含有多取代基的联稀酰胺化合物及其制备方法
文献发布时间:2023-06-19 12:21:13
技术领域
本发明属于有机材料合成技术领域,涉及一种含有多取代基的联稀酰胺化合物及其制备方法。
背景技术
联烯是一类非常重要的含有丙二烯官能团的有机中间体,由于联稀反应位点多,可广泛应用于成很多天然化合物和药物分子合成。多取代联烯类化合物的合成一直以来都是有机化学领域非常重要的研究方向。虽然目前构建联烯化合物的方法有很多,但是大多是芳香基或杂原子取代类型的联烯化合物,这类联烯化合物合成存在的问题以及缺陷;需贵金属催化剂制备多取代联稀化合物,成本相对较高以及贵金属残留较高,对环境不友好。
而多取代联烯酰胺类化合物,因为其结构特性,具有非常高的原子经济型和步骤经济型,合成的多取代联稀酰胺类化合物可能用在药物及材料领域,但是对于构建多取代联烯酰胺类化合物的文献到目前为止仍未见报道。
发明内容
针对上述存在的问题,本发明提供一种含有多取代基的联稀酰胺化合物及其制备方法,原料易得、成本低;合成条件温和、合成产率高、具有较大的工业化潜力。
为了实现上述目的,本发明采用的技术方案是:
一种含有多取代基的联稀酰胺化合物的结构式如式I所示:
其中:n=0、1或2;Ar为苯基、对甲基苯基和对氯基苯基中的任意一种。
一种含有多取代基的联稀酰胺化合物的制备方法包括以下步骤:
1)制备α,β不饱和酰胺衍生物,备用;所述α,β不饱和酰胺衍生物的结构式如式II所示:
其中:n=0、1或2;
2)向步骤1)制备的α,β不饱和酰胺衍生物中,依次加入苯乙炔类化合物、铜盐、无机碱以及2,2’-联吡啶,混合后,在氮气氛围下加入反应溶剂,在温度20~60℃,反应10~24小时;
3)反应结束后,加入加硅胶,经蒸发、浓缩、层析得到白色固体,即为如式I所示的联稀酰胺化合物。
进一步的,所述步骤2)中,α,β不饱和酰胺衍生物与苯乙炔类化合物的摩尔比为1:1~3;铜盐与α,β不饱和酰胺衍生物的摩尔比为1:10~20;α,β不饱和酰胺衍生物与无机碱的摩尔比为1:1~3;所述铜盐与2,2’-联吡啶的摩尔比例为1:1~2;;所述α,β不饱和酰胺衍生物与反应溶剂的摩尔体积比为1mmol:2~3mL。
进一步的,所述苯乙炔类化合物为苯乙炔、对甲基苯乙炔或对氯苯乙炔;所述反应溶剂为四氢呋喃、乙醚、甲苯、正己烷或二氯甲烷。
进一步的,所述铜盐为碘化亚铜、溴化亚铜、氯化亚铜、醋酸亚铜、噻吩-2-甲酸亚铜CuTc、硫氰酸亚铜中的任意一种。
进一步的,所述无机碱为氢氧化钠、氢氧化钾、碳酸钠、碳酸钾、碳酸铯、乙醇钠、甲醇钠、叔丁醇钠、叔丁醇钾中的任意一种。
进一步的,所述步骤1)中,α,β不饱和酰胺衍生物的制备过程是:
1.1)取α,β-不饱和环状酰胺类底物,用氮气交换空气多次后,继续向其中加入有机溶剂,并在冰浴中搅拌15min,得到反应体系A;
1.2)将正丁基锂缓慢滴加至反应体系A中,在冰浴条件下继续搅拌1.5小时,得到反应体系B;
1.3)取N-氟代双苯磺酰胺NFSI溶解于有机溶液中得到混合液,再将混合液逐滴加入至步骤2)的反应体系B中,在冰浴中恢复室温搅拌10h后,得到反应体系C;
1.4)反应体系C经盐酸水溶液淬灭反应后,采用二氯甲烷萃取后,得到的有机层依次经洗涤、干燥、过滤、蒸发、浓缩、层析、纯化得到黄色油状液体,即为α,β不饱和酰胺衍生物。
进一步的,所述步骤1.1)中,所述α,β-不饱和环状酰胺类底物与有机溶剂的质量体积比为1g:15~50mL;所述α,β-不饱和环状酰胺类底物的结构式为
进一步的,所述步骤1.2)中,所述正丁基锂与α,β-不饱和环状酰胺类底物的体积质量比为2.0~3.0mL:1g。
进一步的,所述步骤1.3)中,所述N-氟代双苯磺酰胺与α,β-不饱和环状酰胺类底物的质量比为1.5~5:1;所述N-氟代双苯磺酰胺和有机溶液的质量体积比为1g:15~30mL;所述有机溶液为四氢呋喃、乙醚、乙二醇二甲醚或甲基叔丁基醚。
本发明的有益效果是:
1、本发明以铜盐、无机碱和2,2’-联吡啶为催化剂,以不同结构的α,β不饱和酰胺衍生物和苯乙炔类化合物为原料,高产率的合成多取代基的联烯酰胺衍生物,合成产率高,同时合成的联烯酰胺衍生物是很多天然产物和药物分子的核心骨架,本发明为多取代联烯酰胺衍生物的合成提供了一个新思路。
2、本发明提供的制备方法,原料易得、催化剂价格低廉,反应条件温和,底物适用范围广,产率较好,合成中无金属残留,对环境友好;具有较大的工业化潜力。
附图说明
图1为II-a的
图2为II-a的
图3为II-a的
图4为II-b的
图5为II-b的
图6为II-b的
图7为II-c的
图8为II-c的
图9为II-c的
图10为I-a的
图11为I-a的
图12为I-b的
图13为I-b的
图14为I-c的1H核磁共振谱图;
图15为I-c的
图16为I-d的
图17为I-d的
图18为I-e的
图19为I-e的
具体实施方式
现结合附图以及实施例对本发明做详细的说明。
本发明使用铜盐、无机碱和联吡啶为催化剂,实现α,β不饱和酰胺类衍生物(结构式如式II所示)与苯乙炔类化合物的炔基化反应,从而异构化生成多取代联烯酰胺类衍生物(结构式如式I所示)。
本发明中,式I和式II中,n=0、1、2中的任意一种;式I中,Ar为苯基、对甲基苯基和对氯基苯基中的任意一种。
本发明中式II的结构包括II-a、II-b或II-c,具体结构式分别如下:
本发明中,式II是由α,β-不饱和环状酰胺类底物制备而成的。α,β-不饱和环状酰胺类底物的结构是为1a、1b或1c,分别制备得到II-a、II-b或II-c。
本发明多取代联烯酰胺类衍生物的制备方法,包括以下步骤:
1)采用不同的α,β-不饱和环状酰胺类底物制备α,β不饱和酰胺类衍生物,备用;具体制备过程包括:
1.1)向三口瓶中加入α,β-不饱和环状酰胺类底物后,用氮气交换空气3次,加入有机溶剂,在冰浴中搅拌15分钟,得到反应体系A;
1.2)随后将正丁基锂缓慢低价至反应体系A中,反应在冰浴条件下继续搅拌1.5小时,得到反应体系B;
1.3)将N-氟代双苯磺酰胺溶解在有机溶液中,逐滴加入至反应体系B中,得到反应体系C;
1.4)随后将反应体系C放在冰浴中恢复室温搅拌10小时后,用1M盐酸水溶液淬灭反应并转移到分离漏斗中,加入二氯甲烷萃取三次,随后有机层用饱和NaHCO
本发明中,α,β-不饱和环状酰胺类底物与有机溶剂的质量体积比为1g:15~50mL;正丁基锂与α,β-不饱和环状酰胺类底物的体积质量比为2.0~3.0mL:1g;N-氟代双苯磺酰胺和有机溶液的质量体积比为1g:15~30mL;N-氟代双苯磺酰胺与α,β-不饱和环状酰胺类底物的质量比为1.5~5:1;
2)然后α,β不饱和酰胺类衍生物与苯乙炔类化合物制备得到多取代联烯酰胺类衍生物。
将α,β不饱和酰胺类衍生物、苯乙炔、铜盐、无机碱以及2,2’-联吡啶依次加入反应管中,用氮气交换空气3次,并在氮气氛围下加入反应溶剂,在室温下反应24小时;
本发明中,α,β不饱和酰胺衍生物与苯乙炔类化合物的摩尔比为1:1~3;铜盐与α,β不饱和酰胺衍生物的摩尔比为1:10~20;α,β不饱和酰胺衍生物与无机碱的摩尔比为1:1~3;铜盐与2,2’-联吡啶的摩尔比例为1:1~2;;α,β不饱和酰胺衍生物与反应溶剂的摩尔体积比为1mmol:2~3mL;
3)TCL检测反应结束后,直接加硅胶,旋转蒸发浓缩后,通过硅胶柱层析(洗脱剂为石油醚:乙酸乙酯=20/1,v/v)得到白色固体,即为多取代联烯酰胺类化合物。
本发明中,苯乙炔类化合物为苯乙炔、对甲基苯乙炔或对氯苯乙炔。
本发明中,反应溶剂为四氢呋喃、乙醚、甲苯、正己烷或二氯甲烷。
本发明中,铜盐为碘化亚铜、溴化亚铜、氯化亚铜、醋酸亚铜、噻吩-2-甲酸亚铜CuTc、硫氰酸亚铜中的任意一种。
本发明中,无机碱为氢氧化钠、氢氧化钾、碳酸钠、碳酸钾、碳酸铯、乙醇钠、甲醇钠、叔丁醇钠、叔丁醇钾中的任意一种。
本发明中,有机溶剂为二氯甲烷、三氯甲烷、无水四氢呋喃、1,2-二氯乙烷、乙腈、甲苯、1,4-二氧六环中的任意一种。
本发明中,有机溶液为四氢呋喃、乙醚、乙二醇二甲醚或甲基叔丁基醚。
经过上述可以得到本发明在制备时,原料α,β不饱和酰胺类衍生物、反应溶剂、铜盐、有机溶剂和有机溶液的不同选择,以及各物料之间的配比,使得目标物的制备可以有多种实施路线。部分实施例参见表1所示。
表1为本发明提供的9组制备试验实例
现通过1-5组对本发明提供的多取代联烯酰胺类衍生物制备方法进行详细的说明。
实施例1
本实施例中,选用结构为II-a的α,β不饱和酰胺类衍生物与苯乙炔合成多取代联烯酰胺类化合物I-a。
本实施例提供的多取代联烯酰胺类化合物I-a的制备过程是:
1)制备式II-a的α,β不饱和酰胺类衍生物
本实施例采用α,β-不饱和环状酰胺类底物1a制备结构为II-a的α,β不饱和酰胺类衍生物,式II-a的制备路线为:
本实施例提供的制备步骤是:
1.1)向100mL三口瓶中加入1.95g酰胺底物1a后,用氮气交换空气3次,加入30mL有机溶剂(无水四氢呋喃),在冰浴中搅拌15分钟,得到反应体系A;
1.2)随后将4.6mL正丁基锂缓慢低价至反应体系A中,反应在冰浴条件下继续搅拌1.5小时,得到反应体系B;
1.3)将3.87gN-氟代双苯磺酰胺溶解在20mL有机溶液(四氢呋喃)中,逐滴加入至反应体系B中,得到反应体系C;
1.4)随后将反应体系C放在冰浴中恢复室温搅拌10小时后,用1M盐酸水溶液淬灭反应并转移到分离漏斗中,加入二氯甲烷萃取三次,随后有机层用饱和NaHCO
本实施例制备的结构式如II-a的α,β不饱和酰胺类衍生物,进行核磁共振测试,结果参见图1~3:
2)结构式为II-a的α,β不饱和酰胺类衍生物与苯乙炔合成多取代联烯酰胺类化合物I-a
本实施例中,I-a的制备技术路线为:
本实施例中,I-a的制备过程包括:
将0.2mmol的α,β不饱和酰胺类衍生物II-a(质量42.6mg)、0.4mmol苯乙炔(质量40.8mg)、0.02mmol铜盐(碘化铜)、0.2mmol碳酸铯(质量为65.2mg)以及0.02mmol 2,2’-联吡啶(质量3.12g)依次加入25mL的反应管中,用氮气交换空气3次,并在氮气氛围下加入0.5mL反应溶剂(四氢呋喃),在温度20℃下反应24小时;
3)TCL检测反应结束后,直接加硅胶,旋转蒸发浓缩后,通过硅胶柱层析(洗脱剂为石油醚:乙酸乙酯=20/1,v/v)得到白色固体,即为多取代联烯酰胺类化合物,即为I-a,其结构式为
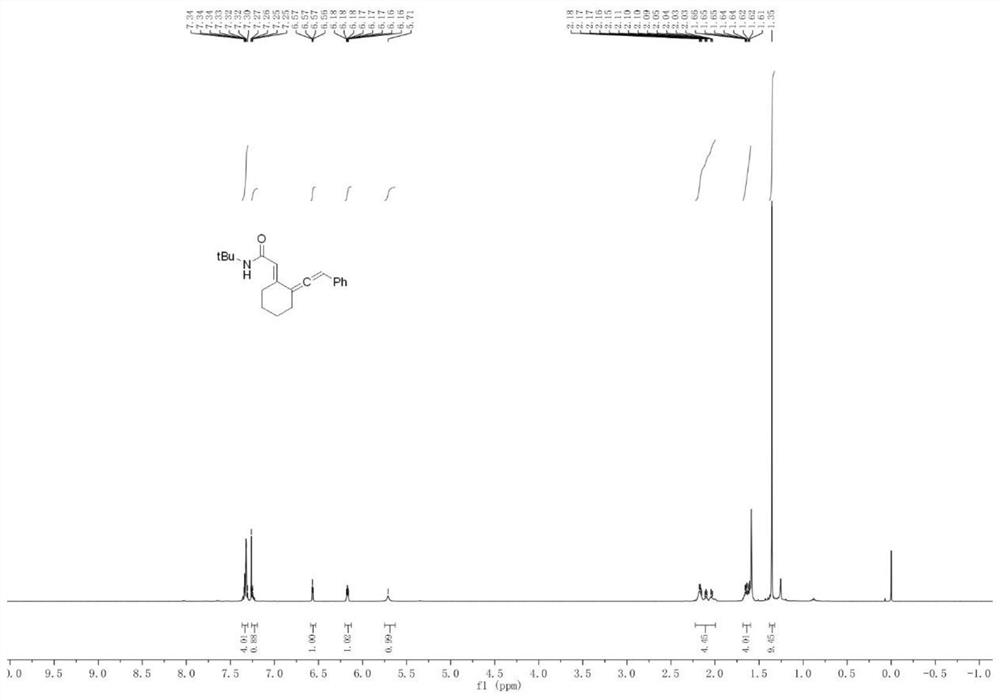
本实施例制备的多取代联烯酰胺类化合物I-a,进行核磁共振测试,结果参见图10和图11。
从图10和11可知:
实施例2
本实施例选用结构为II-a的α,β不饱和酰胺类衍生物与对甲基苯乙炔合成多取代联烯酰胺类化合物I-b。
本实施例提供的制备过程包括以下步骤:
1)制备式II-a的α,β不饱和酰胺类衍生物
本实施中,II-a的α,β不饱和酰胺类衍生物的制备与实施例1的制备方法相同;具体物料和用量选择参见表1中第2组所示;
2)结构式为II-a的α,β不饱和酰胺类衍生物与对甲基苯乙炔合成多取代联烯酰胺类化合物I-b
本实施中,I-b的制备技术路线如下:
本实施中,I-b的制备步骤包括:
2.1)将0.25mmol的α,β不饱和酰胺类衍生物II-a、0.25mmol对甲基苯乙炔、0.017mmol铜盐(碘化铜)、0.5mmol碳酸铯(质量为163mg)以及0.034mmol的2,2’-联吡啶依次加入25mL的反应管中,用氮气交换空气3次,并在氮气氛围下加入0.75mL反应溶剂(选择乙醚),在温度45℃下反应16小时;
3)TCL检测反应结束后,直接加硅胶,旋转蒸发浓缩后,通过硅胶柱层析(洗脱剂为石油醚:乙酸乙酯=20/1,v/v)得到白色固体;即为多取代联烯酰胺类化合物,记为I-b,其结构式为
本实施例制备的多取代联烯酰胺类化合物I-b,进行核磁共振测试,结果参见图12和图13。
从图12和13所示:
实施例3
本实施例选用结构为II-a的α,β不饱和酰胺类衍生物与对氯苯乙炔合成多取代联烯酰胺类化合物I-b,具体制备方法包括以下步骤:
1)制备式II-a的α,β不饱和酰胺类衍生物
本实施中,II-a的α,β不饱和酰胺类衍生物的制备与实施例1的制备方法相同,具体物料和用量选择参见表1中第3组所示;
2)结构式为II-a的α,β不饱和酰胺类衍生物与对氯苯乙炔合成多取代联烯酰胺类化合物I-c
本实施例中,I-c的制备技术路线为:
本实施例中,I-c的制备过程包括:
将0.12mmol的α,β不饱和酰胺类衍生物II-a、0.36mmol对氯苯乙炔、0.006mmol铜盐(碘化铜)、0.36mmol碳酸铯(质量为117.3mg)以及0.009mmol的2,2’-联吡啶依次加入25mL的反应管中,用氮气交换空气3次,并在氮气氛围下加入0.9mL反应溶剂(甲苯),在温度60℃下反应10小时;
3)TCL检测反应结束后,直接加硅胶,旋转蒸发浓缩后,通过硅胶柱层析(洗脱剂为石油醚:乙酸乙酯=20/1,v/v)得到白色固体,即为多取代联烯酰胺类化合物,记为I-c,其结构式为
本实施例制备的多取代联烯酰胺类化合物I-c,进行核磁共振测试,结果参见图14和图15。
从图14和15可知:
实施例4
本实施例中,选用结构为II-b的α,β不饱和酰胺类衍生物与苯乙炔合成多取代联烯酰胺类化合物I-d,制备过程包括以下步骤:
1)制备式II-b的α,β不饱和酰胺类衍生物
本实施例通过α,β-不饱和环状酰胺类底物1b制备结构式II-b的α,β不饱和酰胺类衍生物
本实施例中,II-b制备技术路线如下:
本实施例中,II-b制备过程是:
1.1)向100mL三口瓶中加入1.81g酰胺底物1b后,用氮气交换空气3次,加入30mL有机溶剂(1,2-二氯乙烷),在冰浴中搅拌15分钟,得到反应体系A;
1.2)随后将4.6mL正丁基锂缓慢低价至反应体系A中,反应在冰浴条件下继续搅拌1.5小时,得到反应体系B;
1.3)将3.87gN-氟代双苯磺酰胺溶解在20mL有机溶液(甲基叔丁基醚)中,逐滴加入至反应体系B中,得到反应体系C;
1.4)随后将反应体系C放在冰浴中恢复室温搅拌10小时后,用1M盐酸水溶液淬灭反应并转移到分离漏斗中,加入二氯甲烷萃取三次,随后有机层用饱和NaHCO
本实施例制备的结构式如II-b的α,β不饱和酰胺类衍生物,进行核磁共振测试,结果参见图4-6。
从图4-6可知:
2)结构式为II-b的α,β不饱和酰胺类衍生物与苯乙炔合成多取代联烯酰胺类化合物I-d
本实施例中,I-d的制备技术路线是:
本实施例中,I-d的制备过程是:
将0.2mmol的α,β不饱和酰胺类衍生物II-b(质量39.8mg)、0.4mmol苯乙炔(质量40.8mg)、0.02mmol铜盐(碘化铜)、0.2mmol碳酸铯(质量为65.2mg)以及0.02mmol的2,2’-联吡啶(质量3.12g)依次加入25mL的反应管中,用氮气交换空气3次,并在氮气氛围下加入0.5mL反应溶剂(选取正己烷),在20℃反应24小时;
3)TCL检测反应结束后,直接加硅胶,旋转蒸发浓缩后,通过硅胶柱层析(洗脱剂为石油醚:乙酸乙酯=20/1,v/v)得到白色固体,即为多取代联烯酰胺类化合物,记为I-d,其结构式为
本实施例制备的多取代联烯酰胺类化合物I-d,进行核磁共振测试,结果参见图16和图17。
从图16和17可知:
实施例5
本实施例中,选用结构为II-c的α,β不饱和酰胺类衍生物与苯乙炔合成多取代联烯酰胺类化合物I-e,制备过程包括以下步骤:
1)制备结构式II-c的α,β不饱和酰胺类衍生物
本实施例通过α,β-不饱和环状酰胺类底物1c制备结构式II-c的α,β不饱和酰胺类衍生物
本实施例中,II-c的制备技术路线如下:
本实施例中,,II-c具体的制备步骤是:
1.1)向100mL三口瓶中加入2.0g酰胺底物1c后,用氮气交换空气3次,加入30mL有机溶剂(乙腈),在冰浴中搅拌15分钟,得到反应体系A;
1.2)随后将4.6mL正丁基锂缓慢低价至反应体系A中,反应在冰浴条件下继续搅拌1.5小时,得到反应体系B;
1.3)将3.87gN-氟代双苯磺酰胺溶解在20mL有机溶液(甲基叔丁基醚)中,逐滴加入至反应体系B中,得到反应体系C;
1.4)随后将反应体系C放在冰浴中恢复室温搅拌10小时后,用1M盐酸水溶液淬灭反应并转移到分离漏斗中,加入二氯甲烷萃取三次,随后有机层用饱和NaHCO
本实施例制备的结构式如II-c的α,β不饱和酰胺类衍生物,进行核磁共振测试,结果参见图7-9。
从图7-9可知:
2)结构式为II-c的α,β不饱和酰胺类衍生物与苯乙炔合成多取代联烯酰胺类化合物I-e
本实施例中,I-e的制备技术路线为:
本实施例中,I-e的制备过程是:
将0.2mmol的α,β不饱和酰胺类衍生物II-c(质量45.4mg),0.4mmol苯乙炔(质量40.8mg),0.02mmol铜盐(碘化铜),0.2mmol碳酸铯(质量为65.2mg),0.02mmol的2,2’-联吡啶(质量3.12g)依次加入25mL的反应管中,用氮气交换空气3次,并在氮气氛围下加入0.5mL反应溶剂(二氯甲烷),在60℃下反应12小时;
3)TCL检测反应结束后,直接加硅胶,旋转蒸发浓缩后,通过硅胶柱层析(洗脱剂为石油醚:乙酸乙酯=20/1,v/v)得到白色固体,即为多取代联烯酰胺类化合物,记为I-e,其结构式为
本实施例制备的多取代联烯酰胺类化合物I-e,进行核磁共振测试,结果参见图18和图19。
从图18和19可知:
- 含有多取代基的联稀酰胺化合物及其制备方法
- 含有共轭联烯酰胺结构的化合物、其制备方法、药物组合物和用途