种或多种含有活性化合物的剂型(例如,单位)。包装可以例如包括金属或塑料箔,诸如泡罩包装。包装或分配器设备可以随附单张或其他信息;特别是,描述(向患者和/或主治医师)有关包装中所含药物组合物的重要信息或细节,例如如何给药、推荐剂量、安全性和/或副作用信息。
在一个特定的实施方案中,将本发明的药物组合物配制成用于口服给药,在另一个特定的实施方案中,将本发明的药物组合物配制成用于静脉内给药。
在一个实施方案中,本发明的药物组合物为单位剂型,并且特别地可以为配制用于口服给药的单位剂型。
每个此类单位剂型均可以包含(例如,可以包含)1至950mg的化合物,例如第一方面的激酶抑制剂(例如,具有通式(I)、(Ia)、(II)、(III)、(IV)、(V)、(VI)、(VII)或(VIII)的激酶抑制剂、或溶剂化物盐(特别是药学上可接受的盐)、N-氧化物(特别是R
特别地,在这样的实施方案中,单位剂量形式的本发明药物组合物(特别是配制为口服给药的单位剂量形式的药物组合物)可以包含(例如,可以含有)——对于每种单位剂型——大约选自以下量的此类化合物:2mg、5mg、15mg、20mg、50mg、70mg、80mg、100mg和140mg;特别地,其包含(例如包含)约20mg、50mg、70mg或100mg的量的本发明化合物。
在一个特定的实施方案中,本发明的药物组合物是(例如形成为)片剂、囊片或胶囊;合适地,本发明的药物组合物(例如,其单位剂型)是囊片。形成(例如,制造)片剂和囊片的方法例如在本文其他地方描述。
适用于本发明的药物组合物的赋形剂,特别是当制成片剂或囊片形式时包括,并且本发明的这种药物组合物的具体实施方案包括那些进一步包含一种或多种(例如,全部)选自以下的赋形剂:乳糖(例如乳糖一水合物),微晶纤维素,交联羧甲基纤维素钠,羟丙基纤维素和硬脂酸镁。
在第三方面,本申请提供用作药物例如用于治疗的如上文在“化合物”标题下指定的化合物或如上文在“药用组合物”标题下指定的药物组合物。在这些方面的一个实施方案中,本发明的化合物不包括如上文在“化合物”标题下指定的具有式(I)并且属于群组(1)、(2)、(3)、(4)和/或(5)中的一个或多个的化合物(例如群组(1)(特别是当R
预期上述标题为“化合物”下的化合物可用于抑制:(i)激酶,例如本文所述的激酶,特别是SRC、ABL、BCR-ABL、LCK、SIK1、SIK2、SIK3、FLT3和/或KIT和/或PHA2、EPHA4、CSF-R1、HCK和ACK1和/或NEK11、WEE1、WNK2、Aurora-A、Aurora-B和TBK1;和/或(ii)细胞对(例如细胞介导的)免疫反应的抗性。例如,该化合物可以用于治疗个体(特别是人类患者)的疾病、病症或病状的方法,其中该疾病或病状与这种激酶有关。
本发明的化合物可单独或与一种或多种其他治疗剂联合治疗,例如与本文其他地方定义或公开的治疗剂联合使用,包括EGFR抑制剂、吉西他滨、多西他赛、免疫检查点抑制剂(例如PD1、PDLL、CTLA-4、LAG3或IDO1的抑制剂,特别是选自nivolumab、relatlimab、ipilimumab和BMS-986205的免疫检查点抑制剂)、TNF或TNFR1或TNFR2信号激动剂、过继细胞疗法(包括针对肿瘤抗原的CAR T细胞)、疫苗(包括基于树突细胞(DC)的疫苗接种)或当对对象施用时能够诱导或诱导与增殖性疾病有关的细胞暴露于TNF或TNFR1信号传导激动剂的试剂。
可以在家里、医生办公室、诊所、医院的门诊部或医院提供包括或使用此类化合物的治疗方法。通常在医疗监督下开始治疗,以便医务人员可以密切观察治疗效果并进行必要的调整。治疗的持续时间取决于患者的年龄和状况,以及患者对治疗的反应方式。
患有疾病、病症或疾病的风险较高的人可能会接受预防性治疗,以抑制或延迟该疾病、病症或疾病的症状。
术语“治疗”是本领域技术人员已知的,并且包括对患者的治疗剂(例如,包含所述试剂的药物组合物)的应用或给药或对患者的治疗或应用或给药的治疗(例如,含有所述药剂的药物组合物)或从患有疾病、病症或疾病症状的患者身上分离细胞、细胞培养物、细胞系、样品、组织或器官的程序或对某种疾病、病症或疾病的易感性,目的在于治愈、疗愈、缓解、减缓、改变、补救、改善、改进、影响或预防该疾病、病症或症状的症状或对疾病、病症或疾病的易感性。因此,术语“治疗”可以包括疾病、病症或疾病或者疾病、病症或疾病的症状的预防性治疗。当用于治疗中时,治疗剂包括本发明的激酶抑制剂,并且包括但不限于其他治疗剂,其可以是小分子、肽、拟肽、多肽/蛋白质、抗体、核苷酸(例如DNA或RNA)、细胞、病毒、核酶、siRNA和反义寡核苷酸。
因此,在第四方面,并且如本文可以进一步描述、定义、要求保护或以其他方式公开的,本发明涉及在标题“化合物”下指定的化合物(例如,具有通式(I)、(Ia)、(II)、(III)、(IV)、(V)、(VI)、(VII)或(VIII)或溶剂化物盐(特别是药学上可接受的盐)、N-氧化物(特别是R
在另一个第四方面,并且如本文可以进一步描述、定义、要求保护或以其他方式公开,本发明涉及如上所述的药物组合物(例如,包含标题为“化合物”中指定的化合物的药物组合物(例如,具有通式(I)、(Ia)、(II)、(III)、(IV)、(V)、(VI)、(VII)或(VIII)或溶剂化物盐(特别是药学上可接受的盐)、N-氧化物(特别是R
在相关的第四方面,并且如本文可以进一步描述、定义、要求保护或以其他方式公开的,本发明涉及一种用于治疗对象的增生性疾病的方法,该方法包括向该对象施用(例如治疗有效量的):(X)在“化合物”标题下指定的化合物(例如,具有通式(I)、(Ia)、(II)、(III)、(IV)、(V)、(VI)、(VII)或(VIII)或溶剂化物盐(特别是药学上可接受的盐)、N-氧化物(特别是R
在另一个相关的第四方面,并且如本文可以进一步描述、定义、要求保护或以其他方式公开,本发明涉及标题为“化合物”标题下指定的化合物(例如,具有通式(I)、(Ia)、(II)、(III)、(IV)、(V)、(VI)、(VII)或(VIII)或溶剂化物盐(特别是药学上可接受的盐)、N-氧化物(特别是R
在此类第四方面,这种用途或方法的治疗包括向对象施用(例如,治疗有效量的)本发明的化合物或药物组合物。
在该方面的一个特定实施方案中,所述对象是人类,合适地是成年人类。例如,年龄在18(或16)岁以上的人类,例如年龄在18(或16)至90岁之间,或18(或16)至80岁之间的人类。在某些此类实施例中,成人大约20岁或以上、30岁或以上、35岁或以上、40岁或以上、45岁或以上、50岁或以上或55岁或以上。在这样的实施例的更具体的实施例中,成人是轻年人(例如约18(或16)至45(或40)、或约30至45(或40)岁),中年人(例如约45(或40)至65(或60)、或大约45(或40)至55(或50)、或大约55(或50)至65(或60)岁),或者是老年人(例如约60至90(或更年长,例如92、95或98)、约65到85至约70至88)。
作为此类实施方案的替代方案,所治疗的对象是小儿,例如小于约18岁(或16岁)的小儿。例如,这样的人可以在大约3至18(或16),例如在大约5至16或在大约10至16或12至17。儿科人可以是婴儿(例如在大约两个月至约2岁)、婴幼儿(例如约2岁至约4岁)、幼儿(例如约4岁至约9岁)、青春期前(例如约9岁至约12或13岁(或11或14岁)、或青春期(例如约12或13岁(或11或14岁)至15岁(或16或17岁))。
在该方面的一个实施方案中,所述治疗包括每天向需要其的成年对象施用少于约140mg的本发明的化合物(例如,包含在药物组合物中)。例如,任选地,在增殖性疾病不是(例如,该对象患有的增殖性疾病不是)慢性阶段Ph+CML的情况下。在该方面的替代实施方案中,所述治疗包括向有此需要的成年人类对象施用每天大于约140mg(例如每天大于150mg)的量的此类化合物(例如,包含在药物组合物中)。
在该方面的另一个实施方案中,所述治疗包括每天向需要其的成年对象施用少于约100mg的本发明的化合物(例如,包含在药物组合物中)。例如,任选地,在增殖性疾病不是(例如,该对象患有的)慢性阶段Ph+CML的情况下。在该方面的替代实施方案中,所述治疗包括向有此需要的成年人类对象施用每天大于约100mg(例如每天大于120mg)的量的此类化合物(例如,包含在药物组合物中)。
在一个可供选择的实施方案中,所述治疗包括向有此需要的小儿人对象施用以下量的本发明化合物:
·对于体重在10kg至20kg之间的小儿科患者,每日少于40mg;
·对于体重在20kg至30kg之间的小儿科患者,每日少于60mg;
·对于体重在30kg至45kg之间的小儿科患者,每日少于70mg;或者
·对于体重至少为45kg的小儿科患者,每日少于约100mg。
在另一个可供选择的实施方案中,所述治疗包括向有此需要的小儿人对象施用以下量的本发明化合物:
·对于体重在10kg至20kg之间的小儿科患者,每日大于约40mg;
·对于体重在20kg至30kg之间的小儿科患者,每日大于约60mg;
·对于体重在30kg至45kg之间的小儿科患者,每日大于约70mg;或者
·对于体重至少为45kg的小儿科患者,每天大于约100mg。
对于将一定量的这类化合物(例如待施用)施用至人对象的实施方案,该量的施用频率可能比每天更少。例如,给定的量“每天少于40mg”可以通过每天例如每天35、30或20mg或每两天一次(或更低频率)一次75、65或40mg来实现。
在一个特定的实施方案中,在这样(例如治疗有效量)的本发明化合物的施用时(或之后),该对象不太可能(例如没有)发生(或遭受)不良反应,例如骨髓抑制。
在这样的特定实施方案之一中,在这样(例如治疗有效量)的本发明化合物的施用时(或之后),该对象不太可能(例如没有)发生(或遭受)非血液学不良反应,例如心脏病学不良反应。
在这样的实施方案中更具体地,在向对象施用(例如治疗有效)量的化合物时(或之后),该对象不太可能(例如没有)发生(或遭受)QT延长。
在一个实施方案中,所述对象的特征在于不同时使用强CYP3A4抑制剂。例如,不同时使用酮康唑、伊曲康唑、红霉素、克拉霉素、利托那韦、泰利霉素或摄入葡萄柚汁。
在某些实施方案中,在本文描述的发明的上下文中,所述疾病、病症或病状是增生性疾病(包括与此类病症有关的病状或症状)。
“增殖性疾病”是指以细胞异常增殖为特征的疾病。增生性疾病并不意味着对细胞生长速率的任何限制,而仅表示丧失了影响生长和细胞分裂的正常对照。因此,在一些实施方案中,增殖性疾病的细胞可以具有与正常细胞相同的细胞分裂速率,但是不响应于限制这种生长的信号。在“增生性疾病”的范围内的是肿瘤或肿瘤,这是组织或细胞的异常生长。癌症是本领域已知的,并且包括以细胞增殖为特征的各种恶性肿瘤中的任何一种,这些细胞具有侵袭周围组织和/或转移至新的定殖位点的能力。增生性疾病包括癌症、动脉粥样硬化、类风湿性关节炎、特发性肺纤维化和肝硬化。非癌性增生性疾病还包括皮肤细胞过度增生,例如牛皮癣及其各种临床形式,赖特氏(Reiter's)综合征、红斑性糠疹、角质化失调的过度增生变体(例如光化性角化病、老年性角化病)、硬皮病等。
在更具体的实施方案中,增生性疾病是癌症或肿瘤,特别是实体瘤(包括与这种癌症或肿瘤有关的病症或症状)。这类增生性疾病包括但不限于头颈癌、鳞状细胞癌、多发性骨髓瘤、孤立性浆细胞瘤、肾细胞癌、视网膜母细胞瘤、生殖细胞肿瘤、肝母细胞瘤、肝细胞癌、黑素瘤、肾脏的横纹肌瘤、尤因肉瘤、软骨肉瘤、任何血液系统恶性肿瘤(例如慢性淋巴细胞性白血病、慢性骨髓单核细胞性白血病、急性淋巴母细胞性白血病、急性淋巴细胞性白血病、急性骨髓性白血病、急性成肌细胞白血病、慢性粒细胞性白血病、霍奇金氏病、非霍奇金氏白血病、慢性淋巴细胞性白血病、慢性粒细胞性白血病、骨髓增生异常综合症、毛细胞白血病、肥大细胞白血病、肥大细胞肿瘤、滤泡性淋巴瘤、弥漫性大细胞淋巴瘤、套细胞淋巴瘤、边缘区淋巴瘤、伯基特淋巴瘤、真菌病、丝裂综合征、皮肤T细胞淋巴瘤、外周T细胞淋巴瘤、慢性骨髓增生性疾病、骨髓炎肉芽肿、髓样化生、系统性肥大细胞增多症)和中枢神经系统肿瘤(例如脑癌、胶质母细胞瘤、非胶质母细胞瘤脑癌、脑膜瘤、垂体腺瘤、前庭神经鞘瘤、原始神经外胚层肿瘤、髓母细胞瘤、星形细胞瘤、间质星形胶质细胞瘤、少突胶质细胞瘤、室管膜瘤和脉络丛乳头状瘤)、骨髓增生性疾病(例如真性红细胞增多症、血小板增多症、特发性骨髓纤维化)、软组织肉瘤、甲状腺癌、子宫内膜癌、类癌或肝癌。
在一个特定的实施方案中,本发明的各个方面涉及(例如用于本发明的化合物或药物组合物)增生性疾病的治疗,所述增生性疾病包括本文所述的那些。因此,在这样的实施方案中,增殖性疾病可以是癌症或肿瘤。
在一个特定的实施方案中,所述癌症是造血或淋巴样癌,并且在一个这样的实施方案中,所述增殖性疾病是(例如,对象患有或怀疑患有)费城染色体阳性白血病;例如,费城染色体阳性的慢性髓细胞性白血病(Ph+CML)或费城染色体阳性的急性淋巴细胞性白血病(Ph+ALL)。
在某个实施方案中,该增生性疾病是(例如,该对象(例如成年人类受试者)患有或怀疑患有):
·慢性期的新诊断的(Ph+CML);
·对包括伊马替尼(例如甲磺酸伊马替尼)在内的先前治疗有耐药性或耐受性的慢性、加速或爆炸性CML;或者
·对先前的治疗有耐药性或不耐受性的Ph+急性淋巴细胞白血病(ALL)和淋巴母细胞性CML。
在另一个特定的实施方案中,所述对象是儿科人类并且所述增殖性疾病是(例如,所述受试者患有或怀疑患有):
·对以前的治疗(包括伊马替尼)有抗药性或耐受性的新诊断的慢性期Ph+CML(Ph+CML-CP)或Ph+CML-CP。
在另一个特定的实施方案中,癌症是实体瘤,并且在一个这样的实施方案中,增生性疾病是(例如,所述对象患有或怀疑患有)本文其他各处所述的实体瘤之一的实体瘤,例如胰腺癌、乳腺癌、肺癌、前列腺癌、黑色素瘤、卵巢癌、食道癌、肉瘤和结直肠癌。在某些这样的实施方案中,增殖性疾病是(例如,所述对象患有或怀疑患有)胰腺癌;在这样的实施方案的另一个中,所述增殖性疾病是(例如,所述对象患有或怀疑患有)前列腺癌;在此类实施方案的又一个中,增殖性疾病是(例如,所述对象患有或怀疑患有)肺癌(例如,非小细胞肺癌)。
如其他地方所述,本发明的化合物(或药物组合物)可以通过另一种医学程序(例如另外的治疗剂,例如本文其他地方描述的,手术或放射疗法)施用于对象(例如,作为联合疗法或方案)。然后,这样的组合治疗方案可以包括其中这样的暴露/施用同时发生的实施方案。在替代实施方案中,这样的施用可以是按顺序的;特别是在这样的其他程序之前施用本发明的化合物(或药物组合物)的那些实施方案中。例如,化合物(或药物组合物)可以在其他程序(例如之前)的约14天之内,例如在其他程序(例如之前)的约10天、7天、5天、2天或1天之内按顺序施用;并且进一步包括其中化合物(或药物组合物)可以在其他程序(例如之前)约48小时、24小时、12小时、8小时、6小时、4小时、2小时、1小时、30分钟、15分钟或5分钟内依次给药。
这种联合治疗方案可以包括(例如进一步)对所述对象施用:
·EGFR抑制剂和/或吉西他滨——特别是当增生性疾病是(例如,所述对象患有或怀疑患有)胰腺癌时;
·多西紫杉醇——特别是当增生性疾病是(例如,所述对象患有或怀疑患有)前列腺癌时;和/或
·免疫检查点抑制剂——特别是当增生性疾病是(例如,所述对象患有或怀疑患有)肺癌(例如非小细胞肺癌)时。
可能包含此类联合疗法或方案的示例性免疫检查点抑制剂在其他地方进行了描述,包括PD1、PDL1、CTLA-4、LAG3或IDO1的抗体或小分子抑制剂,尤其是此类免疫检查点抑制剂可以选自由下列组成的列表中的一种:nivolumab,relatlimab,ipilimumab和BMS-986205,特别是nivolumab。
在其他实施方案中,组合方案可包括(例如进一步)向所述对象施用:
·免疫激活剂(例如,激动剂)抗体,例如针对OX40(例如,Yang et al 2012,Blood120:4533)、41BB、CD40或ICOS(例如,Deng et al 2004,Hybrid Hybridomics 23:176)的抗体,特别是通过刺激/刺激T细胞而增加TNF水平的;和/或
·基于树突状细胞(DC)的疫苗接种(例如,Lowe et al 2014,Oncoimmunology 3:e27589)。
在一个特定的实施方案中,增生性疾病(例如,在对象中)已经在(例如尽管)标准疗法下发展,或者在另一实施方案中,对象可能无法接受标准疗法,例如由于对象不对其耐受。在任何一个这样的实施方案中,对象可以被表征(例如,分层)为已经下标准疗法下进展或不能接受(例如,不耐受)标准疗法。
作为标准疗法的例子,可以是伊马替尼(例如,对于CML或ALL)、多西他赛(例如,对于前列腺癌)或免疫疗法,例如描述为ehrein的免疫检查点抑制剂(例如,对于黑素瘤或肺癌)。
对免疫反应和抑制激酶的敏感化
本发明的化合物可使与增殖性疾病有关的细胞对细胞介导的免疫反应敏感。
因此,在一个实施方案中,包括向对象施用本发明的化合物(或药物组合物)的治疗涉及(例如,被介导或被支持)使与增殖性疾病有关的细胞对细胞介导的免疫反应敏感。
在一个替代的实施方案中,包括向对象施用本发明的化合物(或药物组合物)的治疗涉及(例如,被介导或被支持)抑制参与对细胞介导的免疫响应的抗性的激酶(例如抑制SIK3)。
在一个相关的实施方案中,包括向对象施用本发明的化合物(或药物组合物)的治疗涉及(例如,被介导或被支持)抑制参与对细胞介导的免疫响应的抗性的激酶(例如抑制SIK3),并且(例如由此)使与增殖性疾病有关的细胞对细胞介导的免疫应答敏感。
在另一方面,并且如本文中可以进一步描述、定义、要求保护或以其他方式公开的,本发明涉及一种用于在对象的增生性疾病治疗中与增殖性疾病有关的细胞对细胞介导的免疫应答的敏化的方法,该方法包括向对象施用本发明的化合物(或药物组合物);在另一个方面,并且如本文可以进一步描述、定义、要求保护或以其他方式公开的,本发明涉及一种抑制在对象的增生性疾病治疗中与细胞介导的免疫反应(例如抑制)的抗性有关的激酶的方法,该方法包括向所述对象施用本发明的化合物(或药物组合物)。
在相关的进一步方面,并且如本文可以进一步描述、定义、要求保护或以其他方式公开的,本发明涉及用作药物的本发明的化合物(或药物组合物),用于:(i)致敏与细胞介导的免疫反应的增殖性疾病有关的细胞;和/或(ii)抑制涉及对细胞介导的免疫反应的抗性的激酶,例如抑制SIK。
在又一个相关的进一步方面,并且如本文可以进一步描述、定义、要求保护或以其他方式公开的,本发明涉及用作药物(例如免疫肿瘤药物药物)的本发明化合物(或药物组合物),该药物使与增殖性疾病(例如肿瘤或癌症)有关的细胞对细胞介导的免疫反应敏感,例如使与增殖性疾病有关的细胞对可能由细胞介导的免疫反应诱导的杀伤(细胞死亡)敏感。“免疫肿瘤”药物是本领域普通技术人员会认可的药物,并且包括旨在(例如,专门设计用于)增强生物体(例如人)的免疫系统的一种或多种成分抵抗该生物体中存在的癌细胞或肿瘤细胞的能力。免疫肿瘤药物可以是与外部免疫(抑制性)检查点分子(例如本文其他地方所述)结合的药物(例如抗体),并且(例如直接)抑制针对癌细胞或肿瘤细胞的T细胞功能,或免疫肿瘤药物可以是抑制癌细胞或肿瘤细胞所固有的免疫调节剂(如本发明中的SIK3)的药物,其中这种内在的免疫调节剂不能主动(例如直接)抑制T细胞,而是通过抗性机制保护肿瘤或癌细胞免受免疫反应。
在这些方面的特定实施方案中,与增殖性疾病有关的细胞可以对通过细胞介导的免疫应答(例如由其诱导)的杀死(细胞死亡)敏感。
“盐诱导型激酶3”或“SIK3”(同义词QSK和KIAA0999)是丝氨酸/苏氨酸蛋白激酶亚家族的成员,包括属于AMP激活蛋白激酶(AMPK)家族的SIK1、SIK2和SIK3。在本发明的上下文中,SIK3蛋白通常是蛋白激酶。关于人SIK3蛋白的相关信息可在UniProt:Q9Y2K2(2017年3月15日条目版本138)上获得,并且在本发明的上下文中,SIK3蛋白优选具有SIK3 2017年3月15日的条目版本138或2018年3月28日的条目版本144中所示的氨基酸序列,这些序列通过引用合并在此。SIK3是具有丝氨酸/苏氨酸激酶活性的细胞质蛋白,其通过LKB1复合物通过激酶结构域T环中保守的苏氨酸残基(163位)的磷酸化来调节。据报道,磷酸化是SIK3催化活性必不可少的(Lizcano,J.M.et al.;EMBO J.23,833–843(2004))。为了本文公开的发明的目的,术语“磷酸化的SIK3”应表示基本上被磷酸化的SIK3蛋白,因为SIK3蛋白可以被(例如被)LKB1磷酸化,其中优选地,这种磷酸化的SIK3在氨基酸位置163包含磷酸苏氨酸。在本发明的上下文中,磷酸化的SIK3是在其细胞生物学环境中被激活的SIK3蛋白。通过SIK3基因产物的可变剪接产生的至少四种蛋白质同种型(SIK3-001至SIK3-004)是已知的。人SIK3基因位于染色体位置11q23.3(HGNC基因Symbol Acc:HGNC:29165),在许多物种中都具有保守性,例如黑猩猩、恒河猴、狗、牛、小鼠、大鼠、鸡、斑马鱼和青蛙。在本发明的一些实施方案中,术语SIK3也可以涉及具有与上述SIK3的氨基酸序列基本相同或至少80%、优选85%、更优选90、95、96、97、98、99或100%序列同一性的氨基酸序列的人SIK3蛋白的变体,上述同一性使用例如由Tatusova&Madden 1999(FEMS Microbiol Lett 174:247-250)描述的“Blast 2序列”算法确定,并且其(优选地)保持与相应参考SIK3相同或基本相同的生物学活性(例如磷酸化一种或多种II类(例如IIa)HDAC,例如HDAC4)。SIK3蛋白的优选变体包含其序列变体,这是由于各个物种的种群之间和之中的序列多态性,以及与SIK3的野生型序列相比位于活性环或激活环中或紧邻活性环或激活环的突变(T环)SIK3。SIK3蛋白的优选变体是SIK3 T163突变,例如影响SIK3激活的突变。在优选的实施方案中,本发明的SIK3蛋白不是SIK1(同义词:SIK和SNF1LK)蛋白和/或不是SIK2(同义词:QIK、KIAA0781和SNF1LK2)蛋白。通过引用将人SIK1(UniProt:P57059;2017年3月15日的条目版本168)和人SIK2(UniProt:Q9H0K1;2017年3月15日的条目版本153)的氨基酸序列并入本文。如适用于上下文(如果没有更具体指出的话),术语SIK3可以指SIK3蛋白(例如上述的一种)或编码这种SIK3蛋白的mRNA分子。应当理解关于“SIK1”和“SIK2”的类似含义。
作为“SIK3的抑制剂”(或“SIK3抑制剂”)的化合物是抑制SIK3的任何部分,这可能意味着抑制SIK3的活性,尤其是SIK3的蛋白,特别是磷酸化的SIK3的活性。SIK3抑制剂可能会损害(例如,诱导或减少)SIK3一种或多种活性的效率、效力、数量或速率,例如本文所述的活性中的一种或多种,例如,SIK3的活性磷酸化II类(例如IIa)HDAC(例如HDAC4)和/或使与增殖性疾病有关的细胞对细胞介导的免疫反应敏感。
这种抑制SIK3的部分可以直接发挥作用,例如,通过与SIK3结合并降低SIK3的一种或多种特性(例如其功能,特别是其作为激酶的能力)的量或比率(例如磷酸化HDAC4),例如,通过降低细胞中磷酸化的SIK3的活性。
作为SIK3抑制剂的化合物在本文其他地方描述,包括可以通过本文列出的适用的功能和/或结构特征来表征的那些。
在优选的实施方案中,特别地,“对象”还意指包括所有哺乳动物,包括但不限于人类,但也包括非人类的灵长类,例如食蟹猴。它还包括狗、猫、马、绵羊、山羊、牛、兔子、猪和啮齿动物(例如小鼠和大鼠)。应当理解,根据本发明的特别优选的对象是人类对象,例如患有病况、疾病或病状(或有风险患有病况、疾病或病状的人类),例如人类患者。
如本文所用,“疗法”与治疗疾病、病症或病状同义,其包括减轻疾病、病症或病状的症状,抑制疾病、病症或病状的进展,引起疾病、病症或病状的消退和/或治愈疾病、病症或病状。
在优选的实施方案中,本发明中的“治疗”,特别是还包括例如疗法(例如疗法治疗)以及疾病(或病症或病状)的预防或抑制措施。因此,例如,在疾病发作之前成功施用本发明的化合物(或药物组合物)可导致疾病的治疗。“治疗”还涵盖在疾病出现之后给予本发明的化合物(或药物组合物)以改善或根除疾病(或其症状)。在发作后和临床症状后给予本发明的化合物(或药物组合物),可能减轻临床症状并可能改善疾病,也包括疾病的治疗。“需要治疗”的对象包括已经患有该疾病、病症或病状的对象(例如人类受试者),以及易于或怀疑患有该疾病、病症或病状的对象者,包括预防其中患有该疾病、病症或病状的对象。
对细胞介导的免疫应答敏感的细胞合适地是与增殖性疾病有关的一种细胞(例如,与增殖性疾病有关的细胞),在某些实施方案中,这种细胞是与增殖性疾病有关的一种细胞(例如,异常增殖的细胞,例如过度增殖的细胞)。例如,这种细胞可以是特征在于失去影响其生长和细胞分裂的正常对照的细胞,例如肿瘤或肿瘤细胞。在特定的实施方案中,这样的细胞可以是癌细胞,或者是衍生自癌细胞或肿瘤细胞的细胞。在其他实施方案中,这样的细胞可以是皮肤细胞,例如显示过度增殖的细胞,例如涉及牛皮癣、雷特氏(Reiter's)综合征、毛疹糠疹或硬皮病的皮肤细胞。
例如,如果细胞与细胞增殖相关,例如它是该细胞增生性疾病的致病因素,或者受到细胞增生性疾病的影响,则该细胞可能“与细胞增生性疾病有关”。如果该细胞的特征在于诸如异常细胞生长或细胞分裂的异常增殖,并且如果该异常细胞生长或细胞分裂是该增生性疾病的病理学或引起原因,则该细胞特别是“与增殖性疾病有关”。在其中增殖性疾病是肿瘤或癌症的实施方案中,“与增殖性疾病有关”的细胞的非限制性例子可以是肿瘤(或癌细胞)细胞或源自于这样的肿瘤或癌症的(组织)的细胞;特别是实体瘤。
在某些实施方案中,本发明的化合物可以在与增殖性疾病有关的细胞(例如肿瘤细胞)中抑制SIK3。特别地,在这样的实施方案中,化合物可以在这样的细胞中抑制SIK3优先于在这样的细胞中抑制SIK1和/或SIK2;和/或可以在这样的细胞中抑制SIK3优先于在一种或多种类型的免疫细胞中抑制SIK1和/或SIK2和/或SIK3。例如,本发明的化合物可以在与增殖性疾病有关的细胞(例如肿瘤细胞)中抑制SIK3,优先于抑制巨噬细胞和/或树突状细胞中的SIK1和/或SIK2和/或SIK3(特别是能够或产生IL-10的)。
本发明的化合物(或药物组合物)可以特别以有效抑制SIK3和/或有效使与增殖性疾病有关的细胞对细胞介导的免疫反应敏感的量施用至对象。用于这种施用的合适的量、制剂和方式在本文其他地方描述。
在特定的实施方案中,本发明的化合物(或药物组合物)以有效降低SIK3(优选在与增生性疾病相关的细胞中的SIK3)活性的量(例如治疗有效量)施用。在这样的实施方案中,化合物(或药物组合物)的“治疗有效量”可以是能够将SIK3的活性降低至适用水平,但是就化合物(或药物组合物)的其他活性而言,其不会导致明显的(例如,不能忍受的)副作用或过量。
优选地,SIK3的活性被有效地抑制(降低),优选是指与增殖性疾病有关的细胞中的SIK3激酶。例如,“有效”抑制(或降低)可包括其中活性降低具有生理作用(例如降低至治疗有效水平)的程度(或水平)的行为,例如降低各个激酶活性的约10%、20%、50%或大于50%,例如70%或90%。关于SIK3,可能需要这样的减少之一以引起治疗反应。
术语“免疫细胞”是本领域公认的描述与该生物体的免疫系统有关的生物体的任何细胞,特别是哺乳动物如人的细胞。白细胞(白色血液细胞)是先天免疫系统中涉及的免疫细胞,而适应性免疫系统的细胞是特殊类型的白细胞,称为淋巴细胞。B细胞和T细胞是淋巴细胞的主要类型,它们来源于骨髓中的造血干细胞。B细胞参与体液免疫反应,而T细胞参与细胞介导的免疫反应。在本发明的优选实施方案中,免疫细胞可以是髓样细胞,例如T细胞,并且特别地(例如当需要增加细胞介导的免疫应答例如治疗癌症时),T细胞可以是细胞毒性T细胞(也称为TC、细胞毒性T淋巴细胞、CTL、T杀伤细胞、溶细胞性T细胞、CD8+T细胞或杀伤性T细胞)。CTL是一种T细胞,参与杀死癌细胞,被感染的细胞(尤其是被病毒感染)或以其他方式损坏的细胞。用于此类实施方案的其他优选的免疫细胞可包括肿瘤浸润淋巴细胞(TIL)。TIL是已经离开血液并迁移到肿瘤中的白细胞。通常,TIL是可变比例的不同类型细胞(例如,T细胞、B细胞、NK细胞、巨噬细胞)的混合物,T细胞是最丰富的细胞。TIL通常可以在基质和肿瘤本身中发现,并与杀死肿瘤细胞有关。肿瘤中淋巴细胞的存在通常与更好的临床结果相关。
如本文所用,术语“细胞介导的免疫应答”可包括但不限于宿主生物中的反应,其涉及、利用和/或促进T细胞的成熟、增殖、活化、迁移、浸润和/或分化,和/或巨噬细胞、天然杀伤细胞、T淋巴细胞(或T细胞)、辅助性T淋巴细胞、记忆性T淋巴细胞、抑制性T淋巴细胞、调节性T淋巴细胞和/或细胞毒性T淋巴细胞(CTL)的激活、迁移、浸润和/或分化,和/或一种或多种细胞可分泌或细胞分泌因子(例如细胞因子或自体抗体(特别是促炎性细胞因子,例如TNF)的产生、释放和/或作用,和/或任何此类过程的一种或多种组分(例如,细胞因子或自体抗体,特别是促炎性细胞因子,例如TNF)。如本文所用,术语“细胞介导的免疫应答”可以包括涉及基因工程改造、体外培养、自体、异源、修饰和/或转移的T淋巴细胞的细胞应答,或者可以包括可细胞分泌的或通过基因工程产生的细胞分泌因子(例如细胞因子或自体抗体,特别是促炎性细胞因子,例如TNF)。细胞介导的免疫应答优选不是体液免疫应答,例如涉及抗体释放的免疫应答。在某些实施方案中,特别是当增殖性疾病是癌症或肿瘤时,细胞介导的免疫应答是抗肿瘤细胞介导的免疫应答。例如,导致肿瘤(细胞)生长减少的,例如杀死癌细胞或肿瘤的细胞毒性细胞介导的免疫反应(例如细胞毒性T细胞和/或TNF暴露)。
在某些实施方案中,细胞介导的免疫应答可以由能够分泌(例如分泌)促炎性细胞因子的细胞(例如免疫细胞)介导,例如选自:白介素-1(IL-1)、IL-8和IL-12,肿瘤坏死因子(TNF)、干扰素γ(IFN-γ)和粒细胞-巨噬细胞集落刺激因子。特别地,在这样的实施方案中,促炎细胞因子是肿瘤坏死因子(TNF)α。
在其他实施方案中,细胞介导的免疫反应可以是细胞可分泌的或细胞分泌的因子(例如细胞因子或自体抗体),尤其是免疫细胞可分泌或分泌的一种因子。特别地,在此类实施方案中,细胞介导的免疫反应是促炎细胞因子,特别是肿瘤坏死因子(TNF)。
如本文在用于细胞对细胞介导的免疫反应敏感的上下文中所使用的,术语“对……敏感”、“致敏”和“敏感化/敏化”(等等)将被本领域技术人员理解,并且包括这样的含义,即这种细胞对细胞介导的免疫应答可能对这种细胞表现出的一种或多种作用(例如治疗作用)表现出更高的敏感性。特别地,当存在(例如暴露于)细胞介导的免疫反应时,与尚未如此“敏化”的类似细胞相比,如此敏化的细胞可以被更容易地杀死(例如,更快地、更大比例的细胞死亡或被杀死和/或细胞介导的免疫应答的数量或暴露更低)。例如,当暴露于较少数量的T细胞或较低浓度的TNF(例如约10%、20%、30%、40%、50%或多于50%更少的T细胞或更低浓度的TNF)时,如此敏化的细胞可被诱导进入细胞死亡(例如凋亡)。本文例如在实施例中描述了确定此类细胞是否已经对细胞介导的免疫反应敏感(以及程度如何)的方法。因此,在本发明的某些实施方案中,可以通过细胞介导的免疫应答(例如CTL或促炎性细胞因子,例如TNF)使与增殖性疾病有关的细胞对细胞死亡/杀伤(例如通过进入凋亡)敏感。
在本文公开的发明的上下文中,术语“肿瘤坏死因子”和“TNF”(以前并因此可替代地称为肿瘤坏死因子α和TNF-α)应当理解为是指在本领域中以这些名称已知的任何蛋白质。特别地,术语TNF涵盖存在该生物的任何生物的内源性TNF,并且优选地包括动物或哺乳动物,例如人的内源性TNF。通过举例而非限制的方式,人TNF可以涵盖特别是Pennica etal.1984(Nature 312:724-9)以及UniProtKB/Swiss-Prot数据库中的条目号P01375(例如,2017年3月15日的条目版本224)以及由由于人群之间和人群中的正常序列多态性的任何序列变体的内源性蛋白质。借助其他非限制性实例,该术语可涵盖UniProtKB/Swiss-Prot数据库中注释的牛(Q06599)、狗(P51742)、山羊(P13296)、豚鼠(P51435)、猫((P19101)、马(P29553)、小鼠(P06804)、黑猩猩(Q8HZD9)、猪(P23563)、兔(P04924)、大鼠(P16599)等以及由于各个物种种群之间和之内的序列多态性的其任何序列变体的内源性TNF蛋白。此外,术语TNF特别地包括TNF的可溶性、分泌的细胞因子形式,包括其单体形式以及优选地其通常更具活性的三聚体形式(参见,例如Smith&Baglioni 1987.J Biol Chem 262:6951-4)。内源性TNF的可溶形式的一级氨基酸序列在上述UniProtKB/Swiss-Prot数据库条目中针对各个示例性生物指示。另外,术语TNF还可以包括在某些细胞类型的表面上表达的TNF的膜结合形式(参见,例如Kriegler et al.1988.Cell 53:45-53)。此外,术语TNF还可以包括这样的合成或重组蛋白,其一级氨基酸序列与内源性TNF的序列相同或基本相同(“基本相同”,如贯穿本说明书所使用的,通常指≥80%,例如≥85%,优选≥90优选≥95%,甚至更优选≥98%或≥99%序列同一性),如使用例如Tatusova&Madden 1999(FEMS Microbiol Lett174:247-250)描述的“Blast 2序列”算法确定的,其(优选)保留与各自内源性TNF相同或基本相同的生物活性,如使用例如Flick&Gifford 1984(J Immunol Methods 68:167-75)所述的细胞毒性试验确定的。如从本发明的方面和实施方案的上下文中将清楚的,术语TNF在本文中可以特别地指由细胞、组织、器官或生物产生的可溶和/或膜结合的(优选可溶的)内源性TNF,优选人类。然而,术语“TNF”也预见了肿瘤坏死因子的外源形式,特别是通过重组技术产生的那些,并且在某些实施方案中,可以在本发明的各个方面和实施方案中施用于对象,或使其暴露于细胞或与细胞接触。在某些这样的实施方案中,所述TNF可以是用作治疗剂的重组TNF,例如他森敏(BEROMUN)。
在某些实施方案中,细胞介导的免疫反应可以由促炎性细胞因子分泌细胞(例如淋巴细胞(例如T细胞),特别是细胞毒性T淋巴细胞(CTL))介导。
在特定的实施方案中,细胞介导的免疫应答可以诱导与增殖性疾病有关的细胞的杀伤(例如细胞死亡,例如通过凋亡)。例如,治疗(方法)可以包括(例如可以涉及)细胞介导的免疫应答(或由其介导)诱导与增殖性疾病有关的细胞的这种杀伤。
与增殖性疾病有关的细胞可以通过一种或多种细胞毒性过程被杀死(例如,诱导进入细胞死亡),特别是这种细胞内源性的那些,例如程序性细胞死亡(PCD)。细胞死亡过程可包括但不限于坏死(特别是坏死病)、细胞凋亡、神经衰弱、自噬、肥大症、有丝分裂灾难和激活诱导的细胞死亡。在某些优选的实施方案中,与增殖性疾病有关的细胞(例如肿瘤细胞)通过细胞介导的免疫应答(例如通过TNF)被诱导为凋亡。在另一个实施方案中,在不存在细胞介导的免疫应答(例如,不存在TNF)的情况下,施用本发明的化合物(或药物组合物)以不杀死此类细胞。特别地,在这样的其他实施方案中,化合物(或药物组合物)可以在不存在细胞介导的免疫应答的情况下不有效杀死此类细胞的量(例如,剂量)施用。本文的实施例描述了各种测定法,通过这些测定法,可以确定本发明的化合物(或药物组合物)的量,该量仅或优选在细胞介导的免疫应答的存在下有效地杀死此类细胞。
在其他特定的实施方案中,细胞介导的免疫应答可涉及至少一种免疫细胞效应分子,特别是免疫细胞可分泌或分泌的效应分子。特别地,在这样的实施方案中,效应分子可以是促炎细胞因子,优选是肿瘤坏死因子(TNF)。
在某些实施方案中,效应分子不是选自Fas配体(FasL或CD95L)和TNF相关凋亡诱导配体(TRAIL、CD253或TNFSF10)的细胞效应分子。
在本发明的特定实施方案中,本发明的化合物(或药物组合物)可以施用于对象(例如以有效量或剂量),其意图(或从而)(有效地)使与增生性疾病有关的细胞对TNF引起的杀伤敏感。例如,化合物(或药物组合物)可以治疗有效量施用,例如有效使与增殖性疾病有关的细胞对由TNF诱导的杀伤(细胞死亡)敏感的量。
例如,可以将本发明的化合物(或药物组合物)施用于对象(例如以有效量或有效剂量)以诱导由TNF介导的此类细胞的凋亡,例如当此类细胞存在或接触TNF时。在进一步的实施方案中,可以将本发明的化合物(或药物组合物)施用于对象(例如以有效量或剂量)以诱导减少量的细胞毒性(例如凋亡)——例如
TNF可以通过与肿瘤坏死因子受体1(TNFR1)和/或肿瘤坏死因子受体2(TNFR2)结合和/或通过信号传导来诱导促凋亡过程。因此,在某些实施方案中,本发明的化合物(或药物组合物)可以(例如有效的量或剂量)施用于对象以(有效地)使与增殖性疾病有关的细胞对由肿瘤坏死因子受体1(TNFR1)信号和/或肿瘤坏死因子受体2(TNFR2)信号介导的细胞凋亡敏感。优选地,可以将化合物(或药物组合物)施用于对象(例如以有效的量或剂量)以(有效地)使与增殖性疾病有关的细胞对由此介导的细胞凋亡特别是由TNFR1介导的细胞凋亡敏感。例如,可以以有效介导TNFR1-和/或TNFR2-信号转导和/或由此介导的细胞凋亡的治疗有效量来施用化合物(或药物组合物)。
例如,在某些实施方案中,可以施用本发明的化合物(或药物组合物)(例如以有效的量或剂量)以通过TNFR1和/或TNFR2信号传导(例如在活性TNFR1信号传导时)诱导此类细胞的凋亡。特别地,在这样的实施方案中,可以将化合物(或药物组合物)施用至对象(例如以一定量或剂量,例如治疗有效量)以(有效)诱导减少量的细胞毒性(例如细胞凋亡)——例如不诱导此类细胞的凋亡——在不存在TNFR1和/或TNFR2信号传导的情况下,例如在不存在活性TNFR1信号传导的情况下。例如,化合物(或药物组合物)可以以对细胞毒性(例如细胞凋亡)不那么有效的量或剂量给药——例如不能有效地诱导这种细胞凋亡——在没有这种信号传导的情况下。
因此,在某些实施方案中,可以将本发明的化合物(或药物组合物)施用于对象(例如以一定量或剂量)以在没有细胞介导的免疫反应的情况下,对与增殖性疾病有关的细胞诱导减少量的细胞毒性(例如细胞凋亡),例如不具有细胞毒性。
在特定的实施方案中,即使在治疗过程中对象的肿瘤增大,本发明的化合物(或药物组合物)也可以继续给予对象。不受理论的束缚,即使在这样的治疗过程中观察到肿瘤大小增加,也可能表明针对肿瘤细胞的(增强的)免疫反应(例如,细胞已变得对细胞介导的免疫反应敏感;和由于这种免疫应答,肿瘤的大小在增加),因此,在这样的实施方案中,化合物(或药物组合物)的施用可以继续施用,以维持这种敏感性和相关的(增强的)免疫反应。
如PCT/EP2018/060172中所述,SIK3的抑制与许多关键的生物学过程或表型相关,包括令人惊奇地参与控制和/或触发细胞固有的细胞毒性过程,例如细胞凋亡。例如,通过抑制SIK3,肿瘤细胞可以对TNF的凋亡/细胞毒性作用敏感,通过途径及其成分其作用,包括肝激酶B1(LKB1、STK11或NY-REN-19)、组蛋白脱乙酰基酶4(HDAC4)、激活的B细胞核因子κ轻链增强子(NF-kappaB)和受NF-kappaB调节的促凋亡基因,例如Caspase 8和Caspase9。c-JunN末端激酶(JNK)是通过抑制SIK3与TNF的凋亡/细胞毒作用致敏相关的信号传导成分。
在本实施例(和其他适用的实施例)的上下文中,术语“与……相关”可以表示两个组件、变量、效果或表型相互关联,和/或彼此相关(例如关联),和/或第一和第二成分、变量、效果或表型之间存在因果关系(例如第二个是对第一个的响应,第二个是第一个的结果,或第二个是由第一个引起的)。
因此,在一个这样的实施方案中,本发明的化合物(或药物组合物)的施用可以与在与增殖性疾病有关的细胞中NF-κB活性的损害相关(例如,通过增强或增加NF-κB从细胞核外转运)。
特别是在此类实施方案中,此类NF-κB活性受损(例如,由于NF-κB增强或从细胞核外转运)可能与这些细胞中(激活的)TNF-和/或TNFR1介导的信号传导(或TNFR2介导的信号传导)相关。
在某些实施方案中,本发明的化合物(或药物组合物)可以(例如以有效的量或剂量)施用于对象以损害或抑制与增殖性疾病有关的细胞中的NF-κB活性,用于例如增强或增加NF-κB从此类细胞核外转运的例子。例如,化合物(或药物组合物)可以以特定的(例如治疗有效)量给予对象,该量有效(高效)削弱与增殖性疾病有关的细胞中NF-κB活性,特别是有效(高效)增强或增加NF-κB从与增殖性疾病有关的细胞核外转运。
在替代或进一步的实施方案中,本发明的化合物(或药物组合物)的施用可以与增加与增殖性疾病有关的细胞中的II类(例如IIa)HDACs(例如HDAC4)的活性有关(例如,以有效的量或剂量施用化合物(或药物组合物),从而增加),例如其在此类细胞核中的移位或定位或在其细胞核中的活性;尤其是在此类细胞中由TNF和/或TNFR1介导的信号传导(或TNFR2介导的信号传导)时。
在其他替代或进一步的实施方案中,本发明化合物(或药物组合物)的施用可以与核NF-κB的去酰化(例如在其p65亚基上的去酰化)和/或一种或多种抗凋亡因子的反式激活减少有关,特别是在与增殖性疾病有关的细胞中由TNF-和/或TNFR1介导的信号传导(或TNFR2介导的信号传导)时。例如,可以施用化合物(或药物组合物)(例如以有效的量或剂量)以引起核NF-κB(例如在其p65亚基处)的去酰化和/或减少一种或多种抗凋亡因子的反式激活。
在另一个替代或进一步的实施方案中,本发明的化合物(或药物组合物)的施用可以与Caspase 8和/或Caspase 9在与增殖性疾病有关的细胞中的裂解的增加有关(例如以有效的量或剂量施用该化合物(或药物组合物),从而增加),特别是在此类细胞中由TNF-和/或TNFR1介导(或TNFR2介导的信号转导)时。
在其他替代或进一步的实施方案中,本发明化合物(或药物组合物)的施用可以与一种或多种抗凋亡因子的转录降低有关,特别是在与增殖性疾病有关的细胞中TNF-和/或TNFR1介导的信号传导(或TNFR2介导的信号传导)时,例如减少此类细胞中一种或多种NF-κB靶基因的转录。特别地,可以施用化合物(或药物组合物)(例如以有效剂量的剂量)以减少一种或多种这样的抗凋亡因子的转录,特别是在与增殖性疾病有关的细胞中TNF-和/或TNFR1介导的信号传导(或TNFR2介导的信号传导)时。
在一个实施方案中,本发明的化合物(或药物组合物)的施用可以与增加与增殖性疾病有关的细胞中的JNK活化(例如通过磷酸化)有关(例如,以有效的量或剂量施用化合物(或药物组合物),从而增加),特别是在此类细胞中的TNF-和/或TNFR1介导的信号传导(或TNFR2介导的信号传导)时。
在另一个实施方案中,本发明化合物(或药物组合物)的施用可能与CREB途径信号的显著变化和/或CREB和/或CREB调节介导的基因表达的显著变化不相关。
在一个特定的实施方案中,与增殖性疾病有关的细胞中TNF-(TNFR2-)和/或TNFR1介导的信号传导可能与此类细胞中pLKB1水平的增加有关。
如对本领域普通技术人员来说,根据本发明的知识现在将显而易见的是,本发明的治疗方面可以进一步包括施用一个或多个其他部分的步骤,所述其他部分适当地修饰了上述一个或多个这些其他途径成分中的表达、活性、功能或稳定性,以累加或协同地有助于治疗效果。例如,在一个这样的实施方案中,本发明的治疗方面可以进一步包括施用LKB1抑制剂的步骤。在另一个这样的实施方案中,本发明的治疗方面可以进一步包括以下步骤:施用促进、增强或增加与增殖性疾病有关的细胞核中一种或多种II类(例如IIa)HDAC(组蛋白脱乙酰基酶),例如HDAC4。在此类实施方案的又一个中,本发明的治疗方面可以进一步包括施用NF-κB抑制剂(激活)的步骤。本发明还预想了可以将两种或更多种其他这样的部分的组合与本发明的化合物(或药物组合物)一起使用和/或与化合物(或药物组合物)一起使用其他(例如抗癌)治疗活性剂(例如另外的治疗剂,如本文别处所述的治疗剂)。
在另一方面,并且如本文可以进一步描述、定义、要求保护或以其他方式公开的,本发明涉及一种使与增殖性疾病有关的细胞对细胞介导的免疫应答敏感的方法,该方法包括将与增殖性疾病有关的细胞暴露(例如接触)至本发明的化合物(或药物组合物)。通常可以将这种方法实践为体外和/或离体方法。
在一个特定的实施方案中,细胞介导的免疫应答包括杀死与增殖性疾病有关的细胞,例如其中所述杀死涉及TNF(TNF)、TNFR2-和/或TNFR1-介导的信号传导(例如由其介导,或被其支持)。例如,杀死此类细胞可能涉及由TNF、TNFR2和/或TNFR1介导的信号传导诱导的此类细胞的凋亡。在本发明各个方面的该实施方案和其他适用实施方案中,TNFR2和/或TNFR1介导的信号传导可通过任何合适的触发分子(例如TNF,TNF的变体和/或TNFR2、TNFR1激动剂)触发(例如激活);特别是通过将与增生性疾病相关的细胞暴露(例如接触)到触发分子(例如TNF、TNF变体或TNFR1激动剂)。这样的暴露可以导致触发分子(例如TNF、TNF变体或TNFR1激动剂)与TNFR2和/或TNFR1结合,尤其是TNFR1信号的触发(例如激活)。
在另一方面,并且如本文可以进一步描述,定义,要求保护或以其他方式公开的,本发明涉及一种杀死与增殖性疾病有关的细胞的方法,该方法包括暴露(例如接触)该细胞。与增殖性疾病有关的细胞:(i)TNF、TNFR1或TNFR2信号转导的TNF变体和/或激动剂(优选TNFR1信号转导);使与增殖性疾病有关的细胞暴露于(ii)本发明的化合物(或药物组合物)。如本领域技术人员或普通技术人员将理解的那样,通常可以将这种方法实践为体外和/或离体方法。
在相关方面,本发明涉及本发明的化合物(或药物组合物),其用于治疗涉及杀死与所述增殖性疾病有关的细胞的增殖性疾病,所述治疗包括将所述细胞暴露于(i)TNF、TNF变体和/或TNFR1或TNFR2激动剂;(ii)本发明的化合物(或药物组合物)。
在这些方面的特定实施方案中,通过使这种细胞对细胞介导的免疫反应敏感,特别是通过诱导对涉及(例如,由其介导或由其支持)TNF、TNFR2和/或TNFR1介导的信号传导的细胞凋亡的敏感性来介导与增殖性疾病有关的细胞的杀伤。
与增殖性疾病有关的细胞可通过使该细胞与该触发分子接触而暴露于TNF、TNF变体和/或TNFR1或TNFR2激动剂;和/或通过使这种细胞与本发明的化合物(或药物组合物)接触(或引入),可以使这些细胞暴露于本发明的化合物(或药物组合物)。(i)TNF、TNF变体和/或TNFR1或TNFR2激动剂;和/或(ii)本发明的化合物(或药物组合物)的量(或剂量)通常是有效量;这是在例如使细胞对由TNF、TNFR2和/或TNFR1介导的信号传导诱导的细胞凋亡敏感(例如通过杀死细胞)有效的量(或剂量)。在其他地方公开了可以掺入本发明的这些方面的合适量的这些活性剂(或确定它们的方法),本发明化合物(或药物组合物)的其他特定特征也一样。因此,在某些实施方案中:(i)TNF、TNF变体和/或TNFR1或TNFR2激动剂;和(ii)本发明的化合物(或药物组合物)可以施用于患有增生性疾病的对象(例如,治疗可以包括施用:(i)TNF、TNF变体和/或TNFR1或TNFR2激动剂;和(ii)本发明的化合物(或药物组合物)可以施用于对象)。
与增殖性疾病有关的细胞可以是如本文其他地方所述的细胞,并且特别地,这种细胞可以是癌细胞或肿瘤细胞。例如,这样的细胞可以是实体瘤或源自实体瘤的细胞。
在这些方面的某些实施方案中,该方法是体外(和/或离体)方法。在此类方法的替代实施方案中,与增殖性疾病有关的细胞(例如肿瘤细胞)存在于此类对象中,特别是在需要对其进行治疗的对象中。
在这些方面的方法的其他实施方案中,该方法的(例如,对与增殖性疾病有关的细胞)的(治疗)作用可以由(例如,治疗可以包括、涉及或由其介导)抑制SIK3;特别地,通过抑制SIK3或蛋白质(例如,磷酸化的SIK3蛋白质和/或如本文其他地方所述的)的功能和/或活性介导。特别地,在这样的实施方案中,SIK3活性被降低(例如有效),例如降低到治疗有效水平。
在此类方法的某些实施方案中,在不存在(例如有效量或剂量)本发明的化合物(或药物组合物)的情况下,与增殖性疾病有关的细胞(例如肿瘤细胞)不会在TNF、TNFR2和/或TNFR1介导的信号传导和/或暴露于(例如有效量或剂量的)TNF、TNF变体、TNFR2或TNFR1激动剂时被杀死或诱导进入凋亡(例如,它们增殖)。
如上所述,在这些方法的某些实施方案中,本发明的化合物(或药物组合物)可以抑制与增殖性疾病有关的细胞(例如肿瘤细胞)中的SIK3。特别地,在此类实施方案中,化合物(或药物组合物)可优先于抑制此类细胞中的SIK1和/或SIK2而抑制这种细胞中的SIK3;和/或可以优先于抑制一种或多种类型的免疫细胞中的SIK1和/或SIK2和/或SIK3而抑制这种细胞中的SIK3。例如,本发明的化合物(或药物组合物)可以在与增殖性疾病有关的细胞(例如,肿瘤细胞)中抑制SIK3,优先于抑制巨噬细胞和/或树突状细胞(特别是能够或产生IL-10的)中的SIK1和/或SIK2和/或SIK3。在特定的实施方案中,(治疗)作用是由(例如,该治疗包括、涉及、由其介导)抑制与增殖性疾病有关的细胞(例如肿瘤细胞)中的SIK3介导的;并且在这样的实施方案的进一步中,(治疗)作用不是由(或该效果不是由其介导)(例如,该治疗不包括、不涉及或不由其介导)抑制SIK2、特别是在其他细胞(例如,与增殖性疾病或免疫细胞有关的细胞)中/的SIK2介导的,和/或(治疗)作用不是由(或该效果不是由其介导)抑制SIK1(例如,该治疗不包括、不涉及或不由抑制SIK1介导)、特别是在其他细胞(例如,与增殖性疾病或免疫细胞有关的细胞)中/的SIK1介导的。
因此,在一个实施方案中,(例如,通过本发明的化合物或药物组合物)抑制了与增殖性疾病有关的细胞(例如,在其内)的SIK3。在另一个(或进一步的)实施方案中,(例如,在其内)免疫细胞(例如CTL)中的另一种激酶(例如SIK2,特别是SIK2)被抑制的程度小于SIK3(例如在涉及增殖性疾病的细胞中)的抑制程度。在又一个(或进一步的)实施方案中,SIK1,特别是(例如在其内)免疫细胞(例如CTL)中的SIK1被抑制的程度小于这种SIK3的。
在某些此类实施方案中,选自以下的激酶中的一种或多种:SIK3、SIK1、SIK2、JAK1、RET、ERBB4 PDGFR-α和EPHB2被抑制(例如,被本发明的化合物或药物抑制)的程度小于选自以下的激酶中的一种或多种:ABL1、SRC、BCR-ABL、LCK、LYN、YES、FYN、KIT和FLT3。
在某些这样的实施方案中,选自以下的激酶中的一种或多种:PDGFR-α、TGFB-R1、B-RA、p38-β、ACV-R1、BMPR1A和RET被抑制(例如,被本发明的化合物或药物抑制)的程度小于以下的激酶的一种或多种:EPHA2、EPHA4、CSF1-R、HCK和ACK1。
在某些这样的实施方案中,选自以下的激酶中的一种或多种:NEK11、WEE1、WNK2、Aurora-A、Aurora-B和TBK1被抑制(例如,被本发明的化合物或药物抑制)的程度小于以下的激酶的一种或多种:ABL1、SRC、BCR-ABL、LCK、LYN、YES、FYN和KIT。
如果例如另一种激酶(例如SIK3)的抑制量大于给定激酶的约2倍,例如抑制量比给定的激酶多约5、10、20、50、75或100倍,则给定的激酶(例如SIK1或SIK2)与另一种激酶(例如SIK3)相比的抑制“程度更少”。特别地,其他激酶(例如SIK3)的抑制量可以比给定激酶多约5至20倍、20至50或50至100倍。例如,相对于SIK1和/或SIK2(即给定的激酶),SIK3(即另一种激酶)可被抑制约20至50倍。举例来说,本发明的化合物(或药物组合物)可以将另一种激酶(例如,SIK3)抑制80%(即,仅具有其未抑制活性的20%),但是抑制给定的激酶(例如,SIK1)仅4%,SIK2仅8%。因此,另一种激酶(例如SIK3)的抑制比给定激酶(例如SIK1)多约20倍,比另一种给定激酶(例如SIK2)多约10倍。在特定实施方案中,另一种激酶(例如SIK3)可以被抑制至与例如SIK1相同的程度(例如彼此之间约2至53倍之间),并且例如SIK2被抑制的程度小于例如SIK3和SIK1的其中任一个(或两者):例如,在这种实施方案中,例如SIK3和SIK1的抑制比中SIK2(例如在免疫细胞中的)的抑制多约20至50倍。
本发明的化合物显示为一种或多种激酶的有效抑制剂(如“实施例”所示,特别是图3所示)。特别地,图3中的激酶中的任何一种(或它们的任何组合)具有约50%至约25%的残余活性,或小于约25%的残余活性(特别是,具有小于10%的残余活性的激酶),被认为是被本发明的各个化合物抑制的“关键激酶”。其中也考虑了此类激酶的突变体。作为特定实例,关键激酶包括选自以下的一种或多种激酶:SIK1、SIK2、SIK3、ABL1/BCR-ABL、SRC、FLT3、KIT、YES、LYN、FYN和LCK;和/或EPHA2、EPHA4、CSF1-R、HCK、ACK1;和/或PDGFR-α、TGFB-R1、B-RAF和/或p38-β;和/或ACV-R1和/或BMPR1A;和/或RET;和/或NEK11、WEE1和/或WNK2;和/或Aurora-A和/或Aurora-B;和/或TBK1;特别是ABL1/BCR-ABL、ABL1/BCR-ABL和FLT3。
发明人发现,与其他激酶抑制剂相比,本发明的化合物抑制不同组的激酶和/或抑制每种激酶至不同程度。例如,化合物B3和A8是ABL1和SRC(以及ABL1突变体)的等效抑制剂。但是,如“实施例”所示,它们在不同程度上抑制SIK1、SIK2、SIK3,尤其是FLT3、KIT和SYK。
在实施例中还显示,与达沙替尼相比,本发明的化合物对ABL1和(特别是)对SRC激酶具有更高的选择性,并且该选择性也适用于蛋白质-酪氨酸激酶类别。
抑制不同激酶和/或不同程度抑制激酶的化合物在体内具有不同的性质,可用于不同的医学适应症或相同的医学适应症,但在功效和副作用方面表现出不同的性质。可以理解,对激酶具有不同特异性的化合物可以具有令人惊讶的不同性质和应用。
因此,在一个实施方案中,该治疗包括(例如,涉及、通过或由其介导)抑制一种或多种关键激酶(例如,ABL1和/或SRC激酶和/或LCK)。特别地,在这样的实施方案中,所述治疗包括对所述关键激酶的抑制(例如,涉及、通过或由其介导)大于其他键激酶中的一种或多种(例如SIK3和/或SIK1和/或SIK2)的抑制(例如,涉及、通过或由其介导)。例如,治疗可涉及抑制ABL1和/或SRC激酶,和/或一种或多种选自以下的激酶:BCR-ABL、LCK、LYN、YES、FYN和KIT。
在一个特定的(替代的或另外的)实施方案中,该治疗不包括(例如,不涉及、不是或不由其介导)一种或多种关键激酶的抑制。特别地,在这样的实施方案中,所述治疗不包括(例如,不涉及、不是或不由其介导)对SIK3的抑制,和/或治疗不包括(例如,不涉及、不是或不由其介导)抑制SIK1和/或SIK2。
在进一步的实施方案中,所述治疗可以不包含(例如,可以不涉及、不是或不由其介导)抑制以下一种或多种激酶:JAK1、RET、ERBB4、PDGFR-α或EPHB2。
在另一个特定的(替代的或另外的)实施方案中,所述治疗不包括(例如,不涉及、不是或不由其介导)SYK的抑制。例如,化合物B3抑制SYK的IC50超过25uM,而化合物A8抑制SYK的IC50小于5uM。
在又一个特定的(替代的或另外的)实施方案中,所述治疗包括(例如,涉及、是或由其介导)KIT抑制。例如,化合物B3抑制KIT的IC50小于50nM,化合物A8对KIT的IC50也小于50nM。
在另一个特定的(替代的或另外的)实施方案中,所述治疗包括(例如,涉及、是或由其介导)抑制FLT3。例如,化合物B3抑制FLT3的IC50小于10uM,而化合物A8对FLT3的IC50大于25uM。
的确,在某些特定的(替代的或另外的)实施方案中,所述治疗包括(例如,涉及、是由或由其介导)抑制KIT和FLT3;例如,通过向对象施用化合物B3,并且例如,该治疗包括(例如,涉及、是由或由其介导)抑制ABL1、SRC和/或SIK3。
在另一个实施方案中,所述治疗包括(例如,涉及、是由或由其介导)ABL1或KIT激酶的突变体的抑制;例如,对BCR-ABL的抑制,或对ABL1的另一种突变,例如选自以下中的一种:G250E、Q252H、Y253F、E255K、F317I、M351T和H396P。
如本文其他地方所述,在一个(替代或另外的)实施方案中,本发明的化合物使与增殖性疾病有关的细胞对细胞介导的免疫反应(例如TNF)敏感(例如,该治疗包括、涉及、或由其敏化介导)。然而,在替代的(替代的或另外的)实施方案中,化合物不使与增殖性疾病有关的细胞对细胞介导的免疫反应(例如TNF)敏感(例如,该治疗不包括、不涉及、或不由其敏化介导)。
与其他使用激酶(例如SIK)抑制剂的研究相反,在某些实施方案中,用根据本发明的本发明的化合物(或药物组合物)进行的治疗可能与一种或多种抗炎细胞因子的产生(例如抗炎细胞因子可以选自IL-1ra、IL-4、IL-10、IL-11、IL-13和TGF-β)的(有效)增加不相关,尤其可能与IL-10产生的(有效)增加不相关。相应地,在其他或进一步的实施方案中,用根据本发明的本发明的化合物(或药物组合物)进行的治疗可能与一种或多种促炎细胞因子的产生的(有效)减少不相关。例如,选自以下的一种:IL-1-β、IL-6、IL-12和TNF、IFN-γ和粒细胞-巨噬细胞集落刺激因子,并且在特定的实施方案中可能与TNF的产生的(有效)减少不相关。因此,在某些实施方案中,本发明的化合物(或药物组合物)可以以下量施用于对象:(i)对(有效)增加一种或多种(例如此类)抗炎细胞因子的产生无效的(治疗有效)量;和/或(ii)不能有效(有效)减少一种或多种(例如此类)促炎细胞因子的产生的(治疗有效)量。
在某些实施方案中,在本发明的各个方面中,预期与增殖性疾病有关的某些细胞(例如肿瘤细胞)可以对本发明的化合物(或药物组合物)的敏化作用更敏感。例如,这样的细胞可以是表现出(例如经受)TNFR2和/或TNFR1信号传导的激活,特别是活化的TNFR1的细胞。在某些实施方案中,此类细胞是表达TNFR2和/或TNFR1的细胞,特别是表达TNFR1的肿瘤细胞。因此,在某些实施方案中,此类细胞通过活化的TNFR1和/或TNFR2信号传导来区分或表征(或通过如此区分或表征具有与增殖性疾病有关的细胞(例如肿瘤细胞)的方式来区分或表征对象)。本领域普通技术人员将知道用于确定此类细胞(例如对象的细胞)中TNFR1和/或TNFR2活化状态的技术。例如,通过检测或监测TNFR1和/或TNFR2信号通路中的一种或多种下游蛋白。这样的蛋白质在本文其他地方描述,并且包括NF-κB和/或HDAC4。
在一个相关方面,本发明涉及一种用于治疗对象的增生性疾病(例如肿瘤)的方法,该(治疗)方法包括将本发明的化合物(或药物组合物)施用于所述对象,通过抑制激酶/关键激酶(例如SIK3),其中与增殖性疾病有关的细胞的特征在于(例如,表现出或经受)活化的TNFR2和/或TNFR1信号传导(例如,活化的TNFR1信号传导)。在另一个相关的方面,本发明涉及用于治疗增生性疾病的本发明的化合物(或药物组合物),其中与该增生性疾病有关的细胞的特征在于(例如表现出或经受)活化的TNFR2和/或TNFR1信号传导(例如激活的TNFR1信号传导)。
在本发明各个方面的某些实施方案中,与增殖性疾病有关的细胞是暴露于合适的触发或活化分子(例如TNF、TNF的变体和/或TNFR2-或TNFR1-信号激动剂(优选TNFR1信号传导激动剂)的细胞,特别是暴露于有效量的这种触发或活化分子中。
在特定的实施方案中,当触发或激活分子是TNF时,它是人TNF。在某些这样的实施方案中,TNF是重组人TNF(rHuTNF)。然而,在其他实施方案中,TNF是内源性TNF,例如由对象(例如人类患者)产生或以其他方式存在于对象中。
研究表明,多种类型的癌症(包括卵巢癌)中的血浆TNF水平升高(Dobrzycka etal 2009,Eur Cytokine Netw 20:131),例如,如使用Quantikine人类TNF-α免疫测定PDTA00C测得的,健康人群中总TNF的正常上限对象为1.8pg/mL。在其他癌症和检测方法中(例如TNF-alpha-EASIA Kit,DIAsource),食道癌患者和对照组的TNF血浆水平分别为12.35±9.69和4.62±3.06pg/mL(Aydin et al 2012,Turk J Med Sci 42:762)。因此,在其他实施方案中,与增殖性疾病有关的细胞是(例如肿瘤是)对象中的细胞,该对象的TN F血浆浓度大于约1.5、2.5或4pg/mL,例如大于约5pg/mL,特别是大于约10pg/mL(例如,通过Quantikine人TNF-α免疫测定PDTA00C或TNF-α-ELISA试剂盒,DIAsource测量)。
因此,在一个特定的实施方案中,本发明的治疗方法中涉及的对象可以具有(即,该对象可以通过例如适合于本发明的治疗方法的对象来区分,例如通过显示、拥有或展示)大于约2pg/mL或大于约5pg/mL的TNF血浆浓度(例如,与增生性疾病有关的细胞存在于TNF的血浆浓度大于约2pg/mL或5pg/mL的对象中)。
实际上,在增生性疾病是肿瘤的实施方案中,TNF的肿瘤内浓度可能是肿瘤的特征,例如当肿瘤是实体瘤并且可以进行活检时(Reissfelder et al 2015,J Clin Inv125:739)。例如,在本发明的一些实施方案中,肿瘤(例如实体瘤,例如结肠直肠癌)可以具有大于约0.2、0.5或1pg/mL,例如大于约2pg/mL,特别是大于约5pg/mL的TNF的肿瘤内浓度(例如,在肿瘤组织内)(例如,通过Quantikine人TNF-α免疫测定法测量)。
因此,在这样的实施方案中,当增生性疾病是肿瘤(例如实体瘤)时,则实体瘤(例如,对象体内)可以具有(即,该对象可以通过例如适合于本发明的治疗方法的对象来区分,例如通过显示、拥有或展示)TNF的肿瘤内浓度大于(约)0.5pg/mL或大于约1pg/mL。
因此,在一个相关方面,本发明可以涉及一种在通过具有以下特征来区分的对象中治疗增生性疾病(或用于这种治疗的本发明化合物(或药物组合物))的方法:(i)TNF的血浆浓度大于约2pg/mL(优选大于约5pg/mL);和/或(ii)TNF的肿瘤内浓度大于约0.5pg/mL,优选大于约1pg/mL,所述治疗方法包括向所述对象施用本发明的化合物(或药物组合物),其中所述化合物(或药物组合物):(a)抑制与增殖性疾病有关的细胞中的激酶/关键激酶(例如SIK3);和/或(b)使与增殖性疾病有关的对象的细胞对细胞介导的免疫反应敏感。
特别是在这样的实施方案中,本发明的化合物(或药物组合物)的暴露于与增生性疾病有关的细胞或施用于对象的量(或剂量)与TNF的血浆或肿瘤内浓度相关(或关联),其中在TNF的血浆或肿瘤内浓度较高的情况下,较大量(或剂量)的化合物(或药物组合物)暴露于此类细胞(或施用于此类对象)。
在其他或进一步的实施方式中,肿瘤可以存在于外周血或骨髓中具有肿瘤反应性T细胞的对象中,例如可以通过IFN-γELISPOT确定的。在其他或其他实施方案中,肿瘤显示出Treg、CD4+Tconv和/或CD8+T细胞的浸润。
在其他实施方案中,与增殖性疾病有关的细胞在TNF的启动子区域中包含单核苷酸多态性(SNP),其与TNF的表达增加和癌症敏感性相关,例如与TNF的启动子区域中的-308G/A SNP处的AA或GA基因型有关;在替代实施方案中,在每种情况下,在TNF的启动子区,肿瘤均不包含与TNF表达降低和癌症风险降低相关的SNP,例如不包含在-238G/A SNP或-857T等位基因处的AA或GA基因型(Wang and Lin 2008,Acta Pharmacol Sin 28:1275)。
因此,本发明基于本发明人的令人惊讶的发现提供了替代的联合治疗方案,即本发明的化合物对一种或多种激酶(例如一种或多种关键激酶,例如SIK3)的抑制可影响细胞对TNF的凋亡/细胞毒性作用的敏感性。因此,在第五方面,并且如本文可以进一步描述、定义、要求保护或以其他方式公开的,本发明涉及一种用于治疗对象的增生性疾病的方法,该方法包括使对象中与增殖性疾病有关的细胞暴露于(例如接触)(i)TNF、TNF变体和/或TNFR2或TNFR1信号传导激动剂;以及使对象中与增殖性疾病有关的细胞暴露于(ii)本发明的化合物(或药物组合物)。在某些实施方案中,这种方法的步骤(i)不包括使对象中与增殖性疾病有关的细胞暴露(例如接触)TNF变体。
在某些实施方案中,增殖性疾病和/或此类细胞是肿瘤的,并且在其他实施方案中,组分(i)是TNF,特别是人TNF(例如rHuTNF);和/或组分(i)是TNFR1信号传导的激动剂。
在特定的实施方案中,该方法包括(例如,该治疗包括、涉及、是由或由其介导)增加暴露于对象中与增殖性疾病有关的细胞的TNF的量。
在此类方面的某些实施方案中,治疗可包括(例如,涉及、是由或由其介导)增加对象中的与增殖性疾病有关的细胞中TNFR1-和/或TNFR2-信号转导。因此,在相关方面,本发明涉及一种用于治疗对象的增生性疾病的方法,该方法包括:(i)增加与该增生性疾病有关的细胞中的TNFR1和/或TNFR2信号转导;以及(ii)使对象中与增殖性疾病有关的细胞暴露于(例如接触)本发明的化合物(或药物组合物)。
特别地,该方法可以例如通过抑制激酶(例如,关键激酶,例如SIK3)的抑制的结果来实现(例如,抑制磷酸化的SIK3的功能和/或活性),尤其是与TNFR1和/或TNFR2信号激活的结果结合在一起,例如在TNF、TNF变体和/或TNFR1激动剂与TNFR1或TNFR2结合时。
因此,在某些实施方案中,治疗效果可以涉及或通过抑制激酶(例如,关键激酶,如SIK3)和/或通过使与增生性疾病相关的细胞对TNFR1或TNFR2信号转导的细胞毒性(例如凋亡)作用敏感来介导(例如,引起)。特别地,在这样的实施方案中,激酶/关键激酶活性可以被(有效地)降低至例如治疗有效水平。
如上所述,本文也是可设想的实施方案,其中肿瘤细胞中的激酶(例如关键激酶(例如SIK3))被抑制,以及任选地,其中一种或多种其他激酶/关键激酶(例如SIK2和/或SIK1)被抑制的程度较小,例如免疫细胞的其他激酶(例如SIK2或SIK1)。
如上所述,本文也是设想的实施方案,其中所述治疗包括、涉及、是由或由激酶/关键激酶活性的抑制(例如本发明的化合物(或药物组合物)以有例如有效抑制的治疗有效量的量施用)来介导,从而使其(例如有效地)降低,例如降低至治疗有效水平。
在该方面的某些实施方案中,可以给对象施用本发明的化合物(或药物组合物)和/或可以给对象施用TNF、TNF变体或TNFR1或TNFR2信号转导激动剂。
在这样的实施方案中,本发明的化合物(或药物组合物)和TNF、TNF变体或TNFR1或TNFR2激动剂可以暴露于(例如以该量施用)有效量(或剂量)施用,包括在本文其他地方所述的制剂或行政途径中。特别地,设想了其中将TNF、TNF变体或TNFR1或TNFR2激动剂封装为脂质体或其他纳米颗粒制剂的实施方案。
当暴露/施用TNF、TNF变体或TNFR1或TNFR2激动剂并且暴露/施用本发明的化合物(或药物组合物)时,则此类联合治疗方案可包括其中此类暴露/施用伴随的实施方案。在替代实施例中,这样的暴露/给药可以是按顺序的;尤其是在暴露/施用TNF、TNF变体或TNFR1或TNFR2激动剂之前,暴露/施用本发明的化合物(或药物组合物)的那些实施方案。例如,本发明的化合物(或药物组合物)可以在另一组分的约14天之内(例如之前),例如在另一组分的约10天、7天、5天、2天或1天之内(例如之前)按顺序暴露/施用;并且进一步包括化合物(或药物组合物)可以在另一组分的约48小时、24小时、12小时、8小时、6小时、4小时、2小时、1小时、30分钟、15分钟或5分钟内(例如之前)按顺序暴露/施用。
TNF或TNF变体或TNFR1或TNFR2激动剂可通过常规途径给药,例如s.c.、i.v.或i.m.。在某些实施方案中,可以肿瘤内给药或通过孤立的肢体灌注(ILP)(例如孤立的肝脏灌注(IHP))进行给药;和/或可以约5-500μg/m
在特定的实施方案中,可以暴露/施用TNF变体,例如具有比rHuTNF的更高的抗肿瘤活性和更低的全身毒性的TNF变体。例如,TNF变体可以是选自以下的一种:(i)TNF的-K90R变体;(ii)与TNF缀合的肿瘤归巢肽;以及(iii)TNF-抗体缀合物。
在涉及TNF变体的本发明的那些实施方案中,它可以是具有较高细胞毒性活性和较低全身毒性的TNF的变体形式。
在其他实施方式中,可以暴露/施用TNFR1或TNFR2激动剂,例如抗TNFR1单克隆抗体htr-9(Ferrero et al 2001,Am J Physiol Cell Physiol 281:C1173),在其他实施方式中,可以暴露/施用淋巴毒素-α(Etemadi et al 2013,FEBS J 280:5283)或其变体。
在替代实施方案中,与增殖性疾病有关的细胞(例如肿瘤细胞)可通过以下方式暴露于TNF(或增加的TNFR1-和/或TNFR2-信号传导),即通过施用可能导致此类细胞暴露于(例如内源性)TNF、或暴露于另一种触发分子(例如TNF或TNFR1或TNFR2激动剂的变体的试剂(例如,向具有该细胞的对象))。此类试剂可以是例如能够诱导(例如诱导)此类细胞暴露于(例如升高水平的)TNF的试剂,特别是诱导此类细胞暴露于TNF水平的试剂,例如关于有效量的(例如内源性)TNF,例如血浆或肿瘤内TNF的水平大于一种或本文其他各处所述的水平。
因此,本发明包括其中向对象施用能够诱导(例如诱导)与增殖性疾病有关的细胞暴露于(所述)TNF、(所述)TNF变体或TNFR1或TNFR2信号转导的激动剂的试剂的实施方案。本发明还包括其中向对象施用能够增加与所述增殖性疾病相关的细胞的TNFR1信号传导(和/或TNFR2信号传导)和/或增加暴露于其中的TNF的量的试剂的实施方案。
在某些这样的实施方案中,所述试剂是病毒,特别是已经被工程化以产生触发分子的病毒,所述触发分子是TNF、TNF变体或TNFR1或TNFR2激动剂(特别是被工程化以产生人TNF的病毒)。此类实施方案的进一步包括那些这样的病毒,其中这些病毒优先感染与增殖性疾病有关的细胞(例如肿瘤细胞)和/或优先在(例如当其感染)此类细胞的情况下产生触发分子。现在将显而易见的是,这种病毒的施用可以导致与增殖性疾病有关的细胞暴露于这种触发分子,特别是有效量的这种触发分子如TNF。
因此,在某些此类方法中,该试剂可以是能够诱导(例如诱导)与增殖性疾病相关的细胞暴露于TNFR1、TNF变体或TNFR1或TNFR2信号转导激动剂的病毒。
这样的病毒可以是适于诱导触发分子暴露的任何病毒,并且特别地可以是重组病毒;例如一种经工程改造以感染肿瘤细胞和/或表达TNF的病毒(例如在感染肿瘤细胞之后)。可以如此改造的病毒的例子包括溶瘤病毒(例如基于腺病毒、HSV、牛痘病毒、水泡性口炎病毒或新城疫病毒的溶瘤病毒),例如肿瘤内注射腺病毒载体以增加促炎细胞因子以及趋化因子的血浆水平,包括TNF(Bernt et al 2005,Cancer Res 65:4343)。特别地,在这样的实施方式中,溶瘤病毒可以是基于f Kaufman et al 2015(Nature Rev Drug Disc 14:642)的表1中描述的DNA病毒的一种、基于Kaufman et al 2015的表2中描述的RNA病毒的一种,优选地,是在Kaufman et al 2015的表3中描述的溶瘤病毒,其处于临床试验中。
在其他这样的实施方案中,所施用的试剂(并因此导致与增殖性疾病有关的细胞暴露于触发分子TNF、TNF变体或TNFR1或TNFR2激动剂)是免疫细胞。在某些这样的实施方案中,免疫细胞可以不是产生IL10的巨噬细胞,例如免疫细胞可以是促炎性免疫细胞。特别地,在这样的实施方案中,所给予的免疫细胞可以是淋巴样细胞,例如T细胞或天然杀伤(NK)细胞,例如产生TNF的这种细胞。
当在本发明的此类实施方案中作为试剂施用时,可以通过过继细胞转移(ACT)施用免疫细胞;意思是免疫细胞转移到对象体内(例如,通过输注或其他递送技术)。通常以改善对象的免疫功能和特性为目标进行该过程,并且尽管常规上转移的免疫细胞将起源于同一对象,但是它们也可以源自另一(合适的)个体。
当在本发明的该实施方案中使用时,免疫细胞可以是从对象中提取的T细胞,经基因修饰和体外培养并返回相同的对象,例如在本发明的治疗方法中。这样的遗传修饰可以包括增强免疫细胞的特异性或靶向性的那些,例如将免疫细胞靶向(例如提高其特异性)与增殖性疾病有关的细胞(例如肿瘤细胞)的靶向性。例如,可以修饰在此类实施方案中使用的T细胞,以改变T细胞受体(TCR)的特异性或在嵌合抗原受体(CAR)中引入抗体样识别。尤其设想将CAR免疫细胞用于此类实施方案。CAR免疫细胞是展示工程化受体的免疫细胞,其将任意特异性(例如对肿瘤细胞)移植到免疫效应细胞(例如T细胞)上。通常,这些受体用于将单克隆抗体的特异性移植到T细胞上。通过逆转录病毒载体促进其编码序列的转移。CART细胞是一种有前途的癌症治疗方法(Song et al 2015,Oncotarget.6:21533):使用ACT,可从个体(通常是对象)中去除T细胞并对其进行修饰,使其表达针对患者特定癌症的特异性受体。然后可以将这些可以识别对象的癌细胞的T细胞(重新)引入对象中,导致TNF(例如由CAR T细胞产生的)暴露于肿瘤细胞,从而杀死这种细胞,特别通过暴露于本发明的化合物(或药物组合物)(例如向对象施用后)而对此类TNF介导的细胞毒性敏感的此类细胞。因此,特别是在这样的实施方案中,免疫细胞可以是CAR T细胞,例如一种经工程改造以对与增殖性疾病有关的受试者细胞(例如肿瘤细胞)具有增加的特异性的细胞。
在替代实施方案中,与增殖性疾病有关的细胞暴露于TNF(例如内源性TNF)可以通过其他方式或程序来诱导。因此,在这样的实施方案中,与增殖性疾病有关的细胞暴露于(例如有效量的)TNF可以通过增加对象血浆和/或此类细胞环境中TNF的量的药物、治疗或其他方法诱导(和/或TNFR1信号转导(和/或TNFR2信号转导)的增加)引起(和/或与增殖性疾病有关的细胞中TNFR1信号转导(和/或TNFR2信号转导)的增加)。
在某些实施方案中,可以通过施用癌症免疫疗法来引起对TNF的这种诱导暴露。
在一个实例中,这种诱导的TNF暴露是通过抗肿瘤疫苗(例如,癌症疫苗)引起的。此类癌症疫苗包括将抗原(例如,对癌细胞特异或优先由癌细胞表达的抗原)直接或间接引入对象以在设想对癌细胞具有(更多)特异性的对象中增强或增强免疫应答(通常是适应性免疫应答)的疫苗。癌症疫苗可以包括例如减毒病毒,特别是用于抵抗由这种病毒引起的癌症(例如宫颈癌或肝癌)(例如HPV或HBV)。癌症疫苗可以替代地代表特定肿瘤抗原的个体(或组合)(例如,对癌细胞具有特异性或优先表达的抗原),例如用于免疫对象以提高或增强对象的免疫反应的肿瘤相关抗原(TAA)。所述癌症疫苗可以包含代表TAA(例如来自TAA的肽)的重组蛋白,或者可以是肿瘤特异性碳水化合物抗原,并因此在给药后直接引入对象。或者,所述癌症疫苗可包含编码所述蛋白质(或肽)TAA的核酸(例如DNA或mRNA),并且在将所述核酸疫苗施用给所述对象后,编码的TAA由对象中的细胞靶标表达,因此被间接引入对象中。TAA可以分为两类:共有的肿瘤抗原;和独特的肿瘤抗原。共有的抗原由许多肿瘤表达。独特的肿瘤抗原是由物理或化学致癌物(也称为新抗原)诱导的突变产生的;因此,它们仅由个体肿瘤表达。本领域普通技术人员将知道在临床试验中或已批准使用的癌症疫苗的实例,包括PROSTVAC(Bavarian Nordic)、PROVENGE(Dendreon)和CV9104(CureVac),以及知道各种TAA(包括新抗原)和这种肿瘤抗原的方法可用于癌症疫苗。作为进一步的例子:(1)已证明,与受体孔戚血蓝蛋白(KLH)结合的受体来源的克隆骨髓瘤免疫球蛋白独特型(Id))作为肿瘤抗原进行免疫可产生大量或促炎性细胞因子,包括TNF(Foglietta et al 2013,Bone Marrow Transplant 48:269);(2)WT1抗原的合成微共识SynCon DNA疫苗诱导了新的新抗原样反应优于天然WT1 DNA免疫原诱导的反应,例如强CD4和CD8 T细胞反应(包括IFN-γ、CD107a和TNF响应)。
在另一个实例中,可以通过施用配体(例如抗体,例如单克隆抗体)来引起对TNF的这种诱导暴露,所述配体例如是结合至与增殖性疾病相关的细胞(例如肿瘤细胞)的一种,例如通过与TAA或这种细胞表面的受体结合。细胞表面受体是此类配体(抗体)疗法的常见靶标,包括CD52和CD20。一旦与这样的癌症抗原结合,例如抗体就可以诱导抗体依赖性细胞介导的细胞毒性,激活补体系统或阻止受体与其配体相互作用,所有这些均可导致细胞死亡。批准的这类抗体配体包括alemtuzumab、ofatumumab和rituximab。在某些实施方案中,与本发明的化合物(或药物组合物)组合使用的此类配体可以包括激活T细胞或其他细胞介导的免疫应答的配体。例如:(1)抗CD137单克隆抗体可以显著促进细胞因子诱导的杀伤(CIK)细胞的增殖和TNF的表达(Zhu et al 2009,Biomed Pharmacother63:509);(2)激动剂抗OX40单克隆抗体可通过增强T细胞分化来增强抗肿瘤免疫反应(Redmond et al 2014,Cancer Immunol Res.2014,2:142);以及(3)激活T细胞的抗ICOS抗体(例如Deng et al2004,Hybrid Hybridomics 23:176)。
在另一个实例中,施用于对象的配体是与免疫(抑制)检查点分子结合的配体。例如,这种检查点分子可以是选自以下的一种:A2AR、B7-H3、B7-H4、CTLA-4、IDO、KIR、LAG3、PD-1(或其配体PD-L1和PD-L2中的一种)、TIM-3(或其配体galectin-9)、TIGIT和VISTA。特别地,在这样的实施方案中,配体与选自以下的检查点分子结合:CTLA-4、PD-1和PD-L1。在其他更具体的实施方案中,所述配体是选自以下的抗体:伊匹木单抗、尼古拉单抗、派姆单抗、BGB-A317、阿泰唑单抗、阿伐单抗和杜鲁门单抗。特别地、抗体选自:依匹莫单抗(YERVOY)、尼武单抗(OPDIVO)、派姆单抗(KEYTRUDA)和阿佐单抗(TECENTRIQ)。在其他实施方案中,与免疫(抑制)检查点分子结合的配体可以是非抗体肽,例如高亲和力PD-1变体(例如,Maute et al,2015;PNAS112:E6506)、靶向免疫检查点分子的肽(例如AUNP-12ofAurigene Discovery Technologies,US 2011/0318373)或阻断免疫检查点分子之间的相互作用(例如PDL1-PD1相互作用)的D肽和(D)PPA-1,Chang et al,2015;Anyeg Chem Int54:11760)。在其他实施方案中,与免疫(抑制)检查点分子结合的配体可以是小分子,例如靶向PDL1的BMS-202或BMS-8(Zak et al 2016;Oncotarget 7:30323)、称为BMS-1001或BMS-1166的PDL1/D1(Skalniak et al,2017;Oncotarget 8:72167)、正在进行1期试验的Curis/Aurigen的PDL1和VISTA拮抗剂CA-170(Powderly et al,Ann Onc 28:Issue suppl5,mdx376.007)或f Curis/Aurigen的CA-327,其靶向PDL1和TIM3。
在又一个特定的实施方案中,可以通过放射疗法引起这种对TNF的诱导暴露。
放射疗法是一种局部治疗癌症或肿瘤的方法,利用放射线通过阻断癌细胞的繁殖和/或刺激针对它们的免疫反应(例如对已死亡或垂死的癌细胞的存在而引起的反应)来破坏癌细胞。在本发明的上下文中,放射疗法特别地由电离辐射的治疗用途组成。所述放射疗法和相关的电离辐射是本领域技术人员通常使用和已知的那些。放射疗法尤其包括使用电离辐射,例如伽马射线、X射线和/或从放射性同位素发出的辐射。在本发明的上下文中,更特别地是X射线辐射。放射治疗可以在一个或多个周期内,例如可以在1至4周、更特别地3周的周期内,以分级形式给予。该周期定义了施用方案开始和结束之间的时间间隔。当周期需要三周时,放疗可以在三周内进行,间隔一周。放射疗法可以特别地以每天一次的辐射的速率进行,即7天内的5天,持续所需的周数。(光子)放射治疗中使用的放射量以戈瑞(Gy)为单位,并根据所治疗癌症的类型和阶段而有所不同。对于治愈性病例,实体上皮肿瘤的典型剂量范围为60至80Gy,而淋巴瘤的治疗剂量为20至40Gy。
当本发明的化合物(或药物组合物)与任何其他此类其他程序(例如,其他药剂、癌症免疫疗法、癌症疫苗、抗体或放射疗法)一起用于联合治疗时(如本文所述),则这样的组合治疗方案可以包括其中这样的暴露/给药同时发生的实施方案。在替代实施方案中,这样的施用可以是按顺序的。特别是在化合物(或药物组合物)在这样的其他程序之前给药的那些实施方案中。例如,本发明的化合物(或药物组合物)可以在其他程序的约14天之内(例如之前),例如在其他程序的约10天、7天、5天、2天或1天之内(例如之前)按顺序施用;并且进一步包括其中化合物(或药物组合物)可以在约其他程序的48小时、24小时、12小时、8小时、6小时、4小时、2小时、1小时、30分钟、15分钟或5分钟内按顺序施用。
不受理论的束缚,本发明化合物(或药物组合物)的施用(并因此抑制激酶/关键激酶例如SIK3的表达、量、功能、活性或稳定性,例如在肿瘤细胞中)在TNF、TNF变体或TNFR1或TNFR2激动剂的施用之前,或在此类其他程序(例如,其他药物)的施用之前,癌症免疫疗法、癌症疫苗、抗体或放射疗法预期在使与增殖性疾病有关的细胞对细胞介导的免疫反应的细胞毒性作用敏感方面特别有效。
如上所述,涉及给予TNF和/或使用抗TNF分子的现有疗法(或临床试验中的疗法)具有某些已知的缺点;以及特殊的副作用。本发明提供了可用于减轻(或减少)这种缺点和/或特定副作用的方法。
在第六方面,并且如本文可以进一步描述、定义、要求保护或以其他方式公开的,本发明涉及一种用于在用其治疗增生性疾病(例如癌症疾病或肿瘤)的对象中增加用TNF治疗的治疗指数的方法,该方法包括向对象施用本发明的化合物(或药物组合物)。
在相关方面,本发明涉及用于在患有增生性疾病(例如癌症或肿瘤)的对象中支持TNF治疗的方法,该方法包括向对象施用本发明的化合物(或药物组合物)。
在第七方面,并且如本文可以进一步描述、定义、要求保护或以其他方式公开的,本发明涉及一种使患有增生性疾病(例如癌症或肿瘤)的对象对涉及向该对象施用TNF的治疗敏感的方法,该方法包括向所述对象施用本发明的化合物(或药物组合物)。
如本文所用,术语“致敏/使……敏感(化)”(等)在对象对疗法敏感的情况下使用(例如,涉及TNF的给药),本领域普通技术人员将理解该术语,并且包括意思是对象增加对这种疗法可能对对象产生的一种或多种(治疗)效果(特别是功效效果)的敏感性。特别地,如此敏感的对象在经历这样的治疗时,可以显示出比被有被如此“敏感”的类似对象更高的反应(例如更快、更大程度的反应和/或在这种治疗的较低剂量或暴露下)。
在第八方面,并且如本文可以进一步描述、定义、要求保护或以其他方式公开的,本发明涉及用于在用抗TNF剂治疗的对象中降低(发展)血液学增生性疾病(例如继发性疾病)的风险的方法,该方法包括向对象施用本发明的化合物(或药物组合物)。例如,该方面可替代地被认为是用于在用抗TNF剂治疗的对象中预防血液学增生性疾病(作为继发性疾病)的方法,该方法包括向所述对象施用本发明的化合物(或药物组合物)。
本发明的该方面基于如上所述的观察,即有报道接受抗TNF生物制剂发展为淋巴瘤和其他血液系统恶性肿瘤的患者。实际上,此类疾病通常在包装单张/处方信息中描述为使用抗TNF剂治疗的可能的副作用(但很少见)。由于人们普遍认为血液系统恶性肿瘤增加,以及这些药物和其他免疫抑制剂的广泛使用,WHO对肿瘤的分类现在包括“医源性免疫缺陷相关性淋巴增生性疾病”类别。
因此,通常在此类情况下,患者正在接受抗TNF药物治疗增生性疾病以外的适应症,尤其是在此类实施方案中,在开始抗TNF治疗后,患者不进行患有血液学增生性疾病。实际上,通常该对象会患有和/或正在接受抗TNF药物治疗自身免疫疾病。优选自身免疫性疾病选自:类风湿性关节炎,银屑病关节炎,强直性脊柱炎,斑块状银屑病,炎性肠病,溃疡性结肠炎,克罗恩氏病,牛皮癣,化脓性汗道炎和难治性哮喘;例如选自以下的一种:类风湿性关节炎,牛皮癣性关节炎,强直性脊柱炎,斑块状银屑病和克罗恩氏病;特别是类风湿关节炎。
在某些实施方案中,抗TNF剂选自:英夫利昔单抗(infliximab)、阿达木单抗(adalimumab)、戈利木单抗(golimumab)、humicade、依那西普(etanercept)、奥那西普(onercept)和西妥珠单抗聚乙二醇,特别是英夫利昔单抗或humicade。
在某些实施方案中,血液系统恶性肿瘤增生性疾病可以是淋巴增生性疾病,特别是与医源性免疫缺陷相关的淋巴增生性疾病。
在此类第六至第八方面的某些实施方案中,(治疗)效果(例如治疗指数的增加,对象的致敏性或风险的降低)是由(例如,该治疗包括、是由、由其介导或涉及)以下介导的:(i)抑制激酶(例如,关键激酶如SIK3)(例如通过抑制磷酸化SIK3的功能和/或活性),特别是通过在与增殖性疾病有关的细胞中抑制这种激酶(例如关键激酶);和/或(ii)使此类细胞对TNF的杀伤(凋亡/细胞毒性)作用敏感。在进一步的实施方案中,(治疗)作用可以不通过抑制一种或多种其他关键激酶(例如,ABL1和/或SRC,或SIK2和/或SIK1)来介导(例如,治疗可以不包括或不涉及),尤其是不由免疫细胞中的抑制或更多种其他关键激酶(例如SIK2和/或SIK1(和/或SIK3))介导(例如,治疗不包括或不涉及)。
在某些实施方案中,对象是人类志愿者;例如已经选择(例如同意)给予本发明化合物(或药物组合物)用于该化合物的临床试验或其他实验用途的人。在另一个实施方案中,对象是实验动物,特别是选自以下的动物:小鼠、大鼠、兔、猪和猴子。
在这样的(例如实验治疗)实施方案中,可以治疗多个这样的对象;尤其是5个或更多的对象,例如约5到20、10到50、25和200或75到250个对象,或多于约250个对象。
此类实验(或临床试验)治疗可包括:(i)向此类对象中的至少一个施用一剂本发明的化合物和/或本发明的药物组合物的一种制剂;以及(ii)向此类对象中的至少另一个施用将至少不同剂量的化合物和/或不同制剂的药物组合物。
在此类实施方案的进一步中,此类实验(或临床试验)治疗可包括:(i)向此类对象中的至少一个施用一种剂的本发明化合物和/或本发明的药物组合物的一种制剂;以及(ii)向此类对象中的至少另一个施用(a)安慰剂;(b)(i)的对象的化合物的剂量和/或药物组合物的制剂,以及另外的药物、治疗或其他方法。
术语“安慰剂”是本领域公认的,并且包括无预期治疗价值的物质或治疗。在这样的实施方案中,可以使安慰剂类似于另一种给药方式,以使其充当对照,例如在盲法试验中。
在某些此类实施方案中,此类实验(或临床试验)治疗是专门设计用于调查和/或确定本发明化合物的治疗有效剂量和/或鉴定本发明的药物组合物的治疗有效剂量。
在第九方面,本发明涉及一种诊断和治疗疾病、病症或病状的方法,所述疾病、病症或疾病的特征在于对象(例如人类患者)中一种或多种蛋白质适用的生物标记物(例如激酶)的存在或数量和/或特征在于其(例如,异常的)表达或活性,包括:
·从所述对象的生物学样品中检测一种或多种此类适用的生物标记物,从而诊断该对象是否正在遭受(或可能遭受)这种疾病、病症或病状;和
·向如此诊断的对象施用有效量的本发明的化合物(和/或包含该化合物的药物组合物),尤其是对该对象实施本发明的治疗方法。
在这样的实施方案之一中,所述疾病、病症或病状是增生性疾病,例如本文其他地方公开的一种(例如肿瘤或癌症)。
术语“适用的生物标志物”是指与增殖性疾病有关的细胞表达的任何一个(或多个)基因,这些基因与(例如激酶/关键激酶介导的)细胞对免疫反应的抵抗力有关(例如细胞介导的免疫反应(例如TNF)。此类基因包括:(X)一种或多种激酶,特别是本文所述的一种或多种关键激酶,例如SIK1、SIK2、SIK3、ABL1(BCR-ABL)、SRC、FLT3、KIT、YES、LYN、FYN和LCK;特别地,ABL1(BCR-ABL)、ABL1(BCR-ABL)和FLT3、尤其是磷酸化的SIK3;(Y)激酶的突变体,例如突变体ABL1激酶(例如,BCR-ABL)或KIT激酶的突变体;和/或(Z)以下(a)至(f)中的一个或多个:
(a)TNFR1(或TNFR2),例如TNFR1(或TNFR2)(特别是TNFR1)的存在(或含量)或表达和/或活性;
(b)LKB1,例如LKB1的存在(或含量)或表达和/或活性,特别是LKB1或pLKB1的含量或活性增加;
(c)一种或多种II类(例如IIa)HDAC,例如HDAC4,例如这种HDAC的存在(或含量)或表达和/或活性,特别是这种HDAC或pHDAC的含量或活性增加,特别是在肿瘤细胞的细胞质中;
(d)NF-κB的表达,特别是NF-κB的组成型表达;
(e)NF-κB,例如NF-κB的存在(或含量)或表达和/或活性,尤其是NF-κB或乙酰化NF-κB的含量或活性增加,特别是在肿瘤细胞核中;和/或
(f)一种或多种抗凋亡基因,例如一种或多种抗凋亡基因的存在(或含量)或表达和/或活性,特别是一种或多种受NF-κ-B转录控制的此类基因。
在某些实施方案中,适用的生物标记是选自以下的一种或多种关键激酶:EPHA2,EPHA4,CSF1-R,HCK,ACK1;和/或PDGFR-α,TGFB-R1,B-RAF和/或p38-β;和/或ACV-R1和/或BMPR1A;和/或RET;和/或NEK11、WEE1和/或WNK2;和/或Aurora-A和/或Aurora-B;和/或TBK1。
该诊断和治疗方法的给药(或治疗)步骤的其他实施方案在其他地方有更详细的描述;如该方法的检测、确定或诊断方法步骤的方法的特定实施例一样。这样的实施方案的具体包括其中向对象施用的本发明的化合物(和/或药物组合物)的量与血浆或肿瘤内TNF浓度(在受试者中)相关的那些,其中在血浆或肿瘤内TNF的浓度更高的情况下,更大的量(或剂量)化合物(和/或药物组合物)被施用至这样的对象。
在某些实施方案中,生物学样品将(优选地)包含对象的细胞或组织,或此类细胞或组织的提取物,特别是当此类细胞是(通常,典型地;或在这种情况下或在特定情况下)与增生性疾病(例如肿瘤细胞,例如实体瘤细胞)有关的。所述肿瘤或其细胞可以是本文别处所述的肿瘤之一或源自该肿瘤之一。
在该方面的特定实施例中,该方法还将包括以下步骤:
·提供(例如通过获取)来自对象的生物样品,特别是在检测步骤之前执行该步骤的情况下。
在特定实施例中,可以将这种检测和/或确定方法实践为诊断方法,例如诊断哺乳动物对象(例如人类对象或患者)是否患有疾病、病症或病状的方法,特别是增生性疾病(例如癌症或肿瘤)(的存在)(或有风险发展为此类疾病、疾病或病症),其与针对细胞介导的免疫反应的细胞抗性相关和/或与适用的生物标志物(例如SIK3)的表达或活性相关(例如异常);特别是(实体)肿瘤,例如对细胞介导的免疫反应具有细胞抗性的。
在这些检测、确定和/或诊断方法的某些实施方案中,针对细胞介导的免疫应答的细胞抗性是针对T细胞介导的免疫应答的细胞抗性,特别是对TNF和/或TNFR1或TNFR2信号传导的杀伤(细胞凋亡/细胞毒性)作用的细胞抗性。
因此,这些检测和/或诊断方法的特定实施方式还可包括确定样品中TNF的存在或含量的步骤,其中样品中TNF的存在(或含量)表示与对象中与针对细胞介导的免疫反应的细胞抗性有关和/或与激酶(例如SIK3)的(异常)表达或活性有关的增生性疾病(或发展成增生性疾病的风险)。特别地,在此类实施方案中,定性确定样品中TNF的量。优选地,所述对象被区分为具有:(i)来自所述对象的血浆样品中的TNF的血浆浓度大于约2pg/mL或5pg/mL;和/或(ii)来自对象组织样品的TNF的肿瘤内浓度大于约0.5pg/mL或1pg/mL,表明在对象中存在与针对细胞介导的免疫应答的细胞抗性和/或与激酶(例如SIK3)的表达或活性有关的增生性疾病或发展这种增生性疾病的风险。
本文其他地方介绍了确定样品中TNF的存在或含量的方法(尤其是使用ELISA分析(例如QuantitkineTNF-α免疫测定)定量检测TNF);同样如果样本中存在的TNF超出的TNF量,则表明对象中与针对细胞介导的免疫反应的细胞抗性有关和/或与激酶的(异常)表达或活性有关的增生性疾病(例如SIK3)。
在某些实施方案中,生物样品是从哺乳动物对象(例如人类患者)获得的样品。术语“生物样品”以其最广义使用,并且可以指获自对象(例如人类患者)的身体样品。例如,生物样品可以包括临床样品,即源自对象的样品。这样的样品可以包括但不限于:外周体液,其可以包含或可以不包含细胞,例如血液、尿液、血浆、粘液、胆汁、胰液、上清液和血清;组织或细针活检样本;具有已知诊断、治疗和/或结果历史的肿瘤活检样品或切片(或其细胞)以及档案样品。生物样品还可以包括组织切片,例如出于组织学目的而采取的冷冻切片。术语“生物样品”还可以包括通过处理样品得到的任何材料。衍生材料可包括但不限于从生物样品中分离的细胞(或其后代)、从样品中提取的核酸和/或蛋白质。生物样品的处理可涉及以下一项或多项:过滤,蒸馏,提取,扩增,浓缩,固定,干扰组分失活,添加试剂等。
在一些实施例中,这些检测、确定和/或诊断方法可以是计算机实现的方法,或者是由计算机辅助或支持的方法。在一些实施方案中,通过至少一个处理器获得反映样品中待确定(或其活性)的适用生物标志物(例如关键激酶,例如ABL1/BCR-ABL、SRC和/或SIK3)的存在或数量的信息:和/或由另一处理器以用户可读的格式提供反映样品中此类标记物的存在或数量(或其活性)的信息。一个或多个处理器可以耦合到在计算机操作系统的控制下或与计算机操作系统结合操作的随机存取存储器。处理器可以被包括在一个或多个服务器、群集或其他计算机或硬件资源中,或者可以使用基于云的资源来实现。操作系统可以是例如Linux
这样的检测、确定和/或诊断方法可以作为体外(例如离体)方法来进行,并且可以例如使用本发明的试剂盒(或其组分)来实践。体外方法可以在永生化细胞系(例如在动物或人类体外复制、培养或无限期保存的永生化细胞系)上使用、参与或实践,或者可以使用直接或从人类动物体内新鲜获得的细胞(例如原代细胞)来使用、参与或体外实践(例如,实践为所谓的“离体”方法)。
在这些检测、确定和/或诊断方法的一些实施方案中,生物样品是来自对象的组织样品,例如来自对象的肿瘤或癌症的样品。如上所述,这样的组织样本可以是肿瘤或癌症的活检样本,例如针头活检样本,或肿瘤活检切片或其档案样本。这样的组织样品可以包括活的、死的或固定的细胞,例如来自肿瘤或癌症的细胞,并且可以怀疑这种细胞表达(例如异常地或定位地)待确定的适用生物标记物。
在一些实施方案中,本发明的确定和/或诊断方法可以包括,例如在进一步的步骤中,将适用的生物标志物(例如激酶/关键激酶,例如SIK3,尤其是磷酸化的SIK3)(例如其蛋白或mRNA)的检测量(或活性)与标准值或临界值进行比较;其中大于标准值或临界值的检测量表示与对象中针对细胞介导的免疫反应的细胞抗性有关和/或与激酶/关键激酶(例如SIK3)的(异常)表达或活性相关的表型(或发展表型的风险)。这样的标准或截断值可以通过使用对照测定法来确定,或者可以根据从一项研究中获得的一个或多个值或具有已知表型的多个样品来预先确定。例如,可以通过在受控临床研究的背景下对从患者身上采集的样品进行分析,并根据所需(或获得的)测试的灵敏度和/或特异性确定临界值。
在某些实施方案中,可通过检测适用的生物标记物的蛋白质或通过检测编码适用的生物标记物的mRNA来检测适用的生物标记物。检测蛋白质(例如,尤其是通过免疫组织化学的抗体检测)和mRNA(例如,通过杂交、qPCR或测序)的方法是众所周知的。
可用于检测适用的生物标记物(例如SIK3的激酶/关键激酶,特别是磷酸化的SIK3)(例如存在或不存在或其含量)的方法的例子包括免疫测定,例如酶联免疫吸附测定(ELISA)和放射免疫测定(RIA),其采用了抗原结合蛋白(“ABP”),例如抗体或其抗原结合片段,其特异性结合这种可适用的生物标记物。
本文公开的化合物可以如下所述制备或通过与其类似的方法制备,这些方法是有机合成领域的普通技术人员容易知道和可获得的。
在第十方面,本发明涉及化合物(例如中间体)在制备本发明的化合物中的用途(例如制备式(I)、(Ia)、(II)、(III)、(IV)、(V)、(VI)、(VII)或(VIII)的化合物或溶剂化物盐(特别是药学上可接受的盐)、N-氧化物(特别是R
其中R
特别是在这样的实施方案中,化合物(例如中间体)不是化合物B3。
在此类实施方案的某些实施方案中,化合物(例如中间体)为2-溴-4-氯吡啶-3-胺(化合物IaB3)或为4-氯-2-甲基吡啶-3-胺(化合物IbB3)。
在这样的实施方案的另一种确定的形式中,化合物(例如中间体)选自:2,4-二甲基吡啶-3-胺、3-甲基吡啶-2-胺、4-溴-2-甲基吡啶-3-胺、3-氯-5-甲基吡啶-4-胺、3,5-二甲基吡啶-4-胺、2-甲基吡啶-3-胺、2-氯-4-甲基噻吩-3-胺、1,3,5-三甲基-1H-吡唑-4-胺、3,5-二甲基异恶唑-4-胺、奎尼丁-3-胺和2-乙基-4-甲基吡啶-3-胺。
在这样的实施方案的另一个确定中,化合物(例如中间体)选自具有式(I)、(Ia)、(II)、(III)、(IV)、(V)、(VI)、(VII)或(VIII)的结构的化合物(例如中间体),其中R1a是离去基团,例如卤素,优选Cl。
在相关方面,本发明还涉及化合物(例如中间体)(的物质组成)为2-溴-4-氯吡啶-3-胺或4-氯-2-甲基吡啶-3-胺,特别是其中这样的化合物(例如中间体)的量大于约1g或10g,例如大于约100g,和/或呈纯化形式(例如,如本文其他地方所定义)。
在第十一方面,本发明涉及制备本发明化合物(例如式(I)、(Ia)、(II)、(III)、(IV)、(V)、(VI)、(VII)或(VIII)的化合物)(其为(例如基本上)纯的形式)的方法,该方法包括以下步骤:
·提供在具有一种或多种杂质的混合物中的化合物(例如C2、C、C7、C8、C9或C12);和
·从混合物中去除至少一部分杂质。
在该方面的某些实施方案中,除去一部分杂质的合适方法是众所周知的,包括例如柱色谱法、选择性沉淀、用所需溶剂不溶于其中的合适溶剂对杂质进行研磨和洗脱等。
去除的杂质比例可以使得该化合物以基本上纯的形式制备;也就是说,例如以如上所述的百分比纯度。
在其他实施方案中,通过合成不纯形式的化合物来提供混合物。
在第十二方面,本发明还涉及用于制备药物组合物的方法,所述方法包括与药学上可接受的赋形剂(例如本文其他地方所述的赋形剂,例如载体的药学上可接受的稳定剂)一起配制本发明的化合物(例如式(I)、(Ia)、(II)、(III)、(IV)、(IV)、(VI)、(VII)或(VIII)的化合物)的步骤。
在这样的实施方案的一个具体实施方案中,所述药物组合物的制造量大于约10g;例如大于约100g,并且适当地大于约1Kg(或大于10Kg)。
在该方面的一个实施方案中,药物组合物以单位剂型制造。
在某些这样的实施方案中,药物组合物形成为片剂、囊片或胶囊剂,特别是作为片剂或胶囊剂。
在此类实施方案的另一个中,所述药物组合物形成为薄膜衣片或薄膜衣囊片。例如,该方法可以进一步包括用薄膜(特别是用药物有效的薄膜)包衣片剂或囊片的步骤。
在第十三方面,本发明还涉及一种制备药物包装的方法,包括以下步骤:
·将本发明的药物组合物插入包装中,从而形成包含药物组合物的包装;以及任选的
·在包装中插入说明药物组合物处方信息的单张。
在这样的实施方案之一中,药物组合物为成品药物形式。例如,其形式为将被施用(或最终准备被施用)给对象。
包装可以是初级包装和/或二次包装。例如,初级包装可以是玻璃小瓶或泡罩带。典型的(但非限制性)二次包装可以是盒子或纸箱,在某些实施例中,可以用其所包含的药物组合物的名称、强度和/或品牌进行标记。
包装可能还包含单张或其他信息。特别地,描述(向患者和/或主治医师)关于包装中包含的药物组合物的重要信息或细节,例如如何施用、推荐剂量、安全性和/或副作用信息。
在第十四方面,本发明还涉及包含本发明的药物组合物的药物包装。优选地,其中药物组合物为成品药物形式。在该方面的某些实施方案中,药物包装可进一步包含描述药物组合物的处方信息的单张。
在第十五方面,本发明还涉及一种将本发明的药物组合物或药物包装从第一位置递送至第二位置的方法,该方法包括以下步骤:
·在第一位置处接收来自第二位置的递送一定量的药物组合物或药物包装的请求;以及
·在一次或多次递送中,从第一位置向第二位置递送所请求的量的至少一部分(最好是全部),
其中第一位置和第二位置相隔大于约10m的距离,并且通过轮式运输工具将所述递送中的一次或多次运送第一位置与第二位置之间的距离的至少一部分。
在这样的方面的实施例中,可以通过的电子方法来发送、传送和/或接收请求,例如电话、电子邮件或其他计算机消息或信号。
两个位置之间的距离大于大约10m,例如大于大约100m或大于大约1Km。在特定实施例中,第一位置和第二位置之间的距离大于大约10Km,例如更大超过大约100Km或1,000Km。
技术单元是轮式运输工具,用于在两个位置之间的距离的至少一部分上运输药物组合物或药物包装。例如,对于短距离,可以首先从存储架上用脚步行携带药物组合物或药物包装,然后将其放置在轮式手推车上以运输到另一房间或医院的一部分。
在其他实施例中,轮式运输机可以是运输药物组合物或药物包装例如更长距离的车辆(例如货车或货车)。例如,可以使用货车在相距大于约10Km的位置之间运输药物组合物或药物包装。或者,可以使用自行车(或摩托车)在相距小于约10Km的位置之间运输药物组合物或药物包装。然而,即使药物组合物或药物包装被运输更长的距离(例如,从一个国家或大洲到另一个国家),例如大于大约100公里,并且被例如飞机或轮船占据了该距离的大部分,设想药物组合物或药物包装仍将由轮式运输工具(例如货车或大货车)运输到出发机场或港口(和/或从机场或港口运输),或在港口或机场在拖车上的运输集装箱中。
鉴于上述情况,应当理解,本发明还涉及以下逐项实施例:
1.一种化合物,其选自下式的激酶抑制剂:
以及其溶剂化物、盐、N-氧化物、络合物、多晶型物、晶体形式、外消旋混合物、非对映异构体、对映异构体、互变异构体、构象异构体、同位素标记形式、前药及其组合;
其中:
R
R
R
R
R
R
L选自键、C
R
R
A选自S、O、NR
R
R
X独立地选自O、S和N(R
E是O或S;
B是N或CR
R
R
R
R
R
y是0到2的整数;
R
R
其中
R
R
X
附带条件是
(1)当R
(2)当R
(3)当R
(i)R
(ii)R
(4)当E为O,B为CR
(5)当R
2.根据项目1所述的化合物,其中R
3.根据项目1或2所述的化合物,其中R
4.根据项目1-3中任一项所述的化合物,其中R
5.根据项目1-4中任一项所述的化合物,其中R
5a.根据项目1-4中任一项所述的化合物,其中R
5b.根据项目1-4中任一项所述的化合物,其中R
6.根据项目1-5b中任一项所述的化合物,其中R
7.根据项目1-6中任一项所述的化合物,其中R
7a.根据项目1-7中任一项所述的化合物,其中R
7b.根据项目1-7a中任一项所述的化合物,其中R
8.根据项目1-7b中任一项所述的化合物,其中一个R
9.根据项目1-8中任一项所述的化合物,其中R
其中
10.根据项目1-8中任一项所述的化合物,其中R
其中
11.根据项目1-8中任一项所述的化合物,其中R
其中
12.根据项目1-11中任一项所述的化合物,其中L选自:键,C
13.根据项目1-12中任一项所述的化合物,其中L选自:键;任选被一个R
14.根据项目1-13中任一项所述的化合物,其中L是键。
15.根据项目1-14中任一项所述的化合物,其中R
16.根据项目1-15中任一项所述的化合物,其中R
17.根据项目1-16中任一项所述的化合物,其中R
18.根据项目1-17中任一项所述的化合物,其中R
19.根据项目1-18中任一项所述的化合物,其中R
19a.根据项目1-19中任一项所述的化合物,其中R
20.根据项目1-19a中任一项所述的化合物,其中R
21.根据项目1-20中任一项所述的化合物,其中R
22.根据项目1-21中任一项所述的化合物,其中R
23.根据项目1-21中任一项所述的化合物,其中R
24.根据项目1-23中任一项所述的化合物,其中A选自S、O、NH、N(C
25.根据项目1-24中任一项所述的化合物,其中A是S、O或N(CH
26.根据项目1-25中任一项所述的化合物,其中A是S。
27.根据项目1-26中任一项所述的化合物,其中B是N或CR
28.根据项目1-26中任一项所述的化合物,其中B是N。
29.根据项目1-28中任一项所述的化合物,其中E是O。
30.根据项目1-29中任一项所述的化合物,其中R
31.根据项目1-30中任一项所述的化合物,其中R
32.根据项目1-31中任一项所述的化合物,其中R
33.根据项目1-32中任一项所述的化合物,其中R
34.根据项目1-33中任一项所述的化合物,其选自:
以及其溶剂化物、盐、N-氧化物、络合物、多晶型物、晶体形式、互变异构体、构象异构体、同位素标记形式、前药及其组合。
35.根据项目1-33中任一项所述的化合物,其选自:
以及其溶剂化物、盐、N-氧化物、络合物、多晶型物、晶体形式、互变异构体、构象异构体、同位素标记形式、前药及其组合。
36.根据项目1-33中任一项所述的化合物,其选自:
以及其溶剂化物、盐、N-氧化物、络合物、多晶型物、晶体形式、互变异构体、构象异构体、同位素标记形式、前药及其组合。
37.根据项目1-33中任一项所述的化合物,其选自:
以及其溶剂化物、盐、N-氧化物、络合物、多晶型物、晶体形式、互变异构体、构象异构体、同位素标记形式、前药及其组合。
38.根据项目1-33中任一项所述的化合物,其选自:
以及其溶剂化物、盐、N-氧化物、络合物、多晶型物、晶体形式、互变异构体、构象异构体、同位素标记形式、前药及其组合。
38a.根据项目1-33中任一项所述的化合物,其选自:
以及其溶剂化物、盐、N-氧化物、络合物、多晶型物、晶体形式、互变异构体、构象异构体、同位素标记形式、前药及其组合。
38b.根据项目1-33中任一项所述的化合物,其选自:
以及其溶剂化物、盐、N-氧化物、络合物、多晶型物、晶体形式、互变异构体、构象异构体、同位素标记形式、前药及其组合。
38c.根据项目1-33中任一项所述的化合物,其选自:
以及其溶剂化物、盐、N-氧化物、络合物、多晶型物、晶体形式、互变异构体、构象异构体、同位素标记形式、前药及其组合。
39.根据项目1至38中任一项或项目38a至38c中任一项的化合物,其为基本上纯的形式,特别是大于约90%、95%、98%或99%的纯形式。
40.根据项目1至38和39中任一项或项目38a至38c中任一项的化合物,其为水合物。
41.一个或多个容器,分别独立地或全部一起包含多于约10g或50g的项目1至38、39和40中任一项或项目38a至38c中任一项所述的化合物,合适地包含大于或等于约100g的这种化合物,更合适地包含多于约500g的这种化合物。
42.一种药物组合物,其包含项目1至38、39和40中任一项或项目38a至38c中任一项所述的化合物,并且任选地还包含药学上可接受的赋形剂。
43.根据项目42所述的药物组合物,其被配制用于口服给药。
44.根据项目42或43所述的药物组合物,其为单位剂型。
45.根据项目44所述的药物组合物,每单位剂型包含1至950mg的化合物。
46.根据项目45所述的药物组合物,每单位剂型包含2至150mg的化合物。
47.根据项目45所述的药物组合物,每单位剂型包含10至150mg的化合物。
48.根据项目45或46所述的药物组合物,每单位剂型包含约选自以下的量的化合物:2mg,5mg,15mg,20mg,50mg,70mg,80mg,100mg和140mg。
49.根据项目42至48中任一项所述的药物组合物,其为单位剂型,其中所述单位剂型为片剂、囊片或胶囊剂。
50.根据项目42至49中任一项所述的药物组合物,其进一步包含选自以下的赋形剂中的一种或多种,优选全部:乳糖,微晶纤维素,交联羧甲基纤维素钠,羟丙基纤维素和硬脂酸镁。
51.根据项目44至50中任一项所述的药物组合物,其中所述单位剂型是薄膜包衣的片剂或薄膜包衣的囊片。
52.根据项目51所述的药物组合物,其中所述薄膜包衣包含选自以下的赋形剂中的一种或多种,优选全部:羟丙甲纤维素,二氧化钛和聚乙二醇400。
53.项目1至38、39和40中任一项或项目38a至38c中任一项所述的化合物,或项目42至52中任一项所述的药物组合物,其用于治疗。
54.一种用于治疗对象的增生性疾病的化合物或药物组合物,所述治疗包括向所述对象施用所述化合物或药物组合物,其中,所述化合物是项目1-38、39和40中任一项或项目38a至38c中任一项所述的化合物或所述药物组合物是项目42-52中任一项所述的药物组合物。
55.根据项目53或54所述的特定用途的化合物或药物组合物,其中所述对象是成年人;适当地,其中成年人是大约30岁或更大;特别是其中成年人是年轻人、中年人或老年人。
56.根据项目53或54所述的特定用途的化合物或药物组合物,其中所述对象合适地是儿科人类,其中所述儿科人类小于约16岁;特别是其中所述儿科人类是青少年、青春期前、幼儿、婴幼儿或婴儿。
57.根据项目53至56中任一项所述的特定用途的化合物或药物组合物,其中所述治疗不涉及抑制SIK3激酶。
58.根据项目53至57中任一项所述的特定用途的化合物或药物组合物,其中所述治疗:
·不涉及抑制SIK3激酶;
·不涉及抑制SIK1和/或SIK2激酶;
·不涉及抑制JAK1激酶;
·不涉及抑制RET激酶;
·不涉及抑制ERBB4激酶;
·不涉及抑制PDGFR-α激酶和/或
·不涉及抑制EPHB2激酶。
59.根据项目53至58中任一项所述的特定用途的化合物或药物组合物,其中所述治疗:
·不涉及抑制PDGFR-α激酶;
·不涉及抑制TGFB-R1、B-RAF和/或p38-β激酶;
·不涉及抑制ACV-R1和/或BMPR1A激酶;和/或
·不涉及抑制RET激酶。
60.根据项目53至59中任一项所述的特定用途的化合物或药物组合物,其中其不涉及使与增殖性疾病有关的细胞对细胞介导的免疫反应敏感。
61.根据项目53至60中任一项所述的特定用途的化合物或药物组合物,其中所述治疗:
·涉及抑制ABL1和/或SRC激酶;
·涉及抑制BCR-ABL激酶;
·涉及抑制LCK激酶;
·涉及抑制LYN激酶
·涉及抑制YES激酶;
·涉及抑制FYN激酶;和/或
·涉及抑制KIT激酶。
62.根据项目53至61中任一项所述的特定用途的化合物或药物组合物,其中所述治疗:
·涉及抑制EPHA2激酶;
·涉及抑制EPHA4激酶;
·涉及抑制CSF1-R激酶;
·涉及抑制HCK激酶;和/或
·涉及抑制ACK1激酶。
63.根据项目53至62中任一项所述的特定用途的化合物或药物组合物,其中所述治疗涉及抑制ABL1和/或SRC激酶多于抑制SIK3和/或多于抑制SIK1和/或SIK2。
64.根据项目53至63中任一项所述的特定用途的化合物或药物组合物,其中所述治疗涉及抑制FLT3激酶。
65.根据项目53至64中任一项所述的特定用途的化合物或药物组合物,其中所述治疗:
·涉及抑制NEK11、WEE1和/或WNK2激酶;
·涉及抑制Aurora-A和/或Aurora-B激酶;和/或
·涉及抑制TBK1激酶。
66.根据项目53至65中任一项所述的特定用途的化合物或药物组合物,其中所述治疗包括抑制激酶突变;尤其涉及抑制BCR-ABL激酶和/或ABL1激酶的另一突变体,和/或涉及抑制KIT激酶的突变体。
67.根据项目53至66中任一项所述的特定用途的化合物或药物组合物,其中所述治疗包括向有需要的成年对象施用每天少于约140mg的化合物,任选地,其中所述增殖性疾病是非慢性期Ph+CML的增生性疾病。
68.根据项目67所述的特定用途的化合物或药物组合物,其中所述治疗包括每向有需要的成年对象施用每天少于约100mg的化合物,任选地其中所述增殖性疾病是慢性期Ph+CML。
69.根据项目53至66中任一项所述的特定用途的化合物或药物组合物,其中所述治疗包括向有需要的儿科人类对象施用以下量的化合物:
·对于体重为10kg至20kg的儿科患者,每天少于40mg;
·对于体重为20kg至30kg的儿科患者,每天少于60mg;
·对于体重为30kg至45kg的儿科患者,每天少于约70mg;或者
·对于体重为少为45kg的儿科患者,每天少于约100mg。
70.根据项目67至69中任一项所述的特定用途的化合物或药物组合物,其中所述人类对象施用所述化合物的量少于每天一次。
71.根据项目67至70中任一项所述的特定用途的化合物或药物组合物,其中向所述人类对象施用一定量的所述化合物,并以一定的频率使与增殖性疾病有关的细胞对细胞介导的免疫反应敏感,特别是使与增殖性疾病有关的细胞对TNF的杀伤(凋亡/细胞毒性)作用敏感。
72.根据项目53至71中任一项所述的特定用途的化合物或药物组合物,其中所述对象没有不良反应。
73.根据项目72所述的特定用途的化合物或药物组合物,其中所述不良反应是骨髓抑制。
74.根据项目72所述的特定用途的化合物或药物组合物,其中所述不良反应是非血液学不良反应。
75.根据项目72所述的特定用途的化合物或药物组合物,其中所述不良反应是心脏病不良反应。
76.根据项目53至75中任一项所述的特定用途的化合物或药物组合物,其中所述对象不伴随使用强CYP3A4抑制剂。
77.根据项目53至76中任一项所述的特定用途的化合物或药物组合物,其中所述增生性疾病是癌症或肿瘤。
78.根据项目77所述的特定用途的化合物或药物组合物,其中所述癌症是造血或淋巴样癌症。
79.根据项目53至78中任一项所述的特定用途的化合物或药物组合物,其中所述增生性疾病是费城染色体阳性白血病。
80.根据项目53至79中任一项所述的特定用途的化合物或药物组合物,其中所述增生性疾病是费城染色体阳性的慢性髓细胞性白血病(Ph+CML)或费城染色体阳性的急性淋巴细胞性白血病(Ph+ALL)。
81.根据项目53至80中任一项所述的特定用途的化合物或药物组合物,其中所述增生性疾病为:
·慢性期的新诊断的(Ph+CML);
·对先前治疗(包括伊马替尼)有耐药性或耐受性的慢性、加速或爆炸性CML;或者
·对先前治疗有耐药性或不耐受性的Ph+急性淋巴细胞白血病(ALL)和淋巴母细胞性CML。
82.根据项目53至80中任一项所述的特定用途的化合物或药物组合物,其中所述对象是儿科人类,并且所述增生性疾病是:
·新诊断的慢性期Ph+CML(Ph+CML-CP)或对先前治疗(包括伊马替尼)有抗药性或耐受性的Ph+CML-CP。
83.根据项目77所述的特定用途的化合物或药物组合物,其中所述癌症是实体瘤。
84.根据项目83所述的特定用途的化合物或药物组合物,其中所述实体瘤是选自以下的组织的组织的癌症:胰腺、乳腺癌、肺癌、前列腺癌、皮肤癌、卵巢癌、食道癌和结肠/直肠癌。
85.根据项目53至77、83或84中任一项所述的特定用途的化合物或药物组合物,其中所述增生性疾病是胰腺癌。
86.根据项目85所述的特定用途的化合物或药物组合物,其中治疗还包括给予EGFR抑制剂和/或吉西他滨。
87.根据项目53至77、83或84中任一项所述的特定用途的化合物或药物组合物,其中所述增生性疾病是前列腺癌。
88.根据项目87所述的特定用途的化合物或药物组合物,其中治疗进一步包括施用多西他赛。
89.根据项目53至77、83或84中任一项所述的特定用途的化合物或药物组合物,其中所述增生性疾病是肺癌,特别是非小细胞肺癌。
90.根据项目53至89中任一项所述的特定用途的化合物或药物组合物,其中治疗还包括施用免疫检查点抑制剂。
91.根据项目90所述的特定用途的化合物或药物组合物,其中免疫检查点抑制剂是nivolumab、relatlimab、ipilimumab或BMS-986205。
92.根据项目53至91中任一项所述的特定用途的化合物或药物组合物,其中所述增殖性疾病在对象的标准疗法上已经进展。
93.根据项目53至91中任一项所述的特定用途的化合物或药物组合物,其中所述对象者不能接受标准疗法。
94.根据项目92或93所述的特定用途的化合物或药物组合物,其中所述标准疗法是免疫疗法。
95.根据项目53至91中任一项所述的特定用途的化合物或药物组合物,其中所述增生性疾病是具有耐药性的CML或ALL,和/或所述对象对先前的治疗不耐受,特别是对先前的伊马替尼治疗不耐受或不耐受。
96.根据项目53至95中任一项所述的特定用途的化合物或药物组合物,其中所述治疗涉及抑制SIK3。
97.根据项目96所述的特定用途的化合物或药物组合物,其中以有效降低SIK3(优选在与增殖性疾病有关的细胞中的SIK3)活性的治疗有效量施用所述化合物或药物组合物。
98.根据项目96或97所述的特定用途的化合物或药物组合物,其中以对降低ABL1和/或SRC激酶(优选在与增殖性疾病有关的细胞中的ABL1和/或SRC激酶)活性无效的治疗有效量施用所述化合物或药物组合物。
99.根据项目96至98中任一项所述的特定用途的化合物或药物组合物,其中所述治疗是通过抑制SIK3介导的;并且其中所述治疗不是通过抑制ABL1和/或SRC激酶介导的。
100.根据项目96至99中任一项所述的特定用途的化合物或药物组合物,其中所述治疗包括抑制与增殖性疾病有关的细胞中的SIK3;优选其中SIK3的活性降低;并且其中所述治疗包括与增殖性疾病有关的细胞中的ABL1和/或SRC不被抑制;优选其中ABL1和/或SRC的活性不降低。
101.根据项目53至100中任一项所述的特定用途的化合物或药物组合物,其中所述治疗包括使与所述增殖性疾病有关的细胞对细胞介导的免疫反应敏感。
102.根据项目53至101中任一项所述的特定用途的化合物或药物组合物,其中所述治疗包括所述细胞介导的免疫应答诱导杀死与所述增殖性疾病有关的细胞。
103.根据项目53至102中任一项所述的特定用途的化合物或药物组合物,其中将所述化合物或药物组合物施用于所述对象以使与所述增殖性疾病有关的细胞对由TNF诱导的杀伤敏感。
104.根据项目101至103中任一项所述的特定用途的化合物或药物组合物,其中使与所述增殖性疾病有关的细胞暴露于TNF和/或TNFR1信号传导激动剂。
105.根据项目101至104中任一项所述的特定用途的化合物或药物组合物,其中:(x)所述对象的特征在于其血浆TNF浓度大于约5pg/mL;和/或(y)增生性疾病是对象中的实体瘤,其通过肿瘤内TNF的浓度大于约1pg/mL。
106.根据项目53至105中任一项所述的特定用途的化合物或药物组合物,所述治疗包括使所述对象中与增生性疾病有关的细胞暴露于:(i)TNF和/或TNFR1信号传导激动剂;以及(ii)所述化合物或药物组合物。
107.根据项目106所述的特定用途的化合物或药物组合物,其中暴露于对象中与增殖性疾病有关的细胞的TNF的量增加。
108.根据项目106或107所述的特定用途的化合物或药物组合物,其中:(i)将TNF或TNFR1信号传导激动剂施用至对象;(ii)将能够诱导或诱导了与增殖性疾病有关的细胞暴露于TNF或TNFR1信号传导激动剂的试剂施用至对象;(iii)与增生性疾病有关的细胞暴露于TNF是通过药物、治疗或其他方法诱导的,该方法增加了对象血浆和/或此类细胞的环境中TNF的量。
109.根据项目106至108中任一项所述的特定用途的化合物或药物组合物,其中所述治疗包括将所述化合物或药物组合物以有效抑制SIK3(特别是与增生性疾病有关的细胞中的SIK3)的治疗有效量施用于对象。
110.根据项目106至109中任一项所述的特定用途的化合物或药物组合物,其中所述治疗包括以不有效抑制ABL1和/或SRC(特别是与增殖性疾病有关的细胞中的ABL1和/或SRC)的治疗有效量将所述化合物或药物组合物施用于对象。
111.根据项目106至110中任一项所述的特定用途的化合物或药物组合物,其中所述治疗包括向所述对象施用TNF或TNFR1信号传导激动剂。
112.根据项目106至110中任一项所述的特定用途的化合物或药物组合物,其中所述治疗包括将能够诱导或诱导了与所述增殖性疾病有关的细胞暴露于TNF或TNFR1信号传导激动剂的试剂施用于对象。
113.根据项目106至110中任一项所述的特定用途的化合物或药物组合物,其中与增生性疾病有关的细胞暴露于TNF是通过药物、治疗或其他方法诱导的,该方法增加对象血浆中和/或这种细胞的环境中TNF的量。
114.根据项目113所述的特定用途的化合物或药物组合物,其中所述药物、治疗或其他方法包括癌症免疫疗法和/或放射疗法。
115.根据项目106至114中任一项所述的特定用途的化合物或药物组合物,其中所述对象的特征在于具有与以SIK3的表达和/或活性为特征的增殖性疾病有关的细胞,特别是此类细胞表达mRNA和/或SIK3的蛋白,和/或对SIK3表达和/或活性呈阳性。
116.根据项目106至115中任一项所述的特定用途的化合物或药物组合物,其中所述增生性疾病是肿瘤,特别是实体瘤。
117.根据项目116所述的特定用途的化合物或药物组合物,其中所述对象被区分为先前已经用免疫疗法治疗并且其肿瘤已经进展,特别是其肿瘤复发、再次发生或不响应。
118.根据项目116或117所述的化合物或药物组合物,其中所述受试者被区分为患有在放射疗法之前进展,特别是复发、再次发生或不响应的肿瘤。
119.一种化合物或药物组合物,该化合物或药物组合物用于在用抗TNF剂治疗的对象中治疗以减少发展为继发性疾病的血液学增生性疾病的风险,所述治疗包括施用项目1至38、39和40中任一项或项目38a至38c中任一项所述的化合物或项目42至52中任一项所述的药物组合物。
120.根据项目119所述的特定用途的化合物或药物组合物,其中所述对象患有自身免疫疾病和/或所述对象用所述抗TNF剂治疗自身免疫疾病;优选地自身免疫性疾病选自:类风湿性关节炎,牛皮癣性关节炎,强直性脊柱炎,斑块状银屑病和克罗恩氏病,最优选类风湿性关节炎。
121.根据项目53至120中任一项所述的特定用途的化合物或药物组合物,其中所述化合物是或所述药物组合物包含SIK3(或SIK3特异性)抑制剂。
122.根据项目53至121中任一项所述的特定用途的化合物或药物组合物,其中该化合物或药物组合物包含比制ABL1和/或SRC激酶更有效地抑制SIK3的SIK3抑制剂;并且其中SIK3抑制剂比抑制SIK2和/或SIK2更少(或更多)有效地抑制SIK3。
123.根据项目53至122中任一项所述的特定用途的化合物或药物组合物,其中所述对象是人类志愿者。
124.根据项目53至122中任一项所述的特定用途的化合物或药物组合物,其中所述对象是实验动物,特别是选自以下的动物:小鼠、大鼠、兔、猪和猴。
125.根据项目123或124所述的特定用途的化合物或药物组合物,其中治疗了多个对象,特别是5个或更多个对象,例如约5至20、10至50、25至200、或75至250个对象,或超过约250个对象。
126.根据项目125所述的特定用途的化合物或药物组合物,其中所述治疗包括:(i)向所述对象中的至少一个施用一种剂量的化合物和/或一种制剂的药物组合物;以及(ii)向所述对象中的至少另一个施用不同剂量的化合物和/或不同制剂的药物组合物。
127.根据项目125或126所述的特定用途的化合物或药物组合物,其中所述治疗包括:(i)向所述对象中的至少一个施用一种剂量的化合物和/或一种制剂的药物组合物;以及(ii)向所述对象中的至少另一个施用:(a)安慰剂;或(b)(i)的对象的化合物的剂量和/或药物组合物的制剂,以及另外的药物、治疗或其他方法。
128.根据项目125至127中任一项所述的特定用途的化合物或药物组合物,其中所述治疗是专门设计用于研究和/或确定该化合物的治疗有效剂量和/或鉴定该药物组合物的治疗有效制剂。
129.一种诊断和治疗以对象中适用的生物标记物的存在或数量和/或以其表达或活性为特征的增生性疾病的方法,包括以下步骤:
·从所述对象的生物学样品中检测适用的生物标记物,从而诊断所述对象是否患有(或可能患有)这种疾病、病症或病状;以及
·对如此诊断的对象施用有效量的项目1至38、39和40中的任一项或项目38a至38c中的任一项所述的化合物或项目42至52中的任一项所述的药物组合物(在对象上实施本发明的治疗方法)。
130.中间体在制备项目1至38、39和40中任一项或项目38a至38c中任一项所述的化合物中的用途,其中该中间体包含R
其中,R
131.项目130所述的中间体的用途,其中该中间体选自:
·4-氯-2-甲基吡啶-3-胺;
·2,4-二甲基吡啶-3-胺;
·3-甲基吡啶-2-胺;
·4-溴-2-甲基吡啶-3-胺;
·3-氯-5-甲基吡啶-4-胺;
·3,5-二甲基吡啶-4-胺;
·2-甲基吡啶-3-胺;
·2-氯-4-甲基噻吩-3-胺;
·1,3,5-三甲基-1H-吡唑-4-胺;
·3,5-二甲基异恶唑-4-胺;
·奎尼丁-3-胺;以及
·2-乙基-4-甲基吡啶-3-胺。
131a.项目130所述的中间体的用途,其中所述中间体选自:
·2-氯-4-甲基噻吩-3-胺。
131b.项目130所述的中间体的用途,其中所述中间体选自:
·4-氯-2-甲基噻吩-3-胺;
·2,4-二甲基噻吩-3-胺;
·2-甲基噻吩-3-胺;
·4-甲基噻吩-3-胺;
·2-氯噻吩-3-胺;
·2-溴噻吩-3-胺;以及
·2,4-二氯噻吩-3-胺。
132.一种中间体,该中间体选自具有项1至38中任一项或项38a至38c中任一项所定义的式(I)结构的化合物,其中R
133.一种制备项目39的化合物的方法,其包括以下步骤:
·提供在具有一种或多种杂质的混合物中的项目1至38或40中任一项或项目38a至38c中任一项所述的化合物;以及
·从混合物中去除至少一部分杂质。
134.根据项目133所述的方法,其中所述混合物通过合成不纯形式的项目1至38或40中任一项或项目38a至38c中任一项所述的化合物而提供。
135.一种制备药物组合物的方法,所述方法包括以下步骤:将项目1至38、39和40中的任一项或项目38a至38c中的任一项的化合物与药学上可接受的赋形剂一起配制。
136.根据项目135的方法,其中所述药物组合物以单位剂型制造。
137.根据项目135或136所述的方法,其中所述药物组合物形成为片剂、囊片或胶囊剂,特别是形成为薄膜包衣的片剂或薄膜包衣的囊片。
138.一种制备药物包装的方法,包括以下步骤:
·将项目42至52中任一项所述的药物组合物(优选以成品药物形式)插入包装中,从而形成包含该药物组合物的包装;以及任选的
·在包装中插入说明药物组合物处方信息的单张。
139.一种药物包装,其包含项目42至52中任一项所述的药物组合物;优选地,其中药物组合物为成品药物形式。
140.根据项目139所述的药物包装,还包含描述该药物组合物的处方信息的单张。
141.一种将项目42至52中任一项或项目139或140所述的药物包装的药物组合物从第一位置递送到第二位置的方法,该方法包括以下步骤:
·在第一位置处接收来自第二位置的递送一定量的药物组合物或药物包装的请求;以及
·在一次或多次递送中,从第一位置向第二位置递送至少一部分(优选全部)所请求的量的药物组合物或药物包装,
其中第一位置和第二位置相隔大于约10m的距离,并且通过轮式运输工具将所述递送中的一次或多次运送第一位置与第二位置之间的距离的至少一部分。
鉴于上述情况,将理解,本发明还涉及以下另外的逐项实施例:
A1.一种化合物,其选自下式的激酶抑制剂:
以及其溶剂化物、盐、N-氧化物、络合物、多晶型物、晶体形式、外消旋混合物、非对映异构体、对映异构体、互变异构体、构象异构体、同位素标记形式、前药及其组合;
其中:
R
R
R
R
R
R
L选自键、C
R
R
A选自S、O、NR
R
R
X独立地选自O、S和N(R
E是O或S;
B是N或CR
R
R
R
R
R
y是0到2的整数;
R
R
其中
R
R
X
附带条件是
(1)当R
(2)当R
(3)当R
(i)R
(ii)R
(4)当E为O,B为CR
(5)当R
A2.根据项目A1所述的化合物,其中R
A3.根据项目A1或A2所述的化合物,其中R
A4.根据项目A1至A3中任一项所述的化合物,其中R
A5.根据项目A1至A4中任一项所述的化合物,其中R
A6.根据项目A1至A5中任一项所述的化合物,其中一个R
A7.根据项目A1至A6中任一项所述的化合物,其中L选自:键;任选被一个R
A8.根据项目A1至A7中任一项所述的化合物,其中L选自键。
A9.根据项目A1至A8中任一项所述的化合物,其中R
A10.根据项目A1至A9中任一项所述的化合物,其中R
A11.根据项目A1至A10中任一项所述的化合物,其中R
A12.根据项目A1至A11中任一项所述的化合物,其中A是S、O或N(CH
A13.根据项目A1至A12中任一项所述的化合物,其中A是S。
A14.根据项目A1至A13中任一项所述的化合物,其中B是N或CR
A15.根据项目A1至A13中任一项所述的化合物,其中B是N。
A16.根据项目A1至A15中任一项所述的化合物,其中E是O。
A17.根据项目A1至A16中任一项所述的化合物,其中R
A18.根据项目A1至A17中任一项所述的化合物,其选自:
以及其溶剂化物、盐、N-氧化物、络合物、多晶型物、晶体形式、互变异构体、构象异构体、同位素标记形式、前药及其组合。
A19.一种药物组合物,其包含项目A1至A18中任一项所述的化合物,并且任选地还包含药学上可接受的赋形剂。
A20.一种化合物或药物组合物,其用于治疗对象的增生性疾病,所述治疗包括向所述对象施用所述化合物或药物组合物,其中,该化合物为项目A1至A18中任一项所述的化合物,或所述药物组合物为项目A19所述的药物组合物。
实施例
在本发明的范围内或在本发明的方法中使用的化合物的选择和/或代表各种示例性或优选的R
实施例示出了:
实施例1:激酶抑制剂的合成,包括式(I)的激酶抑制剂。
通用方法和材料:
使用Biotage Isolera Four系统进行MPLC纯化,使用KP-Sil柱和工业级有机溶剂,即二氯甲烷和甲醇、3-4N NH
使用以下系统在Shimadzu HPLC系统上进行反相HPLC[溶剂A:乙腈,溶剂B:0.1%的甲酸水溶液]。甲酸用作HPLC级。所有分离均在环境温度下进行。对于分析型RP-HPLC分析[Interchim:Uptisphere Strategy
使用以下系统在Dionex Ultimate 3000系统上记录LC-MS光谱[溶剂A:乙腈,溶剂B:0.1%甲酸的水溶液]。甲酸用作HPLC级。所有分离均在环境温度下进行。对于分析型RP-HPLC分析[Interchim:Uptisphere Strategy C18,2.6μm,50x4.6 mm],流速为1.0ml.min
程序:
I.2-((6-氯-2-甲基嘧啶-4-基)氨基)噻唑-5-甲酸乙酯:
向4,6-二氯-2-甲基嘧啶(10g,61.4mmol,1.0当量)和2-氨基噻唑-5-羧酸乙酯(10.6g,61.4mmol,1.0当量)在DMF(210ml)中的溶液中,在惰性气氛下,于0℃,分批加入氢化钠(5.40g,135mmol,2.0当量),并将反应混合物缓慢温热至室温,并搅拌3天。通过加入饱和氯化铵溶液淬灭过量的NaH,并将反应混合物倒入水(3000ml)中,并在室温下搅拌1h。滤出获得的沉淀物并风干以得到2-((6-氯-2-甲基嘧啶-4-基)氨基)噻唑-5-甲酸乙酯(18g,60.0mmol,98%产率),为米色固体。
II.2-((6-氯-2-甲基嘧啶-4-基)氨基)噻唑-5-羧酸:
向2-((6-氯-2-甲基嘧啶-4-基)氨基)噻唑-5-甲酸乙酯(36.0g,121mmol,1.0当量)在甲醇(350ml)和水(150ml)中的悬浮液中,在室温下,加入氢氧化钠(38.6g,964mmol,8.0当量),并将混合物在室温下搅拌16小时。LCMS分析表明原料完全转化为产物。浓缩反应混合物以除去大部分溶剂,然后使用6M HCl水溶液酸化水层。滤出所得沉淀物,并用水洗涤,在高真空下干燥3天,得到2-((6-氯-2-甲基嘧啶-4-基)氨基)噻唑-5-羧酸(27.0g,99.7mmol,83%产率),为米色粉末。
通用程序A1–GP A1–酰胺的形成:
向2-((6-氯-2-甲基嘧啶-4-基)氨基)噻唑-5-羧酸(1.0当量和苯胺(1.1当量)在乙腈(0.2M)中的悬浮液中,在室温下在氮气氛下,加入N,N-二异丙基乙胺(3.5当量)和四甲基氯甲脒六氟磷酸盐(1.2当量)。将反应混合物在室温搅拌过夜。在真空下除去乙腈,然后将反应倒入水中(100mL/mmol)。滤出所得沉淀物,用水[5x]洗涤,风干并在高真空下干燥,得到相应的酰胺,为黄色粉末,其无需进一步纯化即可直接用于下一步。所有物质均通过
通用程序B-GP B:
向氯嘧啶衍生物(1.0当量)在正丁醇(2ml)中的悬浮液中,在室温下在氮气氛下加入2-(哌嗪-1-基)乙-1-醇(5.0当量)和N,N-二异丙基乙胺(0.4当量)。将管密封并在微波条件下(Biotage Initiator
化合物A8:
N-(2-氯-6-甲基苯基)-2-((6-(4-(2-羟乙基)哌嗪-1-基)-2-甲基嘧啶-4-基)氨基)噻唑-5-羧酰胺
步骤1按照GP A1进行,使用2-((6-氯-2-甲基嘧啶-4-基)氨基)噻唑-5-羧酸(750mg,2.77mmol)、2-氯-6-甲基苯胺(959mg,6.77mmol)、四甲基氯甲脒六氟磷酸酯(959mg,3.42mmol)、N,N-二异丙基乙胺(1.25g,9.70mmol)和乙腈(6.0mL,0.42M)。从水中沉淀出来后,获得2-((6-氯-2-甲基嘧啶-4-基)氨基)-N-(2-氯-6-甲基苯基)噻唑-5-羧酰胺(480mg,1.22mmol),为黄色固体,无需进一步纯化即可用于下一步。
第2步是按照GP B进行的,使用2-((6-氯-2-甲基嘧啶-4-基)氨基)-N-(2-氯-6-甲基苯基)噻唑-5-羧酰胺(250mg,0.634mmol)、2-(哌嗪-1-基)乙-1-醇(413mg,3.17mmol)、N,N-二异丙基乙胺(33mg,0.25mmol)和1-丁醇(2.0mL,0.3M)。A8(图1A)。
化合物B3:
N-(4-氯-2-甲基吡啶-3-基)-2-((6-(4-(2-羟乙基)哌嗪-1-基)-2-甲基嘧啶-4-基)氨基)噻唑-5-羧酰胺
方案1:B3的合成
步骤1:2-溴-4-氯吡啶-3-胺(化合物IaB3):
向4-氯吡啶-3-胺(7.00g,54.0mmol,1.0当量)在无水TFA(200mL,0.27M)中的溶液中,在室温氮气氛下,加入NBS(10.7g,60.1mmol,1.1当量)。将该溶液在室温下搅拌18小时。减压除去溶剂,并将残余物溶于2N NaOH(200mL)。用EtOAc(3×200mL)萃取水层。合并的有机层经Na2SO4干燥,过滤并在减压下浓缩。粗产物通过快速色谱法纯化(340g biotage柱,在10CV上用cHex 100%1CV至40%EtOAc,2CV 40%EtOAc),得到2-溴-4-氯吡啶-3-胺(3.07g,14.8mmol,27%),为米色固体。IaB3。
R
步骤2:4-氯-2-甲基吡啶-3-胺(化合物IbB3):
在10mL微波小瓶中,放入2-溴-4-氯吡啶-3-胺(200mg,0.96mmol,1.0当量)、三甲基环硼氧烷(141μL,1.01mmol,1.05当量)、K
R
步骤3和4:N-(4-氯-2-甲基吡啶-3-基)-2-((6-(4-(2-羟乙基)哌嗪-1-基)-2-甲基嘧啶-4-基)氨基)噻唑-5-羧酰胺(化合物B3,图1B):
步骤3按照GP A1进行,使用2-((6-氯-2-甲基嘧啶-4-基)氨基)噻唑-5-羧酸(530mg,1.96mmol)、4-氯-2-甲基吡啶-3-胺(363mg,2.55mmol)、四甲基氯甲脒六氟磷酸酯(659mg,2.35mmol)、N,N-二异丙基乙胺(886mg,6.85mmol)和乙腈(5.8mL,0.3M)。在从H
步骤4按照GP B进行,使用N-(4-氯-2-甲基吡啶-3-基)-2-((6-氯-2-甲基嘧啶-4-基)氨基)噻唑-5-羧酰胺(320mg,0.810mmol)、2-(哌嗪-1-基)乙-1-醇(527mg,4.05mmol)、N,N-二异丙基乙胺(42mg,0.32mmol)和1-丁醇(2.5mL,0.3M)。在用EtOAc萃取,快速纯化(100%DCM到85%DCM/3N NH
化合物C1:
N-(2,4-二甲基吡啶-3-基)-2-((6-(4-(2-羟乙基)哌嗪-1-基)-2-甲基嘧啶-4-基)氨基)噻唑-5-羧酰胺
步骤1按照GP A 1进行,使用2-((6-氯-2-甲基嘧啶-4-基)氨基)噻唑-5-羧酸(266mg,0.982mmol)、2,4-二甲基吡啶-3-胺(120mg,0.982mmol,1.0eq.)、四甲基氯甲脒六氟磷酸酯(331mg,1.18mmol),N,N-二异丙基乙胺(444mg,3.44mmol)和乙腈(5.0mL,0.2M)。在快速纯化后(在10CV上100%DCM至75%DCM/MeOH),获得2-((6-氯-2-甲基嘧啶-4-基)氨基)-N-(2,4-二甲基吡啶-3-基)噻唑-5-羧酰胺(123mg,0.328mmol,33%),为黄色固体。
步骤2按照GP B进行,使用2-((6-氯-2-甲基嘧啶-4-基)氨基)-N-(2,4-二甲基吡啶-3-基)噻唑-5-羧酰胺(118mg,0.315mmol)、2-(哌嗪-1-基)乙-1-醇(0.205g,1.57mmol)、N,N-二异丙基乙胺(16mg,0.13mmol)和1-丁醇(2.5mL,0.1M)。快速纯化后(10CV上100%DCM至75%DCM/MeOH),获得C1(图1C,133mg,0.284mmol,2个步骤的产率为21%),为米色固体状。
纯度(HPLC,254nm):90%(不能通过研磨、第二硅胶柱或RP柱除去)。
化合物C2:
N-((6-(4-(2-羟乙基)哌嗪-1-基)-2-甲基嘧啶-4-基)氨基)-N-(3-甲基吡啶-2-基)噻唑-5-羧酰胺
步骤1按照GP A1进行,使用2-((6-氯-2-甲基嘧啶-4-基)氨基)噻唑-5-羧酸(530mg,1.96mmol)、3-甲基吡啶-2-胺(275mg,2.55mmol)、四甲基氯甲脒六氟磷酸酯(659mg,2.35mmol)、N,N-二异丙基乙胺(886mg,6.85mmol)和乙腈(5.8mL,0.3M)。用EtOAc萃取后,获得2-((6-氯-2-甲基嘧啶-4-基)氨基)-N-(3-甲基吡啶-2-基)噻唑-5-羧酰胺(326mg,0.903mmol),为黄色固体,不经进一步纯化用于下一步。
步骤2按照GP B进行,使用2-((6-氯-2-甲基嘧啶-4-基)氨基)-N-(3-甲基吡啶-2-基)噻唑-5-羧酰胺(510mg,1.41mmol)、2-(哌嗪-1-基)乙-1-醇(920mg,7.07mmol)、N,N-二异丙基乙胺(73mg,0.57mmol)和1-丁醇(4.3mL,0.3M)。快速纯化后(在10CV上于MeOH中100%DCM至85%DCM/3N NH
化合物C3:
N-(4-溴-2-甲基吡啶-3-基)-2-((6-(4-(2-羟乙基)哌嗪-1-基)-2-甲基嘧啶-4-基)氨基)噻唑-5-羧酰胺
步骤1按照GP A1进行,使用2-((6-氯-2-甲基嘧啶-4-基)氨基)噻唑-5-羧酸(145mg,0.535mmol)、4-溴-2-甲基吡啶-3-胺(100mg,0.535mmol)、四甲基氯甲脒六氟磷酸酯(180mg,0.642mmol)、N,N-二异丙基乙胺(242mg,1.87mmol)和乙腈(2.7mL,0.2M)。用EtOAc萃取后,获得N-(4-溴-2-甲基吡啶-3-基)-2-((6-氯-2-甲基嘧啶-4-基)氨基)噻唑-5-羧酰胺(67mg,0.15mmol),为黄色固体,无需进一步纯化即可用于下一步骤。
步骤2按照GP B进行,使用N-(4-溴-2-甲基吡啶-3-基)-2-((6-氯-2-甲基嘧啶-4-基)氨基)噻唑-5-羧酰胺(65mg,0.15mmol)、2-(哌嗪-1-基)乙-1-醇(96mg,0.74mmol)、N,N-二异丙基乙胺(7.6mg,0.059mmol)和1-丁醇(0.50mL,0.3M)。快速纯化(在10CV上MeOH中100%DCM到85%DCM/3NNH
化合物C4:
N-(3-氯-5-甲基吡啶-4-基)-2-((6-(4-(2-羟乙基)哌嗪-1-基)-2-甲基嘧啶-4-基)氨基)噻唑-5-羧酰胺
步骤1按照GP A1进行,使用2-((6-氯-2-甲基嘧啶-4-基)氨基)噻唑-5-羧酸(530mg,1.96mmol)、3-氯-5-甲基吡啶-4-胺(363mg,2.55mmol)、四甲基氯甲脒六氟磷酸酯(659mg,2.35mmol)、N,N-二异丙基乙胺(886mg,6.85mmol)和乙腈(5.8mL,0.3M)。用EtOAc萃取后,获得2-((6-氯-2-甲基嘧啶-4-基)氨基)-N-(3-氯-5-甲基吡啶-4-基)噻唑-5-羧酰胺(113mg,纯度约60%),为黄色固体,其无需进一步纯化即可用于下一步骤。
步骤2按照GP B进行,2-((6-氯-2-甲基嘧啶-4-基)氨基)-N-(3-氯-5-甲基吡啶-4-基)噻唑-5-羧酰胺(110mg,0.278mmol)、2-(哌嗪-1-基)乙-1-醇(181mg,1.39mmol)、N,N-二异丙基乙胺(14mg,0.11mmol)和1-丁醇(0.84mL,0.3M)。快速纯化(在10CV上100%DCM到80%DCM/MeOH)并在Et
化合物C5:
N-(3,5-二甲基吡啶-4-基)-2-((6-(4-(2-羟乙基)哌嗪-1-基)-2-甲基嘧啶-4-基)氨基)噻唑-5-羧酰胺
步骤1按照GP A1进行,使用2-((6-氯-2-甲基嘧啶-4-基)氨基)噻唑-5-羧酸(500mg,1.85mmol)、3,5-二甲基吡啶-4-胺(271mg,2.22mmol)、四甲基氯甲脒六氟磷酸酯(622mg,2.22mmol)、N,N-二异丙基乙胺(836mg,6.46mmol)和乙腈(10mL,0.2M)。从H
步骤2按照GP B进行,使用2-((6-氯-2-甲基嘧啶-4-基)氨基)-N-(3,5-二甲基吡啶-4-基)噻唑-5-羧酰胺(270mg,0.720mmol)、2-(哌嗪-1-基)乙-1-醇(469mg,3.60mmol)、N,N-二异丙基乙胺(37mg,0.29mmol)和1-丁醇(2.5mL,0.3M)。快速纯化(在10CV上从100%DCM至80%DCM/MeOH)并在Et
2-((6-(4-(2-羟乙基)哌嗪-1-基)-2-甲基嘧啶-4-基)氨基)-N-(2-甲基吡啶-3-基)噻唑-5-羧酰胺
步骤1按照GP A1进行,使用2-((6-氯-2-甲基嘧啶-4-基)氨基)噻唑-5-羧酸(530mg,1.96mmol)、2-甲基吡啶-3-胺(275mg,2.55mmol)、四甲基氯甲脒六氟磷酸酯(659mg,2.35mmol)、N,N-二异丙基乙胺(886mg,6.85mmol)和乙腈(5.8mL,0.3M)。从H
步骤2按照GP B进行,使用2-((6-氯-2-甲基嘧啶-4-基)氨基)-N-(2-甲基吡啶-3-基)噻唑-5-羧酰胺(440mg,1.41mmol)、2-(哌嗪-1-基)乙-1-醇(920mg,7.07mmol)、N,N-二异丙基乙胺(73mg,0.57mmol)和1-丁醇(4.3mL,0.3M)。从H
化合物C7:
N-(2-氯-4-甲基噻吩-3-基)-2-((6-(4-(2-羟乙基)哌嗪-1-基)-2-甲基嘧啶-4-基)氨基)噻唑-5-羧酰胺
方案2:C7的合成
步骤1:叔丁基(4-甲基噻吩-3-基)氨基甲酸酯:
向甲基3-氨基-4-甲基噻吩-2-羧酸盐(1.71g,10.0mmol)在H
R
步骤2:叔丁基(2-氯-4-甲基噻吩-3-基)氨基甲酸酯:
向(4-甲基-噻吩-3-基)-氨基甲酸叔丁酯(1.07g,5.00mmol,1.0e当量)在无水EtOAc(8mL)中的溶液中,在室温下在氮气氛下,加入N-氯琥珀酰亚胺(0.700g,5.25mmol,1.05当量)和在EtOH中的盐酸溶液(1.25N,200uL,0.05当量)。5小时后,TLC指示原料仍然存在(cHex/EtOAc 9:1:稍微多的极性斑点:0.76原料,产物:0.68,更长的反应时间未带来更好的转化率)。通过添加1.0N氢氧化钠(5.50mL,5.50mmol,1.1当量)和亚硫酸氢钠(40%,0.05当量)的溶液淬灭反应。丢弃下面的水层。有机相用水(10mL)洗涤。有机层经Na2SO4干燥,过滤并在减压下浓缩。粗产物通过快速色谱法纯化(100g biotage柱,cHex 100%1CV,在10CV上至15%EtOAc,在5CV上至100%EtOAc),获得叔丁基(2-氯-4-甲基噻吩-3-基)氨基甲酸酯(734mg,2.96mmol,59%),为橙色固体。
R
步骤3:2-氯-4-甲基噻吩-3-胺盐酸盐:
向叔丁基(2-氯-4-甲基噻吩-3-基)氨基甲酸酯(567mg,2.29mmol,1.0当量)在无水二恶烷(4.6mL)中的溶液中,在室温下在氮气氛下,加入HCl溶液(二恶烷中的4.0N,1.70mL,6.80mmol,3.0当量)。将反应混合物在室温搅拌16小时。TLC(cHex/EtOAc 9:1,UV254nm)显示仍然存在起始原料。HCl溶液(二恶烷中的4.0N,1.20mL,4.80mmol,2.1当量)和悬浮液在室温下搅拌5小时。减压除去溶剂,并将粗产物在Et
R
步骤4按照GP A1进行,使用2-((6-氯-2-甲基嘧啶-4-基)氨基)噻唑-5-羧酸(367mg,1.35mmol)、2-氯-4-甲基噻吩-3-胺盐酸盐(200mg,1.16mmol)、四甲基氯甲脒六氟磷酸酯(456mg,1.63mmol)、N,N-二异丙基乙胺(700mg,5.42mmol,4.0eq.)和乙腈(6.9mL,0.3M)。用EtOAc萃取后,获得2-((6-氯-2-甲基嘧啶-4-基)氨基)-N-(2-氯-4-甲基噻吩-3-基)噻唑-5-羧酰胺(462mg,1.15mmol,纯度约为70%),为黄色固体,无需进一步纯化即可用于下一步骤。
步骤5按照GP B进行,使用2-((6-氯-2-甲基嘧啶-4-基)氨基)-N-(2-氯-4-甲基噻吩-3-基)噻唑-5-羧酰胺(230mg,0.575mmol)、2-(哌嗪-1-基)乙-1-醇(374mg,2.87mmol)、N,N-二异丙基乙胺(30mg,0.23mmol)和1-丁醇(2.0mL,0.3M)。快速纯化(在10CV上从100%DCM到25%DCM/MeOH)并在Et
化合物C8:
2-((6-(4-(2-羟乙基)哌嗪-1-基)-2-甲基嘧啶-4-基)氨基)-N-(1,3,5-三甲基-1H-吡唑-4-基)噻唑-5-羧酰胺
步骤1按照GP A1进行,使用2-((6-氯-2-甲基嘧啶-4-基)氨基)噻唑-5-羧酸(530mg,1.96mmol)、1,3,5-三甲基-1H-吡唑-4-胺(319mg,2.55mmol)、四甲基氯甲脒六氟磷酸酯(659mg,2.35mmol)、N,N-二异丙基乙胺(886mg,6.85mmol)和乙腈(5.8mL,0.3M)。从H
步骤2按照GP B进行,使用2-((6-氯-2-甲基嘧啶-4-基)氨基)-N-(1,3,5-三甲基-1H-吡唑-4-基)噻唑-5-羧酰胺(175mg,0.463mmol)、2-(哌嗪-1-基)乙-1-醇(301mg,2.32mmol)、N,N-二异丙基乙胺(24mg,0.19mmol)和1-丁醇(1.4mL,0.3M)。快速纯化(在10CV上从100%DCM到85%DCM/3N NH
化合物C9:
N-(3,5-二甲基-1,2-异恶唑基)-3-((6-(4-(2-羟乙基)哌嗪-1-基)-2-甲基嘧啶-4-基)氨基)噻唑-5-羧酰胺
步骤1按照GP A1进行,使用2-((6-氯-2-甲基嘧啶-4-基)氨基)噻唑-5-羧酸(530mg,1.96mmol)、3,5-二甲基异恶唑-4-胺(285mg,2.55mmol)、四甲基氯甲脒六氟磷酸酯(659mg,2.35mmol)、N,N-二异丙基乙胺(886mg,6.85mmol)和乙腈(5.8mL,0.3M)。从H
步骤2按照GP B进行,使用2-((6-氯-2-甲基嘧啶-4-基)氨基)-N-(3,5-二甲基异恶唑-4-基)噻唑-5-羧酰胺(530mg,1.45mmol)、2-(哌嗪-1-基)乙-1-醇(946mg,7.26mmol)、N,N-二异丙基乙胺(75mg,0.58mmol)和1-丁醇(4.4mL,0.3M)。快速纯化后(10CV上100%DCM到75%DCM/MeOH,75%DCM/MeOH 5CV),获得C9(图1C,280mg,0.611mmol,2个步骤的产率为31%),为灰白色固体。
化合物C10:
N-(4-氯-2-甲基吡啶-3-基)-2-((2-甲基)-6-(4-甲基哌嗪-1-基)嘧啶-4-基)氨基)噻唑-5-羧酰胺
根据B3的程序进行步骤1-3。
步骤4按照GP B进行,使用N-(4-氯-2-甲基吡啶-3-基)-2-((6-氯-2-甲基嘧啶-4-基)氨基)噻唑-5-羧酰胺(206mg,0.521mmol)、1-甲基哌嗪e(261mg,2.61mmol)、N,N-二异丙基乙胺(27mg,0.21mmol)和1-丁醇(1.5mL,0.3M)。用EtOAc萃取,快速纯化(在10CV上从100%DCM到80%DCM/3N NH
N-(4-氯-2-甲基吡啶-3-基)-2-((6-((2-羟乙基)氨基)-2-甲基嘧啶-4-基)氨基)噻唑-5-羧酰胺
根据B3的程序进行步骤1-3。
步骤4按照GP B进行,使用N-(4-氯-2-甲基吡啶-3-基)-2-((6-氯-2-甲基嘧啶-4-基)氨基)噻唑-5-羧酰胺(200mg,0.506mmol)、2-氨基乙-1-醇(155mg,2.30mmol)、N,N-二异丙基乙胺(26mg,0.20mmol)和1-丁醇(1.5mL,0.3M)。用EtOAc萃取,快速纯(在10CV上从100%DCM到80%DCM/3N NH
化合物C12:
(R)-2-((6-(4-(2-羟乙基)哌嗪-1-基)-2-甲基嘧啶-4-基)氨基)-N-(奎尼丁-3-基)噻唑-5-羧酰胺
步骤1按照GP A1进行,使用2-((6-氯-2-甲基嘧啶-4-基)氨基)噻唑-5-羧酸(500mg,1.85mmol)、(R)-奎尼丁-3-胺二盐酸盐(441mg,2.22mmol)、四甲基氯甲脒六氟磷酸酯(622mg,2.22mmol)、N,N-二异丙基乙胺(1.31g,10.2mmol,5.5eq.)和乙腈(10mL,0.2M)。从H
步骤2按照GP B进行,使用2-((6-氯-2-甲基嘧啶-4-基)氨基)-N-(奎尼丁-3-基)噻唑-5-羧酰胺(222mg,0.540mmol)、2-(哌嗪-1-基)乙-1-醇(553mg,4.25mmol)、N,N-二异丙基乙胺(44mg,0.34mmol)和1-丁醇(2.5mL,0.3M)。制备型TLC(DCM/MeOH 4:1,R
化合物C13:
N-(2-乙基-4-甲基吡啶-3-基)-2-((6-(4-(2-羟乙基)哌嗪-1-基)-2-甲基嘧啶-4-基)氨基)噻唑-5-羧酰胺
步骤1按照GP A1进行,使用2-((6-氯-2-甲基嘧啶-4-基)氨基)噻唑-5-羧酸(170mg,0.628mmol)、2-乙基-4-甲基吡啶-3-胺(103mg,0.754mmol)、四甲基氯甲脒六氟磷酸酯(211mg,0.754mmol),N,N-二异丙基乙胺(284mg,2.20mmol)和乙腈(5.0mL,0.1M)。用EtOAc萃取后,获得2-((6-氯-2-甲基嘧啶-4-基)氨基)-N-(2-乙基-4-甲基吡啶-3-基)噻唑-5-羧酰胺(90mg,0.23mmol),为绿色固体后,无需进一步纯化即可用于下一步。
步骤2按照GP B进行,使用2-((6-氯-2-甲基嘧啶-4-基)氨基)-N-(2-乙基-4-甲基吡啶-3-基)噻唑-5-羧酰胺(87mg,0.22mmol)、2-(哌嗪-1-基)乙-1-醇(150mg,1.10mmol)、N,N-二异丙基乙胺(12mg,0.089mmol)和1-丁醇(0.7mL,0.3M)。快速纯化后(10CV上100%DCM至75%DCM/MeOH),获得C13(图1C,48mg,0.099mmol,2个步骤的产率为16%),为灰白色固体。
化合物C1至C15、B3和化合物A8(达沙替尼)的特征如下表1A所示。
表A:激酶抑制剂的合成。
化合物D1至D10:
通过方案2与化合物C7类似地制备化合物D1至D10(图1D),不同之处在于在步骤4(GP A1)中使用了适用的胺,在步骤5(GP B)中使用了适用的哌嗪基衍生物。
由表B合成的化合物的特征如下表1B所示。
表1B:进一步的激酶抑制剂的合成。
*使用Bruker AV300机器在300MHz处确定。
基于达沙替尼(A8)的构效关系(SAR),本发明的其他激酶抑制剂:
合成本发明的另外的激酶抑制剂以举例说明式(I)的另一组不同的取代基(特别是在R
例如:(i)参照图8A(来自Lombardo et al 2004,J Med Chem 47:6658的表1),制备式(I)的化合物,其在R
实施例2:通过式(I)的激酶抑制剂和达沙替尼抑制SIK、Abl和Src激酶。
发明人令人惊讶地证明,式(I)的激酶抑制剂(例如C1至C12和B3,以及表B的化合物D1至D10)有效抑制SIK、ABL1和/或SRC激酶(表3A和表3A1),许多化合物以较低的一位数nM IC50抑制SRC,并以较低的两位数nM IC50抑制SIK(特别是SIK3和/或SIK2)和/或ABL1。类似地测试了化合物C13。
达沙替尼、C7和B3抑制ABL1的IC50约为1.5nM、5.1nM和1.6nM,而SRC的为1.5nM、1.5nM和1.5nM;每个分别用达沙替尼、C7和B3(表3A)。化合物C12还是SRC的强抑制剂(<100nM IC50),并且与ABL1相比对SRC具有令人惊讶的选择性。
简而言之,使用放射性蛋白质激酶测定法(
·25uL测定缓冲液(标准缓冲液/[γ-33P]-ATP)
·10uL ATP溶液(在水中)
·5uL的测试化合物(在10%DMSO中)
·20uL酶/底物混合物。
所有蛋白激酶的测定均包含70mM HEPES-NaOH pH7.5、3mM MgCl
每孔使用以下量的酶和适用的底物:
表2A:用于测试的蛋白激酶的测定参数。
*最大摩尔酶测定浓度,意味着酶制剂仅含有100%的活性酶
反应混合物在30℃下孵育60分钟。用50uL的2%(v/v)的H3PO4终止反应,吸出板并用200μL的0.9%(w/v)的NaCl洗涤两次。用微量滴定板闪烁计数器(Microbeta,Wallac)确定33Pi的掺入。所有测定均使用BeckmanCoulter/SAGIAN(TM)核心系统进行。
实施例3:式(I)的激酶抑制剂的改善的激酶选择性。
然而,更令人惊讶的是,发明人发现式(I)的激酶抑制剂(例如C1至C12和B3,以及表B的化合物D1至D10)似乎是与酪蛋白激酶抑制剂达沙替尼相比,具有选择性(尤其是SRC选择性)的蛋白酪氨酸激酶抑制剂(有利于抑制例如ABL1,特别是SRC激酶)。类似地测试了化合物C13。
特别是,尽管许多这些激酶抑制剂抑制了SIK激酶家族(SIK1、SIK2和SIK3)的每个(或一个或多个),一些即使在亚微摩尔浓度下,但是与达沙替尼相比,它们对这些蛋白-丝氨酸/苏氨酸激酶的抑制作用较弱(表3A和表3A1)。
特别地,举例来说,与达沙替尼相比,化合物C3、C8和C12是至少SIK1、SIK2和SIK3的活性更少的抑制剂,但仍是SRC和ABL1尤其是SRC的强抑制剂(IC50分别:<2nM,<25nM和<100nM)。
表3A:式(I)的激酶抑制剂的生物活性
表3A1:式(I)的进一步的激酶抑制剂的生物活性
如实施例2中所述测定化合物对蛋白丝氨酸/苏氨酸激酶SIK家族(SIK1、SIK2和SIK3)的IC50,并且B3和A8的抑制曲线如图2(A至E)所示,以及图2(F至J)中的C3和C12。例如(图2F至J),化合物C3和C12保留了对SRC的有效抑制活性(分别为IC50:<2nM和<100nM),但与达沙替尼相比(参见图2A至E,右栏)是针对ABL1的明显更少活性的抑制剂,尤其是对SIK1、SIK2和SIK3的活性较低。
此外,对一组320种野生型蛋白质(ProQinase“Kinase Profiler”;ProQinase,Freiburg,Germany)进行单点(1uM)抑制试验(一式两份)显示,式(I)的化合物比达沙替尼更具选择性(图3和4,表3AA)。类似地测试了式(I)的其他化合物(例如,化合物D1至D10中的一种或多种)。实际上,总的(以及式(I)的一个示例化合物)激酶抑制剂B3在数值上比达沙替尼(A8)更具选择性,其中B3的选择性得分为0.163,而达沙替尼为0.188。根据Karaman etal(2008;Nat Biotech 26:127),选择性得分是一种化合物浓度依赖性参数,描述了被抑制超过预定程度(例如,超过50%)的激酶部分,相对于特定项目的所有测试激酶。
表3AA:对于式(I)和达沙替尼(A8)的化合物在320种激酶中的选择性得分。
*TK:酪氨酸激酶;TKL:酪氨酸激酶样;CAMK:钙/钙调蛋白依赖性蛋白激酶。
计算残留浓度<50%时,测试浓度下化合物的选择性得分;即,抑制率>50%。使用以下公式计算特定浓度下特定化合物的选择性得分:
选择性得分=(数据点数<50%)/(数据点总数)
实际上,几乎所有测试过的式(I)化合物都显示出比达沙替尼显著提高的选择性得分(即,它们抑制的激酶数量减少了50%以上)。不仅在所有320种激酶中都显示出这种选择性,而且还通过激酶家族特异的选择性得分得以显示。并且,例如,几乎所有测试的式(I)化合物甚至在蛋白酪氨酸和类似蛋白酪氨酸的激酶家族中都比达沙替尼具有更高的选择性,并且所有(包括C7)对CAMK激酶家族都具有比达沙替尼更高的选择性。实际上,已显示化合物C9不会抑制任何CAMK激酶(即使SIK1、SIK2或SIK3)超过50%。
作为式(I)的一种特定激酶抑制剂的进一步实例,化合物B3,特别是,除了少数例外(例如,FLT3,在用1μM的化合物B3处理后仅显示32%的残留活性,但是在用1uM达沙替尼治疗后,其活性的80%仍保留),与两种化合物均同等抑制的激酶(例如ABL1和SRC)不同,这种式(I)的激酶抑制剂在大多数测试的这些激酶中抑制效力较弱(图3和4)。例如,WNK2不受B3抑制(在1μMB3时残留活性为100%),但是在用1μM达沙替尼治疗后,仅保留了62%的WNK2活性;同样,JAK1不受1μM B3的抑制,但用1μM达沙替尼治疗后仅保留41%的活性;用1μM B3处理后,几乎100%的RET活性仍然保留,而用1μM达沙替尼处理后,其活性的40%保留。同样,用1μM的达沙替尼处理丝氨酸/苏氨酸激酶MAP4K5显示残留活性仅为3%,而用1μM的B3处理则显示28%的残留活性。确实,许多激酶在用1μM达沙替尼抑制后具有不到20%的残留活性,但是在用1μM B3抑制时仍保留了近50%的活性。例如,当用1μM B3抑制时,B-RAF保留几乎50%的活性,而当使用1μM达沙替尼抑制时,仅保留15%的活性。
式(I)的激酶抑制剂对达沙替尼抑制的许多其他激酶和激酶家族成员显示出不同的抑制作用;例如,并且特别是通过式(I)的激酶抑制剂化合物B3抑制KIT和EphA/B亚家族的某些成员(表3B)。
表3B:某些激酶/激酶亚家族在式(I)的化合物和达沙替尼(A8)之间显示出不同的抑制作用。
*TK:酪氨酸激酶;STE:Yeast Sterile 7、Sterile 11、Sterile 20激酶的同系物
总体而言,1μM达沙替尼抑制23种激酶至残留活性低于1%,而1μM B3仅抑制13种激酶至残留活性低于1%。同样,在残留活性阈值<5%的情况下,达沙替尼抑制38种激酶,而1μM B3仅抑制30种激酶。而对于残留活性<10%的阈值,达沙替尼抑制43种激酶,而1μM B3仅抑制34种激酶。
表3C显示了在用一种化合物处理时保持大于50%的活性,而在用另一种化合物处理时显示出小于或等于50%的残留活性的激酶。化合物A8(达沙替尼)抑制9种激酶超过50%,其中当用式(I)的激酶抑制剂化合物B3处理时,这种激酶保留其活性的50%以上。相反,在所有测试的激酶中,式(I)的激酶抑制剂(B3)仅抑制了一种激酶(FLT3)大于50%,而达沙替尼也未抑制该激酶大于50%。
表3C:显示在式(I)的化合物B3和达沙替尼(A8)之间的差异抑制(残留活性为50%)的激酶。
*TK:酪氨酸激酶;STE:Yeast Sterile 7、Sterile 11Sterile 20激酶的同系物;TKL:酪氨酸激酶样;CMGC:包含CDK、MAPK、GSK3和CLK家族。
实际上,与达沙替尼(A8)相比,已证实式(I)的激酶抑制剂(例如C2、C4、C7、C8和C9和B3)显示出对一种或多种以上激酶的不同生物学抑制活性,当在生化测定中测试以确定IC 50时(表3D),相应的IC 50曲线示于图5A至D(类似地测试了式(I)的其他化合物)。的确,尽管化合物B3和A8都是(同时也是)LCK和KIT激酶的强抑制剂,但与达沙替尼(A8)相比,化合物B3抑制FLT3的作用远强于SYK(对于B3,IC50分别为6.9μM和26.0μM;对于A8,分别为25.7μM和4.8μM)。
因此,尽管B3(式(I)的激酶抑制剂)通常比激酶抑制剂A8(达沙替尼)具有更高的选择性,但B3被证明比化合物A8更有力地抑制FLT3(并反映在图4A中单个离点上,其代表FLT3)。
表3D:与达沙替尼(A8)相比,式(I)化合物对其他激酶的生物活性
与上述实施例2中所述的IC50分析类似,对每种化合物针对这些激酶的IC50进行了测试,不同之处在于每孔使用以下量的酶和适用底物(表3E):
表3E:其他测试的蛋白激酶的测定参数。
*最大摩尔酶测定浓度,意味着酶制剂仅含有100%的活性酶
实际上,一种优选的本发明化合物(C7)具有另一种激酶抑制特征(图3);表现出不同于化合物A8和化合物B3的特性。图9显示了一组320种野生型蛋白激酶(如上所述的ProQuinase“Kinase Profiler”,)的残留活性%(均值,重复1uM单点抑制测定的)的散点图,其中在每个Y轴上显示化合物C7的残留活性%和,分别在图9A和9B的X轴上显示化合物A8和B3的残留活性%。
令人惊讶的是,化合物C7在抑制多种激酶方面比化合物A8(达沙替尼)和B3分别显著更有效(分别见表3F.1和3F.2),例如,虚线框中包含的激酶在图9A和9B中标识。此外,化合物C7与化合物B3的显著区别在于C7在抑制FLT3方面的相对无活性(C7的残留活性为60%,而B3的残留活性为32%)。
表3F.1:某些激酶/激酶亚家族在化合物C7和达沙替尼(A8)之间显示出不同的抑制作用。
表3F.2:某些激酶/激酶亚家族在化合物C7和化合物B3之间显示出不同的抑制作用。
实际上,特别重要的是,化合物C7与化合物A8和B3相比对以下激酶的抑制作用差异模式(表3G)。事实证明,化合物C7比现有技术化合物A8(达沙替尼)对表3G中列出的许多关键激酶更有效,特别是作为TKL的此类激酶(例如,激酶的ACV家族,RAF1和BRAF和TBFB-R1),以及对CMGC激酶p38-α和-β。此外,化合物C7(式(I)的化合物作为这类激酶的抑制剂明显比化合物B3(也是式(I)的化合物)更有效。还应特别注意的是,化合物C7是NEK11的强效抑制剂(仅10%的残留活性),而这种激酶仅被达沙替尼中度抑制(超过50%的残留活性),而几乎不受化合物B3的抑制(几乎90%的残留活性)。
表3G:与达沙替尼(A8)和化合物B3相比,化合物C7表现出的不同抑制作用
IC50测试反映了这些(或其他)差异中的一个或多个,并证实与化合物A8(达沙替尼)[和/或与化合物B3]相比,式(I)的化合物C7显示出对许多上述激酶具有不同的抑制活性。例如,确定化合物C7对激酶TGFB-R1的IC50对C7为约490nM,对A8为880nM(International Centre for Kinase Profiling,Dundee)。
实施例4:式(I)的激酶抑制剂和达沙替尼对Abl1激酶突变体的抑制。
发明人证明了式(I)的激酶抑制剂(例如C、C4和C7和B3)也能够抑制临床相关的ABL1突变体,例如与CLL对伊马替尼(一种对CLL的护理标准)的耐药性有关的突变体(表4A)。例如,不仅式(I)的抑制剂是比伊马替尼更有力的ABL1野生型(wt)抑制剂(2.60μM对1,060nM),它们还是ABL所有这些相关突变体的有效抑制剂(T315I除外),并显示出可与A8媲美的IC50。对式(I)的其他化合物进行类似的测试。
表4A:Abl 1激酶突变体的抑制。
简而言之,如实施例1所述进行了ABL1放射蛋白激酶测定,不同的是[γ-33P]-ATP活性约为每孔7x10
表4B:ABL1突变蛋白激酶的测定参数。
*最大摩尔酶测定浓度,意味着酶制剂仅含有100%的活性酶
实施例5.1:式(I)的激酶抑制剂的改善的ADMET特性。
发明人证明,式(I)的激酶抑制剂(例如C2、C4、C8、C9和C12和B3)出人意料地在许多体外ADMET(吸收、分布、代谢、排泄、毒性)分析中表现出改善的药物样性质,包括溶解度、稳定性、血浆蛋白结合、CYP450抑制和hERG抑制分析(表5A、5B、5C和5D)。对式(I)的其他化合物进行了类似的测试。
对于口服药物,达沙替尼是一种难溶性化合物,测得的动力学溶解度甚至低于5μM。但是,激酶抑制剂B3显示了82μM的显著提高的溶解度。
在另一个实验中,对式(I)的化合物C2、C4、C7、C8、C9和C12进行了类似的测试,所有化合物(C7除外)显示了进一步提高的动力学溶解度,介于128至195μM之间(对于化合物C12)。
而且,如上所述,达沙替尼在人体内的半衰期非常短(总体平均终末半衰期仅为3-5小时;SPRYCEL完整处方信息第12.3节“药代动力学”)。这反映在人体(h)和小鼠(m)的活体微粒(LM)稳定性测定中,显示出非常短的半衰期。相比之下,激酶抑制剂B3在hLM和mLM分析中均显示出显著改善的稳定性,在孵育40分钟后,半衰期、固有清除率和化合物残留百分比显著提高,在hLM分析中,与达沙替尼相比,半衰期延长了近6倍,而在40分钟时10倍以上的药物剩余。
在一个单独的实验中,对化合物C2、C4、C7、C、C9和C12进行了类似的测试,所有化合物(C7除外)在人和小鼠肝微粒体中均显示出更高的稳定性,其中化合物C8、C9和C12的半衰期超过100分钟。
已证明达沙替尼和式(I)的激酶抑制剂(例如C2、C4、C8和C9和B3)显示出与人和鼠血浆蛋白均适度结合,其中与未结合的达沙替尼相比,某些式(I)的激酶抑制剂(例如C2、C8、C9和B3)在人血浆中的游离药物量(即未与血浆蛋白结合,并且可能可用于药理活性)显著增加(最多2至5倍),在鼠血浆中这种作用甚至更为明显。实际上,某些式(I)的化合物显示出非常低的血浆-蛋白质结合:化合物C12在人和鼠血浆中都保持超过90%的未结合。
因此,在某些情况下(例如C2、C8、C9和C12和B3),对于这些体外ADMET参数,式(I)的激酶抑制剂比达沙替尼具有高度改善的类药物特性(表5A)。类似地研究了其他式(I)化合物,例如化合物D1至D10中的一种或多种。
表5A:包括式(I)的激酶抑制剂的改善的溶解度和稳定性。
溶解度和稳定性测试是由Charles River Inc.在其UK Discovery站点(Cambridge,UK)根据适用的标准操作程序进行的(ADME-SOP-01用于溶解度;AMDE-SOP-100用于微粒体稳定性;AMDE-SOP-90用于血浆蛋白结合)。
已知达沙替尼会抑制某些细胞色素P450(CYP450)酶;包括参与其新陈代谢的那些。实际上,尽管达沙替尼主要通过细胞色素P450酶3A4(CYP3A4)在人体内代谢,但它也是CYP3A4的时间依赖性抑制剂。的确,如果患者同时服用强效CYP3A4抑制剂(见上文),则必须显著降低达沙替尼的剂量(例如,从每天100mg降至每天20mg)。但是,已证明激酶抑制剂B3不能显著抑制所测试的任何CYP450酶,重要的是B3并非已知也被达沙替尼抑制的CYP3A4或CYP2C8抑制剂(表5B)。
在相同、相似或类似的CYP450抑制测定中测试了式(I)的其他激酶抑制剂(例如C1至C13和/或D1至D10中的一个或多个)。
表5B:激酶抑制剂B3不抑制CYP450酶2C8和3A4。
*从NDA 21-986 SPRYCEL(达沙替尼)临床前评价第33页获得的值
CYP450抑制测定法(非针对A8)是由Charles River Inc在其UK Discovery站点(Cambridge,UK)根据适用的标准操作程序(ADME-SOP-97)进行的。达沙替尼的CYP450抑制值取自NDA 21-986的SPRYCEL(达沙替尼)的临床前综述的第33页。
达沙替尼的一种“警告和注意事项”(SPRYCEL的完整处方信息)是它有可能延长心脏心室复极化(QT间隔)。实际上,据其中报道:在临床试验中,多达1%的CML患者经历了QT延长,在258名服用达沙替尼的患者中,有5.8%的人报告了心脏不良反应,其中包括1.6%的患有心肌病、充血性心力衰竭、舒张功能不全、致命的心肌梗塞和左心室功能不全的患者。
在NDA过程中已经认识到达沙替尼的潜在心脏风险,其第3页总结了NDA 21-986的药理/毒性审查和评估:“基于体外hERG和兔Purkinje纤维测定,达沙替尼可能导致QT延长”,其中第31页报道达沙替尼在3、10和3uM时分别抑制hERG电流约6%、36%和77%,计算出的IC50为14.3uM。
形成鲜明对比的是,如表5C所示,激酶抑制剂B3本质上不显示hERG依赖性,产生的IC50值大于或等于测试的最高浓度(30uM),而在30uM时,hERG的抑制率仅为7.7%(+/-1.0%),相比之下据报道在30uM达沙替尼时对hERG的抑制率为76.8%(+/-4.5%)。
在相同、相似或类似的试验中测试了其他式(I)的激酶抑制剂(例如,C1至C13和/或D1至D10中的一个或多个)的hERG抑制。
表5C:激酶抑制剂B3不抑制hERFG。
*从NDA 21-986 SPRYCEL药理/毒性审查和评估第31页获得的值
hERG抑制试验(非针对A8)是由Charles River Inc.在其UK Discovery站点(Cambridge,UK)进行的。简而言之,使用Sophion Qube自动化电生理平台上的CharlesRiver
测定MDR1-MDCK有效流出比的分析表明,达沙替尼是MDR1泵的底物,并从表达该流出泵的细胞中活跃(有效流出比为18.3)。尽管无法确定B3的相对流出量,但由于在MDCK-MDR1细胞中A>B方向的检测极限,式(I)的化合物(有效流出率>9.6),有迹象表明,与达沙替尼相比,激酶抑制剂B3在该试验中显示出明显不同的参数(表5D)。
在相同、相似或类似的测定法中,测试了式(I)的其他激酶抑制剂(例如,C1至C13和/或D1至D10中的一个或多个)的MDR1-MDCK有效流出比。
表5D:在MDCK野生型和MDCK-MDR1细胞中的平均流出比。
MDR1-MDCK有效流出试验是由Charles River Inc在其UK Discovery站点(Cambridge,UK)根据适用的标准操作程序(ADME-SOP-56)进行的。
实施例5.2:本文公开的激酶抑制剂的药代动力学和耐受性研究。
发明人惊讶地发现,尽管与达沙替尼或化合物B3(也是式I化合物)相比,化合物C7(式I化合物)在体外ADMET方面显示出较差的体外ADMET性能,但在体内测试时,与化合物B3相比,化合物C7显示出显著改善的药代动力学性质(以及与达沙替尼有关的合适的药代动力学特征)。例如,表5A(实施例5.1)表明,在与人或小鼠肝微粒体体外测试时,化合物C7具有降低的代谢稳定性,并且在每种情况下与达沙替尼(以及与化合物B3相比)与人和小鼠血浆蛋白的结合更高。然而,当进行体内测试时,化合物C7表现出的药物代谢和药代动力学(DMPK)特性比结构上相关的化合物B3显著提高(表5.2A)。
发明人还惊讶地发现,与达沙替尼(A8)相比,化合物C7的合适药代动力学特性仍然不同,并且与使用达沙替尼的机会相比,这为给药该活性化合物C7提供了新的机会。例如,口服给药后,化合物C7显示出与达沙替尼相似的半衰期和生物利用度,但显示出与达沙替尼相比更高的清除率和约1/3的AUC(表5.2A)。
众所周知,剂量方案,尤其是与其他疗法的组合(其顺序和时机),是许多重要且成功的抗癌疗法的基础。因此,拥有一种具有与达沙替尼相似的激酶活性但又不同的药代动力学特征的化合物(例如C7),可以设计、研究和(临床)测试新的(可能)治疗有效的癌症治疗方案。
特别地,最近已显示达沙替尼可以作为CAR T细胞的药理开/关开关(Mestermannet al 2019,Sci Transl Med 11:eaau5907)。CAR T细胞作为单次“活体药物”治疗进行施用。它们能够在患者体内持续数年,并在(再)暴露于抗原后在体内进行顺序的扩增、收缩和再扩增,因此,CAR T细胞疗法已与大量的急性和慢性副作用相关,在高度专业化的癌症中心限制了临床利用,以使患者适应医学需求。与CAR T细胞疗法相关的最常见的急性毒性是细胞因子释放综合征(CRS),这是由CAR T细胞以及随后产生关键CRS细胞因子白介素6(IL-6)的先天免疫细胞释放炎性细胞因子触发的。梅斯特曼(Mestermann)显示,在体外和体内,使用达沙替尼治疗可中止细胞溶解活性、细胞因子产生和CAR T细胞增殖。达沙替尼的剂量可以进行滴定,以实现对CAR T细胞功能的部分或完全抑制。停用达沙替尼后,抑制作用迅速而完全逆转,CAR T细胞恢复了其抗肿瘤功能。作为与达沙替尼具有类似抑制特征但具有不同药效特性的激酶抑制剂,可以利用化合物C7实时控制CAR T细胞在“功能-开-关-开”序列中的活性,并具有与达沙替尼相比不同解析度的时机。通过进一步的(临床前和临床)测试,将确定化合物C7的哪种剂量对这种CAR T细胞的“功能转换”作用有效。并且该剂量可能与C7抑制单药疗法(以及免疫反应的相关修饰)中的肿瘤生长所需的剂量相差很大(例如,更低),如实施例8所示。
表5.2A:与达沙替尼和B3相比,化合物C7的DMPK特性。
在雄性CD1小鼠(Charles River Inc,UK Discovery站点)中,每管每次饲喂每种化合物一次后,研究了C7、B3和A8的药代动力学特征。将化合物在丙二醇:水1:1的混合物(v:v)中稀释。用LC-MS/MS分析C7、A8和B3的血浆水平(药物给药后的0,25h;0,5h;1h;1,5h;2h;4h;7h;24h取样),并使用非隔室分析和30mg/kg的名义剂量水平确定药代动力学参数。每个时间点分析三只动物,每只小鼠总共用于三次血浆采样。
类似地测试了本发明的其他激酶抑制剂(例如,来自表B、D1至D10的那些)的DMPK特性,并且可以将其与C、A8(达沙替尼)和B3的进行比较。例如,可以在给药后的某些时间点测定30mg/kg po(每os)给药剂量的小鼠后化合物的游离浓度(表5.2B)。
表5.2B:与达沙替尼和B3相比,筛选某些式(I)激酶抑制剂的PK特性。
ND=不可确定
在雌性C57Bl/6小鼠中连续7天通过强饲法对不同浓度(33和100mg/kg)的C7进行每天一次(QD)或每天两次(BID)给药,研究了化合物C7的最大耐受剂量(MTD)。将化合物C7稀释在4%(v/v)的DMSO、72%(v/v)的丙二醇+24%(v/v)的dd-水中。在雌性C57BL/6动物中,化合物C7的最高耐受BID为100mg/kg,可耐受7天。化合物C7是安全的,没有任何不耐受的迹象(姿势、发声、易于操作、流泪、色带、流涎、完整的皮毛/外层、饲养、唤醒、立毛、正常的运动、尾巴捏、腹泻)。与未治疗的对照动物相比,在所有给药组中均观察到体重的轻度降低,这是由第2天的血浆采样引起的(图10A和B)。通过LC-MS/MS测量血浆C7水平,结果显示血浆浓度呈剂量依赖性增加(图10C)。
实施例6:激酶抑制剂的基于体外细胞的抗癌和抗白血病功效。
在一种或多种基于细胞的试验中,测试了式(I)的激酶抑制剂(例如C1至C13或B3和/或D1至D10中的一种或多种)的抗癌活性,及其在这种测定法中将其活性与达沙替尼(A8)进行比较以确定其不同和/或优越性。
在一种方法中,使用
实际上,已经对化合物C7在代表大量血液癌症和细胞系的细胞组中测试了其抗癌活性(Oncolead,Karlsfeld Germany),发现该化合物能够抑制许多此类细胞系的生长。在约5个这样的细胞系上显示出亚纳摩尔的GI50活性,在另外10个这样的细胞系上显示出小于10nM的GI50活性。例如,在96小时后,化合物C7在WSU-NKL B细胞淋巴瘤细胞系的生长上显示出大约8nM的GI50(图15A),而在DIHH-2B细胞淋巴瘤细胞系的生长上显示出大约9nM的GI50(图15B)。类似地研究了式(I)的其他激酶抑制剂(例如,C1至C13或B 3和/或D1至D10中的一个或多个)。
在其他或替代方法中,测试一种或多种此类化合物的抗白血病活性,尤其是在由BCR-ABL或其突变体驱动的一种或多种细胞系中进行测试(例如Gibbons et al 2014,PNAS111:3550所述)。如先前所述(Donato et al 2004,Cancer Res 64:672),构建野生型(wt)和T315I突变体Bcr-Abl(C末端用eGFP标记)的基因构建体。wt或T351I单突变体pMX/eGFP-Bcr-Abl模板用于为BCR-ABL的其他或双突变体生成模板,这些模板也与多种白血病的TKI对药物治疗的耐药性相关(例如,T315I/V299L、T315I/F317L、T315I/F359V或表6A)中列出的其他突变)(通过定点诱变)(Stratagene)。诱变后,对携带突变体(或双重突变体)的4-kbXhoI/SgrAI序列进行测序,并将其亚克隆到pMX/eGFP-BcrAbl模板中(在XhoI/SgrAI位点),以确保在诱变过程中不引入其他突变。
表6A:*BCR-ABL突变与对TKI的耐药性和治疗选择有关。从报告使用细胞系模型得出的IC50值的研究中编译。
*取自Pophali&Patnik 2016,Canc J 22:40
将亲代Ba/F3细胞(鼠IL-3依赖的鼠pro B细胞系)维持在含有10%FBS和1ng/mL小鼠IL-3的RPMI培养基中。使用Amaxa Nucleofector系统用2.5μg质粒对2.5×10
类似地,可以测试式(I)的激酶抑制剂(例如C1至C13或B3和/或D1至D10中的一个或多个)或达沙替尼在携带KIT突变的白血病细胞模型中的作用。例如,可以将携带KIT wt或突变(例如与耐药相关的)的载体转染到转染的FDC-P1鼠骨髓细胞中,并研究化合物对FDC-P1/KIT(wt或突变)增殖和克隆活性的影响,例如基于Liu et al(2010,Cancer Cell17:333)所描述的细胞。
实施例7[预见的]:式(I)的激酶抑制剂和达沙替尼的体内抗白血病功效。
2006年欧洲、中东和非洲(EMEA)的人用药用产品委员会(CMHP)关于达沙替尼的科学讨论中描述了达沙替尼的体内药理活性以及用于证明其活性的实验。特别是,如下:
在SCID小鼠皮下生长的人CML异种移植模型中评估了达沙替尼的抑瘤活性。治愈被定义为在停止治疗后大于肿瘤体积加倍时间十倍的时间不存在可检测到的肿瘤。
使用“5天给药2天停止”治疗方案,每天一次口服10天后,一定剂量水平(8-50mg/kg)的达沙替尼在携带K562人CML肿瘤的小鼠中具有治愈作用。当肿瘤达到200至500mg时开始治疗。高剂量的伊马替尼未能产生比较反应。达沙替尼的最小有效剂量确定为2.5mg/kg。此外,静脉内注射达沙替尼在该模型中非常有效。当以50mg/kg/天的剂量治疗5天时,达沙替尼对患有大KU812肿瘤(肿瘤大至>1g)的小鼠具有治愈作用。
在从携带人类前列腺癌细胞(PC-3)的小鼠中收集的外周单核细胞(PBMC)中研究了SRC磷酸化,该小鼠接受了1、5、15或50mg/kg达沙替尼的治疗。达沙替尼治疗导致剂量依赖性抑制SRC磷酸化,并在15和50mg/kg给药后5小时内几乎完全抑制。给予5mg/kg导致SRC磷酸化的抑制率为44%,而1mg/kg几乎没有活性。根据AUC 0-24h值,5mg/kg和15mg/kg剂量组的动物:人血浆暴露比率分别为0.8和1.9。根据药代动力学数据,估计PBMC中抑制50%磷酸-SRC所需的达沙替尼血浆浓度为91nM。
达沙替尼治疗(10mg/kg/天)增加了颅内注射K562 CML细胞的小鼠的存活率,其存活功效优于伊马替尼(300mg/kg/天)中观察到的。在一项类似的研究中,观察到用10和30mg/kg/天的达沙替尼治疗后存活率增加,而200mg/kg/天的伊马替尼治疗不能抑制颅内肿瘤的生长。因此,在颅内CML的治疗中,达沙替尼似乎比伊马替尼具有治疗优势。
无论每天连续治疗10天,还是在给药方案中引入了短暂的治疗中断(5天给药和2停止),达沙替尼均对小鼠的人CML模型具有活性。此外,每天两次的给药方案产生的功效优于每天一次的给药方案。因此,在带有K562人CML肿瘤的小鼠中,与每天一次方案上给药2.5mg/kg达沙替尼的剂量组相比(抑制生长),在每天两次给药1.25mg/kg的剂量组中,观察到了优异的抗肿瘤活性(治愈)。
一种或多种式(I)的激酶抑制剂(例如C1至C13或B3和/或D1至D10)在一项或多项此类研究中进行了测试(例如剂量为1、1.25、2.5、5、10、25、30或50mg/Kg/天),以确定其与达沙替尼(A8)的区别和/或优越性;特别地,如此测试C2、C8、C9或C12或B3中的一个或多个。
另外或作为替代,在Puttini et al(2006,Cancer Res 66:11314)描述的动物模型中对一种或多种此类化合物进行了比较,简述如下:
五至七周大的CD1 nu/nu雌性小鼠购自Charles River Breeding Laboratories,并保持在标准实验室条件下。将人KU812Bcr-Abl+细胞悬浮于0.5mL PBS中的5000万个细胞中,并在每只动物的左胁经皮下注射该细胞。在另一组小鼠中,将表达Bcr-Abl WT或伊马替尼抗性点突变体(Y253F、E255K、D276G和T315I)的鼠Ba/F3 pro-B细胞悬浮于0.4mL PBS中的1000万个细胞中,并皮下注射至同系裸鼠。每周两次或三次监测肿瘤重量和体重。肿瘤重量通过公式肿瘤重量(mg)=(d2 D/2)计算,其中d和D分别是以毫米为单位测量的肿瘤的最短直径和最长直径。仅考虑荷瘤动物来计算肿瘤重量。达沙替尼(A8)或式(I)的激酶抑制剂(例如C2、C8、C9或C12或B3)从细胞输注后第二天开始通过口服管饲法给予(例如剂量为1.25、2.5、5、10、30或50mg/Kg/天),在肿瘤进入生长阶段时注射细胞后的第8天,或对患有可测量肿瘤的小鼠在第15天时给予。安慰剂治疗的动物仅使用媒介物的相同的治疗方案。
作为另一种附加或替代方法,在Fauvel et al(2013,Am J Can Sci 2:28)描述的动物模型中比较了一种或多种此类化合物,简述如下:
通过将Ba/F3 BCR-ABLT315I(1x10
实施例8:式(I)的激酶抑制剂和达沙替尼的体内抗癌(实体瘤)功效
在体内同系小鼠模型中研究了式(I)的激酶抑制剂(例如C1至C13或B3)和达沙替尼(A8)对实体瘤的抗癌活性,从而将鼠结肠直肠癌细胞MC38皮下植入C57Bl/6N小鼠的腹侧,并用受试化合物(例如C2、C8、C9或C12或B3或A8)处理,剂量范围为例如2.5mg/mL/天直至50、60或100mg/Kg/天*)。详细地,将雌性C57Bl/6N小鼠(4-6周大)植入1x10
表8A:示例性治疗组
*根据上一次体重测量;**po=经os,通过管饲口服
#至少持续3周,最长约5-8周。C2、C8、C9和C12的较低剂量(例如10、15或20mg/Kg)
可以根据其ADMET/PK特性进行调整。
**丙二醇:水1:1
每周两次用卡尺测量小鼠的体重和肿瘤体积(mm
此外,在首次治疗的第9天后,每组杀死5只小鼠,以分析肿瘤和血液样本中的各种免疫反应标记,例如下表8B所示的标记(以及使用Aqua Zombie(BioLegend))确定活/死细胞)。
简而言之,通过在小鼠尾巴上切开一个切口,将外周血样本从这些小鼠收集(然而,在处死前一天)到肝素涂层的试管中。用氯化铵-钾(ACK)裂解缓冲液(LifeTechnologies,Cat.A10492-01)处理血样后,将细胞用荧光染料偶联的mAb染色至一种或多种示例性免疫表型标记物(例如,表8B)。
为了对肿瘤进行免疫表型分析,在处死当天用手术刀将肿瘤切除,然后一分为二。对于一种或多种示例性免疫表型标记物,将肿瘤的一部分固定在4%多聚甲醛(PFA)中以进行免疫组化。对于IHC,将PFA固定的肿瘤组织嵌入石蜡块中,并切成4mm厚。固定在载玻片上和抗原回收步骤后,将切片用抗CD8抗体染色并用Mayer-苏木精复染。肿瘤浸润性CD8 T细胞的计数是从胶囊区域开始以50倍的放大倍数进行的,并向组织的核心计数3个视野。重复相同的过程3次。将所有计数相加,并计算中位数(如Hekim et al 2017,Can Imm Res 5:157中所述)。
将另一半肿瘤转移到装有RPMI 1640培养基的1.5mL试管中,然后使用微管沉淀杵手动匀浆。以300xg离心后,弃去上清液,将细胞重悬于等于肿瘤重量的RPMI培养基中。将肿瘤匀浆在PBS中以1:1稀释,并用荧光染料标记的抗体对一种或多种示例性免疫表型标记进行染色。对于所有Foxp3特异性染色(周围或肿瘤内),用抗CD4抗体标记细胞,然后按照制造商(eBioscience)的建议使用细胞内固定和通透试剂盒与抗Foxp3抗体一起孵育。
表8B:示例免疫表型标记。
发明人证实了与达沙替尼(A8)相比,激酶抑制剂C7在体内同基因小鼠模型中对实体瘤的抗癌活性以及该式(I)化合物的免疫肿瘤学作用。式(I)化合物(例如C7)的剂量和给药方案,根据其各自的SIK3激酶抑制和DMPK特性(参见前面的实施例)进行调整,以使它们的血浆游离药物浓度相对于其SIK3 IC50值与达沙替尼相当。如先前所述(Hekim等人2017)处理化合物A8(达沙替尼)。类似地研究了一种或多种其他式(I)的激酶抑制剂(例如,C1至C13或B3和/或D1至D10)。
每天两次用C7(100mg/kg)治疗显示出显著的肿瘤生长延缓(44%TGI),而每天一次C7治疗显示出与媒介物对照相比29%的肿瘤生长抑制作用(图11A)。相比之下,每天用A8(30mg/kg)进行治疗可抑制38%的肿瘤生长。每周至少两次对动物称重,直到完成研究。没有观察到与治疗有关的副作用和临床症状,例如体重减轻(图11B)。
此外,已证明化合物C7对存在于肿瘤微环境中的免疫细胞具有显著作用(图12)。每天两次用C7进行治疗,可诱导出显著的抗肿瘤免疫表型。细胞毒性T淋巴细胞(CD3+CD8+)与调节性T细胞(CD3+CD4+CD25+FoxP3+)的比率(图12A)显著增加,以及CD25+CD69+(图12B)和颗粒酶B(图12C)表达表示细胞毒性T淋巴细胞活化增加。此外,每天用C7和A8处理一次和两次,免疫抑制性M2巨噬细胞(CD206+MHC-II+)明显减少(图12D)。
体内同源小鼠模型如下进行:雌性C57Bl/6N小鼠(4-6周龄)植入5x10e5个MC38结直肠癌细胞(PBS中100ul)。将小鼠分为治疗组(表8C),以达到100mm
表8C:治疗组
*根据上一次体重测量;**p.o.=经os,通过管饲口服;#4%(v/v)DMSO,72%(v/v)丙二醇和24%(v/v)dd-水
在第18天的末次给药后两小时,通过流式细胞术分析了每组六只动物的至少100uL全血以及肿瘤组织中的CD4+和CD8+T细胞、Treg、粒细胞和单核细胞MDSC、M1和M2巨噬细胞以及NK细胞。采血后立即切除肿瘤并进行流式细胞术分析。分析了肿瘤和血液样品中的各种免疫反应标记物(表8D),以及使用了Zombie染料(BioLegend)来确定活/死细胞。用PMA/离子霉素/布雷菲德菌素A体外刺激T细胞后,检测到淋巴样细胞内的细胞内细胞因子。
表8D:免疫表型标记。
流式细胞术的样品制备方法如下:通过在环境温度下添加10倍体积的氯化铵钾(ACK)缓冲液处理全血样品,轻轻混合并在室温下孵育3-5分钟。温育后,立即加入10倍体积的冷PBS以终止裂解反应,并将细胞以400g沉淀5分钟,并再次在PBS中洗涤。使用gentleMACSTM方案“Tumor Dissociation Kit”,按照制造商的说明解离小鼠肿瘤样品。简要地,切除肿瘤,切成小块(2-4mm),置于酶缓冲液中,并在gentleMACS Dissociator上处理,在37℃下连续旋转孵育20分钟。样品通过70um细胞过滤器过滤,并在PBS/2.5%FBS缓冲液中漂洗两次以除去酶缓冲液。所有的单细胞悬浮液均以PBS中的~1x10e7个细胞/mL制备,并保存在冰上。用PMA/ionomycin/Brefeldin A.对T细胞标志物面板群体进行离体刺激样品。将100uL单细胞悬液添加到96孔板中,染色并用LSRFortessaTM(BD)分析并用FlowJo分析软件(Tree Star,Inc.;版本10.0.7r2)分析。
实施例9:通过式(I)的激酶抑制剂和达沙替尼使肿瘤细胞对体外TNF攻击的敏感化
发明人研究了式(I)的激酶抑制剂(例如C2、C8、C9或C12或C4或C7或B3;或者特别是C7;和/或D1至D10中的一个或多个)以及用于比较的达沙替尼(A8)对于TNF在体外对肿瘤细胞的杀灭(凋亡/细胞毒性)作用的作用,并确定了式(I)的激酶抑制剂对修饰的PANC1或M579-A2细胞系对TNF的攻击的敏感性。类似地测试了其他式(I)化合物,例如C 5。
用激酶抑制剂(例如C7或B3)进行的治疗(以一种或多种浓度测试,例如约1nM至10,000nM,例如约10nM、25nM、50nM、100nM、150nM、500nM、1,000nM、2,500nM和/或5,000nM,在DMSO中)用于研究PANC-1-luc或M579-A2-luc肿瘤细胞对重组人TNF(rHuTNF;R&DSystems)的细胞毒性作用的敏感化,以及这种效应的发作速度。PANC1-luc是表达HLA-A2.1+荧光素酶的胰腺腺癌细胞(PDAC)肿瘤细胞系,而M579-A2-luc是表达HLA-A2.1+荧光素酶的黑色素瘤细胞系。
rHuTNF处理对所指示的PANC-1-luc细胞活力的剂量反应显示为在用各自浓度的rHuTNF治疗后,分别用相应的测试化合物(例如B3或A8)或对照(例如DMSO)处理的肿瘤细胞的相对(细胞毒性/生存力)图(图6),以及Y轴是使用基于萤光素酶的杀灭测定法进行测量的+TNF(细胞毒性)/相对于没有TNF(生存力)的标准化RLU(作为细胞毒性的量度):将细胞、测试化合物然后将rHuTNF(10ng/mL)在37℃,5%CO2下孵育24h,在培养后去除上清液,并用萤光素酶细胞裂解裂解剩余的PANC-1-Luc细胞试剂(0.3%Triton-X在水中)10分钟。裂解后,添加萤光素酶测定缓冲液,并立即使用TECAN-Spark微孔板读数器测量萤光素酶强度。
为确定细胞死亡的发作速度,将肿瘤细胞掺入YOYO-1染料进行预先核标记,并按上述方法用抑制剂和TNF处理。通过实时活细胞显微镜和IncuCyte Zoom(EssenBioScience)评估肿瘤死亡动力学,该图显示刺激6h后图像的YOYO-1+细胞/孔面积(μm
PANC1-luc细胞系的构建如下:从American Type Cell Culture(ATCC)获得PANC-1细胞。使用TransIT-LT1(Mirus)作为转染试剂,用pEGFP-Luc质粒转染肿瘤细胞。用1mg/mL的G418/Geneticin选择转染的细胞,并且在选择后14天,使用例如BD FACSARIA II细胞分选仪分选EGFP+细胞。
肿瘤免疫肿瘤作用被证明是由SIK家族成员,特别是SIK3介导的。使用上述基于荧光素酶的肿瘤细胞生存力读数,在TNF(10ng/mL)和浓度约10至100nM(圆圈)的pan-SIK和ABL1&SRC抑制剂(化合物B1)存在下,与单独存在抑制剂的情况相比(正方形),肿瘤细胞显示出更高的细胞毒性(图7A)。相反,与之相比,化合物B8是有效的ABL1和SRC抑制剂,但对SIK家族成员(特别是SIK3)的抑制活性较低,与单独使用抑制剂(正方形)相比,与TNF(圆圈)组合在这样的浓度范围内未能表现出M579 A2细胞的细胞毒性(图7B)。实际上,与单独使用抑制剂(正方形)相比,与TNF组合的化合物B4(圆圈)在该浓度范围内也没有表现出M579 A2细胞的细胞毒性(图7C),尽管化合物B4不仅是ABL1和SRC的有效抑制剂也是SIK1和SIK2的强抑制剂,而只是SIK3的弱抑制剂。
总之,这些数据暗示SIK家族成员(特别是SIK3)是抗肿瘤免疫肿瘤作用的介质,而不是其他激酶ABL1或SRC。
化合物B1、B4和B8如PCT/EP2018/060172中所述,包括其合成,并在表9A中显示。
表9A:化合物B1、B4和B8
其他式(I)的激酶抑制剂(例如,C1至C13中的一种或多种,例如C7,和/或表B中的一种或多种化合物,D1至D10)用于研究肿瘤细胞对重组人TNF的细胞毒性作用的敏感化,包括在使用MC38鼠肿瘤细胞系的TNF致敏(100ng/mL)细胞活力测定中的作用(表9B)。实际上,化合物C7使MC38肿瘤细胞对TNF的细胞毒性作用敏感(图13B)。
表9B:由式(I)化合物引起的MC38细胞对TNF介导的杀伤的敏感化
++=<100nM;+=<200nM;/=>200nM
证明式(I)的化合物使MC38细胞对TNF介导的杀伤敏感的测定法如下进行。根据制造商的程序,使用CellTiter-Glo(CTG)发光细胞活力测定(Promega,Madison,USA)测量MC38肿瘤细胞的细胞活力。简而言之,将1x10
还显示化合物C7以剂量依赖性方式抑制TNF诱导的NFkB活性(图14A),并且还抑制NFkB活性的关键介质HDAC4的磷酸化(图14B)。研究了本文公开的其他化合物,例如达沙替尼(A8)和/或选自化合物C2至C12和/或D1至D10的化合物的类似活性(表9C)。。
表9C:式(I)化合物对NFKB活性和HDAC4磷酸化的抑制
+=显著抑制;++=强烈且显著抑制
使用NFKB依赖的萤光素酶活性测量了PANC-1细胞中TNF诱导的NFKB活性。在NFKB启动子的控制下产生PANC-1克隆以表达萤光素酶。将NFKB报告基因PANC-1细胞(每孔1,250个)接种在384孔板中24小时。然后,在37℃和5%CO
使用Meso Scale Discovery(MSD)测定法测量PANC-1细胞中的HDAC4磷酸化水平。将PANC-1细胞(6x10
实施例10[预见的]:式(I)的激酶抑制剂的体内免疫肿瘤活性
为了研究TNF诱导疗法(例如抗PD1抗体,例如鼠类抗PD1克隆:RMP1-14,BioLegend)与式(I)的激酶抑制剂(例如C2、C7、C8、C9或C12或B3;和/或化合物D1至D10中的一种或多种)以及作为比较的达沙替尼(A8)(剂量范围为例如2.5mg/Km/天,直至50、60或100mg/Kg/天*)的协同效应,进行实施例9中所述的另一种体内同基因小鼠模型,但使用治疗组处理适用的组合(和对照),例如治疗组可包括例如以下下表10A所述的。
表10A:示例性治疗组
*根据上一次体重测量;
**po=经os,通过管饲口服,ip=腹膜内
#化合物给药至少3周,最多5至8周。抗PD-1和媒介治疗持续时间。C2、C7、C8、C9和C12的较低剂量(例如10、15或20mg/Kg)或较高剂量(例如50、75或100mg/Kg)可以根据其ADMET/PK特性进行调整。
**丙二醇:水1:1
如实施例8中所述:(i)每周两次用卡尺测量小鼠的体重和肿瘤体积(mm
实施例11[预见的]:用于口服给药的式(I)的激酶抑制剂的单位剂量形式的制剂和制备。
简短地如下制备本发明的药物组合物的囊片单位剂型。
首先,通过将一定量的C7或C8(或B3)与一种或多种赋形剂一起干法制粒来制备包含式(I)的激酶抑制剂(例如,C7,C8或B3)的压片混合物。压片混合物中的赋形剂实例可以包括粘合剂,例如乳糖(一水合物)、微晶纤维素和/或羟丙基纤维素,以及任选地与崩解剂(例如淀粉)一起。该混合物还可以包含润滑剂,例如硬脂酸镁。替代地,通过湿法制粒然后干燥来制备压片混合物。
第二,使用旋转式压片机,将混合物从上方填充到合适形状的模具中,并通过将上冲头降低到模具中而将其压缩至约5%至20%的孔隙率。压缩可以分一个阶段或两个阶段进行(主压缩,以及可选的预压缩或夯实),对于规模化生产,压缩会迅速发生(例如,每片500ms以内)。拉起上冲头,将其从模具中取出(减压),然后将囊片从模具中弹出。
第三,使用自动涂布机涂布囊片。该涂层可以包含羟丙甲纤维素、二氧化钛、聚乙二醇和纯净水。
压片混合物包含一定量的激酶抑制剂(如使用的C7、C8或B3),以使每个囊片均包含治疗有效量的C7或C8(或B3),并且不同剂量的囊片可以可以帮助给予正确的C7或C8(或B3)总剂量。例如,每个囊片可以包含约20mg、50mg或70mg的C7或C8(或B3,如适用),或可以包含少于这些量的诸如约5mg、10mg或40mg的C7或C8(或B3,如适用的)。
- 杂环激酶抑制剂及其用途
- 作为蛋白激酶抑制剂的杂环类化合物及其制备方法和用途