一种1,2,3-噻二唑衍生物的合成方法
文献发布时间:2023-06-19 12:22:51
技术领域
本发明属于化合物制备技术领域,尤其涉及一种1,2,3-噻二唑衍生物的合成方法。
背景技术
目前,1,2,3-噻二唑衍生物是一种及其重要的杂环化合物,在功能材料、医药和农药领域具有广泛的应用。((a)Amirhamzeh,M.Vosoughi,A.Shafiee andM.Amini,Med.Chem.Res.,2013,22,1212.(b)I.Cikotiene,E.Kazlauskas,J.Matuliene,V.Michailoviene,J.Torresan,J.Jachno and D.Matulis,Bioorg.Med.Chem.Lett.,2009,19,1089;(c)Q.Zheng,N.Mi,Z.Fan,X.Zuo,H.Zhang,H.Wang and Z.Yang,J.Agric.FoodChem.,2010,58,7846;(d)S.M.S.Atta,D.S.Farrag,A.M.K.Sweed andA.H.Abdel-Rahman,Eur.J.Med.Chem.,2010,45,4920;(e)M.Wu,Q.Sun,C.Yang,D.Chen,J.Ding,Y.Chen,L.Linand Y.Xie,Bioorg.Med.Chem.Lett.,2007,17,869;(f)H.Dai,S.Ge,G.Li,J.Chen,Y.Shi,L.Ye andY.Ling,Bioorg.Med.Chem.Lett.,2016,26,4504;(g)P.Zhan,X.Liu,Y.Cao,Y.Wang,C.Pannecouque and E.De Clercq,Bioorg.Med.Chem.Lett.,2008,18,5368;(h)Q.Du,W.Zhu,Z.Zhao,X.Qian and Y.Xu,J.Agric.Food Chem.,2012,60,346;(i)Y.Xu,Z.Zhao,X.Qian,Z.Qian,W.Tian and J.Zhong,J.Agric.Food Chem.,2006,54,8793;(j)Z.-H.Wang,Y.-Z.Guo,J.Zhang,L.Ma,H.-B.Song and Z.-J.Fan,J.Agric.Food Chem.,2010,58,2715;(k)X.Zuo,N.Mi,Z.Fan,Q.Zheng,H.Zhang,H.Wang and Z.Yang,J.Agric.Food Chem.,2010,58,2755.)。
截止目前,已报道的1,2,3-噻二唑衍生物的合成方法主要有两种途径:(1)在电催化下,N-对甲苯磺酰腙苯乙酮与硫单质构建1,2,3-噻二唑及其衍生物(S.-K.Mo,Q.-H.Teng,Y.-M.Pan and H.-T.Adv.Synth.Catal.,2019,361,1756);(2)在碘和路易斯酸,氯化亚铜的共同作用下,N-对甲苯磺酰腙苯乙酮与硫氰酸钾在加热条件下构建1,2,3-噻二唑及其衍生物(C.Wang,X.Geng,P.Zhao,Y.Zhou,Y.-D.Wu,Y.-F.Cui andA.-X.Wu,Chem.Commun.,2019,55,8134.)。以上方法普遍存在需要高温条件,路易斯酸做添加剂,溶剂不绿色,底物范围不广的缺点。
通过上述分析,现有技术存在的问题及缺陷为:已报道的1,2,3-噻二唑衍生物的合成方法存在需要高温条件,路易斯酸做添加剂,溶剂不绿色,底物范围不广的缺点。
解决以上问题及缺陷的难度为:首先,在常温的条件下进行反应,这对该反应底物在该温度中的溶解程度有着一定的考验;另外,无需加多余的酸碱,即可进行得到较为不错产率的目标产物;最后,需要寻找一种溶剂相比之前已有反应中所用的更为绿色环保的,并且保证反应在该溶剂中能如期进行并且得到较高的产率。
解决以上问题及缺陷的意义为:无需加入多余的过渡金属以及酸碱类化合物,在无金属条件下,以N-对甲苯磺酰腙苯乙酮为原料,廉价的碘单质参与介导,过硫酸钾作为氧化剂,无水乙醇作为溶剂,室温下高效合成1,2,3-噻二唑衍生物,这一合成方法与已报道的1,2,3-噻二唑衍生物的制备方法相比较,绿色环保、无需惰性气体保护和加热、底物适用范围广、官能团兼容性好、原料易得、操作简便。
发明内容
针对现有技术存在的问题,本发明提供了一种1,2,3-噻二唑衍生物的合成方法。
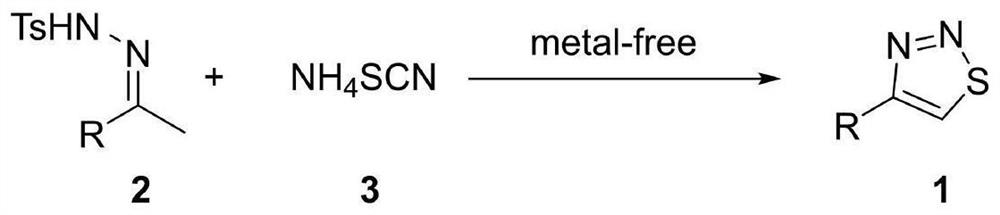
本发明是这样实现的,一种1,2,3-噻二唑衍生物,所述1,2,3-噻二唑衍生物的结构式为:
取代基R为氢或R为卤素F、卤素Cl、卤素Br、卤素I或是烷基、烷氧基、苯基、硝基、噻吩基、呋喃基、萘基、(E)-苯乙烯基、氟代烷基、氟代烷氧基中的一种或两种;取代基的个数为1~2个。
本发明的另一目的在于提供一种如权利要求1所述1,2,3-噻二唑衍生物的1,2,3-噻二唑衍生物的合成方法,所述1,2,3-噻二唑衍生物的合成方法为:以N-对甲苯磺酰腙苯乙酮衍生物2为原料,在无金属条件下,与硫氰酸铵进行[4+1]环化反应,生成1,2,3-噻二唑衍生物1。
进一步,所述1,2,3-噻二唑衍生物的合成方法包括以下步骤:
步骤一,在反应试管中,首先加入0.25mmol的N-对甲苯磺酰腙苯乙酮或N-对甲苯磺酰腙苯乙酮衍生物2与0.25mmol(19mg)硫氰酸铵3;
步骤二,向试管中加入氧化剂:0.25mmol(68mg)过硫酸钾;
步骤三,进行反应温度、反应时间的设定,在试管中加入3ml的无水乙醇溶剂后放入油浴中进行反应;
步骤四,反应结束后进行产物分离:首先在混合溶液中加入硫代硫酸钠除去混合液中多余的碘,加入30ml的水之后分别向水中加入三次10ml的乙酸乙酯对有机物进行萃取,在所得萃取液中加入无水硫酸镁除去溶液中多余的水分,通过旋转蒸发仪将溶液旋蒸干后,用柱层析的方法将产物分离,展开剂比例为:乙酸乙酯:石油醚(8:1),最终得到1,2,3-噻二唑衍生物1。
进一步,所述N-对甲苯磺酰腙苯乙酮或N-对甲苯磺酰腙苯乙酮衍生物2与硫氰酸铵3的摩尔比为1:1~1:3;所述摩尔比优选为1:1。
进一步,所述添加剂为碘化钾、碘化铵、碘化亚铜、四丁基碘化铵、二乙酰基碘苯和碘中的一种;所述添加剂优选为碘。
进一步,所述氧化剂为过硫酸钾、过硫酸铵、叔丁基过氧化氢中的一种;所述氧化剂优选为过硫酸钾。
进一步,所述溶剂为无水乙醇、二甲基亚砜、N,N-二甲基甲酰胺、N,N-二甲基乙酰胺乙腈中的一种;所述溶剂优选为无水乙醇。
进一步,所述反应时间为1~12小时;所述反应时间优选为10~12小时。
进一步,所述反应温度为室温至100℃;所述反应温度优选为室温。
进一步,所述1,2,3-噻二唑衍生物的合成方法的反应通式为:
取代基R为氢或R为卤素F、卤素Cl、卤素Br、卤素I或是烷基、烷氧基、苯基、硝基、噻吩基、呋喃基、萘基、(E)-苯乙烯基、氟代烷基、氟代烷氧基中的一种或两种以上;取代基的个数为1~2个。
结合上述的所有技术方案,本发明所具备的优点及积极效果为:本发明提供的经碘单质介导的无金属制备1,2,3-噻二唑衍生物的方法。在无金属条件下,以N-对甲苯磺酰腙苯乙酮为原料,廉价的碘单质参与介导,过硫酸钾作为氧化剂,无水乙醇作为溶剂,室温下高效合成了1,2,3-噻二唑衍生物。本合成尚未被报道,与已报道的1,2,3-噻二唑衍生物的制备方法相比较,本发明绿色环保、无需惰性气体保护和加热、底物适用范围广、官能团兼容性好、原料易得、操作简便。
本发明在无金属条件下,以N-对甲苯磺酰腙苯乙酮与硫氰酸铵为原料,廉价的碘单质为添加剂,高效合成了1,2,3-噻二唑。反应操作简便,环境友好,室温进行,无需金属催化剂。本发明在国家自然基金(22061040)和新疆维吾尔自治区自然科学基金(2020D01C024)的资助下完成。
本发明在无金属条件下有机反应,使用环境友好的无水乙醇为溶剂。反应在常温常压下进行,不需要额外加碱和惰性气体保护,操作及后处理简便。以廉价的碘单质为添加剂,避免了过渡金属的使用。原料简单易得。
本发明经碘单质介导的无金属制备1,2,3-噻二唑衍生物的方法。在无金属条件下,以N-对甲苯磺酰腙苯乙酮为原料,廉价的碘单质参与介导,过硫酸钾作为氧化剂,无水乙醇作为溶剂,室温下进行了[4+1]环化反应构建了1,2,3-噻二唑衍生物。本发明绿色环保、无需惰性气体保护和加热、底物适用范围广、官能团兼容性好、原料易得、操作简便。
附图说明
为了更清楚地说明本申请实施例的技术方案,下面将对本申请实施例中所需要使用的附图做简单的介绍,显而易见地,下面所描述的附图仅仅是本申请的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下还可以根据这些附图获得其他的附图。
图1是本发明实施例提供的1,2,3-噻二唑衍生物的合成方法流程图。
图2是本发明实施例提供的4-苯基-1,2,3-噻二唑的
图3是本发明实施例提供的4-苯基-1,2,3-噻二唑的
图4是本发明实施例提供的4-(4-甲基-苯基)-1,2,3-噻二唑的
图5是本发明实施例提供的4-(4-甲基-苯基)-1,2,3-噻二唑的
图6是本发明实施例提供的4-(2-甲基-苯基)-1,2,3-噻二唑的
图7是本发明实施例提供的4-(2-甲基-苯基)-1,2,3-噻二唑的
图8是本发明实施例提供的4-(3,4-二甲基-苯基)-1,2,3-噻二唑的
图9是本发明实施例提供的4-(3,4-二甲基-苯基)-1,2,3-噻二唑的
图10是本发明实施例提供的4-(2,4-二甲基-苯基)-1,2,3-噻二唑的
图11是本发明实施例提供的4-(2,4-二甲基-苯基)-1,2,3-噻二唑的
图12是本发明实施例提供的4-(4-叔丁基-苯基)-1,2,3-噻二唑的
图13是本发明实施例提供的4-(4-叔丁基-苯基)-1,2,3-噻二唑的
图14是本发明实施例提供的4-(4-异丙基-苯基)-1,2,3-噻二唑的
图15是本发明实施例提供的4-(4-异丙基-苯基)-1,2,3-噻二唑的
图16是本发明实施例提供的4-(2-甲氧基-苯基)-1,2,3-噻二唑的
图17是本发明实施例提供的4-(2-甲氧基-苯基)-1,2,3-噻二唑的
图18是本发明实施例提供的4-(2-羟基-苯基)-1,2,3-噻二唑的
图19是本发明实施例提供的4-(2-羟基-苯基)-1,2,3-噻二唑的
图20是本发明实施例提供的4-(4-N,N-二甲基-苯基)-1,2,3-噻二唑的
图21是本发明实施例提供的4-(4-N,N-二甲基-苯基)-1,2,3-噻二唑的
图22是本发明实施例提供的4-([1,1'-联苯]-4-基)-1,2,3-噻二唑的
图23是本发明实施例提供的4-([1,1'-联苯]-4-基)-1,2,3-噻二唑的
图24是本发明实施例提供的4-(苯并[d][1,3]二氧杂-5-基)-1,2,3-噻二唑的
图25是本发明实施例提供的4-(苯并[d][1,3]二氧杂-5-基)-1,2,3-噻二唑的
图26是本发明实施例提供的4-(4-三氟甲基-苯基)-1,2,3-噻二唑的
图27是本发明实施例提供的4-(4-三氟甲基-苯基)-1,2,3-噻二唑的
图28是本发明实施例提供的4-(4-三氟甲氧基-苯基)-1,2,3-噻二唑的
图29是本发明实施例提供的4-(4-三氟甲氧基-苯基)-1,2,3-噻二唑的
图30是本发明实施例提供的4-(4-氰基-苯基)-1,2,3-噻二唑的
图31是本发明实施例提供的4-(4-氰基-苯基)-1,2,3-噻二唑的
图32是本发明实施例提供的4-(4-氯基-苯基)-1,2,3-噻二唑的
图33是本发明实施例提供的4-(4-氯基-苯基)-1,2,3-噻二唑的
图34是本发明实施例提供的4-(3,4-二氯基-苯基)-1,2,3-噻二唑的
图35是本发明实施例提供的4-(3,4-二氯基-苯基)-1,2,3-噻二唑的
图36是本发明实施例提供的4-(4-氟基-苯基)-1,2,3-噻二唑的
图37是本发明实施例提供的4-(4-氟基-苯基)-1,2,3-噻二唑的
图38是本发明实施例提供的4-(3-溴基-苯基)-1,2,3-噻二唑的
图39是本发明实施例提供的4-(3-溴基-苯基)-1,2,3-噻二唑的
图40是本发明实施例提供的4-(4-溴基-苯基)-1,2,3-噻二唑的
图41是本发明实施例提供的4-(4-溴基-苯基)-1,2,3-噻二唑的
图42是本发明实施例提供的4-(4-碘基-苯基)-1,2,3-噻二唑的
图43是本发明实施例提供的4-(4-碘基-苯基)-1,2,3-噻二唑的
图44是本发明实施例提供的4-(噻吩-2-基)-1,2,3-噻二唑的
图45是本发明实施例提供的4-(噻吩-2-基)-1,2,3-噻二唑的
图46是本发明实施例提供的4-(呋喃-2-基)-1,2,3-噻二唑的
图47是本发明实施例提供的4-(呋喃-2-基)-1,2,3-噻二唑的
图48是本发明实施例提供的4-(萘-1-基)-1,2,3-噻二唑的
图49是本发明实施例提供的4-(萘-1-基)-1,2,3-噻二唑的
图50是本发明实施例提供的(E)-4-苯乙烯基-1,2,3-噻二唑的
图51是本发明实施例提供的(E)-4-苯乙烯基-1,2,3-噻二唑的
图52是本发明实施例提供的4-(4-(苯基乙炔基)苯基)-1,2,3-噻二唑的
图53是本发明实施例提供的4-(4-(苯基乙炔基)苯基)-1,2,3-噻二唑的
图54是本发明实施例提供的4-([1,1'-联苯])-4-基-1,2,3-噻二唑的
图55是本发明实施例提供的4-([1,1'-联苯])-4-基-1,2,3-噻二唑的
图56是本发明实施例提供的1,2,3-噻二唑衍生物合成路线图。
图57是本发明实施例提供的1,2,3-噻二唑衍生物合成路线示意图。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
针对现有技术存在的问题,本发明提供了一种1,2,3-噻二唑衍生物的合成方法,下面结合附图对本发明作详细的描述。
本发明实施例提供的1,2,3-噻二唑衍生物的结构式为:
取代基R为氢或R为卤素F、卤素Cl、卤素Br、卤素I或是烷基、烷氧基、苯基、硝基、噻吩基、呋喃基、萘基、(E)-苯乙烯基、氟代烷基、氟代烷氧基中的一种或两种以上;取代基的个数为1~2个。
本发明实施例提供的1,2,3-噻二唑衍生物的合成方法为:以N-对甲苯磺酰腙苯乙酮衍生物2为原料,在无金属条件下,与硫氰酸铵进行[4+1]环化反应,生成1,2,3-噻二唑衍生物1。基于先前已有的合成方法,我们提出在反应中不另外加入任何酸碱,无需对该反应进行加热,并且所用溶剂相对绿色环保的条件下高效的合成一种1,2,3-噻二唑类化合物。
如图1所示,本发明实施例提供的1,2,3-噻二唑衍生物的合成方法包括以下步骤:
S101,在反应试管中,首先加入0.25mmol的N-对甲苯磺酰腙苯乙酮或N-对甲苯磺酰腙苯乙酮衍生物2与0.25mmol(19mg)硫氰酸铵3;
S102,向试管中加入氧化剂:0.25mmol(68mg)过硫酸钾;
S103,进行反应温度、反应时间的设定,在试管中加入3ml的无水乙醇溶剂后放入油浴中进行反应;
S104,反应时间到12小时时停止反应,之后进行产物分离:首先在混合溶液中加入硫代硫酸钠除去混合液中多余的碘,加入30ml的水之后分别向水中加入三次10ml的乙酸乙酯对有机物进行萃取,在所得萃取液中加入无水硫酸镁除去溶液中多余的水分,通过旋转蒸发仪将溶液旋蒸干后,用柱层析的方法将产物分离,展开剂比例为:乙酸乙酯:石油醚(8:1),最终得到1,2,3-噻二唑衍生物1。
本发明实施例提供的N-对甲苯磺酰腙苯乙酮或N-对甲苯磺酰腙苯乙酮衍生物2与硫氰酸铵3的摩尔比为1:1~1:3;所述摩尔比优选为1:1。
本发明实施例提供的添加剂为碘化钾、碘化铵、碘化亚铜、四丁基碘化铵、二乙酰基碘苯和碘中的一种;所述添加剂优选为碘。
本发明实施例提供的氧化剂为过硫酸钾、过硫酸铵、叔丁基过氧化氢中的一种;所述氧化剂优选为过硫酸钾。
本发明实施例提供的溶剂为无水乙醇、二甲基亚砜、N,N-二甲基甲酰胺、N,N-二甲基乙酰胺乙腈中的一种;所述溶剂优选为无水乙醇。
本发明实施例提供的反应时间为1~12小时;所述反应时间优选为10~12小时。
本发明实施例提供的反应温度为室温至100℃;所述反应温度优选为室温。
本发明实施例提供的1,2,3-噻二唑衍生物的合成方法的反应通式为:
取代基R为氢或R为卤素F、卤素Cl、卤素Br、卤素I或是烷基、烷氧基、苯基、硝基、噻吩基、呋喃基、萘基、(E)-苯乙烯基、氟代烷基、氟代烷氧基中的一种或两种以上;取代基的个数为1~2个。
下面结合实施例对本发明的技术方案作进一步的描述。
实施例1
本发明在常温、空气氛围下,往10mL带有搅拌子的石英反应管中依次加入N-对甲苯磺酰腙苯乙酮2a(72.0mg,0.25mmol),硫氰酸铵3(19mg,0.25mmol),碘(63.5mg,0.25mmol),过硫酸钾(67.6mg,0.25mmol)和2mL无水乙醇,搅拌反应12小时;反应结束后,先向混合反应物中滴加1~2滴饱和的硫代硫酸钠溶液,之后用乙酸乙酯进行萃取(3×10mL),合并有机层,用无水硫酸钠干燥,过滤、减压下除去挥发组份,然后用硅胶柱层析分离(洗脱液为石油醚(60-90℃馏份)/乙酸乙酯,v/v=8:1),得到白色固体目标产物1a(35.3mg,产率87%);目标产物通过核磁共振谱和高分辨质谱得到确认。
实施例2
本发明反应步骤与操作条件同实施例1,与实施例1不同之处在于,反应中加入碘化钾替代碘单质;停止反应,经同上述相同后处理得到目标产物1a(25.5mg,产率63%);说明碘化钾为碘盐时,不能以最佳收率得到目标产物。
实施例3
本发明反应步骤与操作条件同实施例1,与实施例1不同之处在于,反应中加入碘化铵替代碘单质;停止反应,经同上述相同后处理得到目标产物1a(32.8mg,产率81%),说明碘化铵为碘盐时,不能以最佳收率得到目标产物。
实施例4
本发明反应步骤与操作条件同实施例1,与实施例1不同之处在于,反应中加入碘化亚铜替代碘单质;停止反应,经同上述相同后处理得到目标产物1a(29.2mg,产率72%),说明碘化亚铜为碘盐时,不能以最佳收率得到目标产物。
实施例5
本发明反应步骤与操作条件同实施例1,与实施例1不同之处在于,反应中加入四丁基碘化铵替代碘单质;停止反应,经同上述相同后处理得到目标产物1a(31.6mg,产率78%),说明四丁基碘化铵为碘盐时最佳收率得到目标产物。
实施例6
本发明反应步骤与操作条件同实施例1,与实施例1不同之处在于,反应中加入二乙酰基碘苯替代碘单质;停止反应,经同上述相同后处理得到目标产物1a(28.4mg,产率70%),说明二乙酰基碘苯为碘盐时最佳收率得到目标产物。
实施例7
本发明反应步骤与操作条件同实施例1,与实施例1不同之处在于,将氧化剂过硫酸钾更换为过硫酸铵;停止反应,经同上述相同后处理得到目标产物1a(29.9mg,产率74%),说明过硫酸铵为氧化剂时,不能以最佳收率得到目标产物。
实施例8
本发明应步骤与操作条件同实施例1,与实施例1不同之处在于,将氧化剂过硫酸钾更换为过氧化氢;停止反应,经同上述相同后处理得到目标产物1a(21.5mg,产率53%),说明过氧化氢为氧化剂时,不能以最佳收率得到目标产物。
实施例9
本发明反应步骤与操作条件同实施例1,与实施例1不同之处在于,将氧化剂过硫酸钾更换为叔丁基过氧化氢;停止反应,经同上述相同后处理得到目标产物1a(23.1mg,产率57%),说明叔丁基过氧化氢为氧化剂时,不能以最佳收率得到目标产物。
实施例10
本发明反应步骤与操作条件同实施例1,与实施例1不同之处在于,反应中加入N,N-二甲基甲酰胺替代无水乙醇;停止反应,经同上述相同后处理得到目标产物1a(30.8mg,产率76%),说明以二甲基亚砜为溶剂,不能以最佳收率得到目标产物。
实施例11
本发明反应步骤与操作条件同实施例1,与实施例1不同之处在于,反应中加入二甲基亚砜替代无水乙醇;停止反应,经同上述相同后处理得到目标产物1a(31.6mg,产率78%),说明以二甲基亚砜为溶剂,不能以最佳收率得到目标产物。
实施例12
本发明反应步骤与操作条件同实施例1,与实施例1不同之处在于,反应中加入N,N-二甲基乙酰胺替代无水乙醇;停止反应,经同上述相同后处理得到目标产物1a(25.1mg,产率62%),说明以N,N-二甲基乙酰胺为溶剂,不能以最佳收率得到目标产物。
实施例13
本发明反应步骤与操作同实施例1,与实施例1不同之处在于,反应中加入乙腈替代无水乙醇;停止反应,经同上述相同后处理得到目标产物1a(19.0mg,产率47%),说明以乙腈为溶剂,不能以最佳收率得到目标产物。
实施例14
本发明反应步骤与操作同实施例1,与实施例1不同之处在于,反应中不加入过硫酸钾;停止反应,经同上述相同后处理得到目标产物1a(14.6mg,产率36%),说明过硫酸钾在实验中起到重要的氧化作用,不加过硫酸钾时抑制了反应的发生。
实施例15
本发明反应步骤与操作同实施例1,与实施例1不同之处在于,反应中不加入硫氰酸铵3;停止反应,经同上述相同后处理未得到目标产物1a,说明过硫酸钾中的硫原子并没有代替硫氰酸铵作为硫源参与反应。
实施例16
本发明反应步骤与操作同实施例1,与实施例1不同之处在于,将反应时间延长到16小时;停止反应,经同上述相同后处理得到目标产物1a(32.4mg,产率80%),说明延长反应时间并不能提高产物收率。
实施例17
本发明反应步骤与操作同实施例1,不同之处在于,反应中加入的N-对甲苯磺酰腙苯乙酮衍生物是对甲基N-对甲苯磺酰腙苯乙酮2b(44mg,0.25mmol);停止反应,经后处理得到白色固体目标产物1b(34.8mg,产率85%),目标产物通过核磁共振谱测定得到确认。
实施例18
本发明反应步骤与操作同实施例1,不同之处在于,反应中加入的N-对甲苯磺酰腙苯乙酮衍生物是邻甲基N-对甲苯磺酰腙苯乙酮2c(44mg,0.25mmol);停止反应,经后处理得到黄色液体目标产物1c(29.5mg,产率72%),目标产物通过核磁共振谱测定得到确认。
实施例19
本发明反应步骤与操作同实施例1,不同之处在于,反应中加入的N-对甲苯磺酰腙苯乙酮衍生物是3,4-二甲基N-对甲苯磺酰腙苯乙酮2d(47.5mg,0.25mmol);停止反应,经后处理得到棕色油状目标产物1d(34.2mg,产率72%),目标产物通过核磁共振谱测定和高分辨质谱得到确认。
实施例20
本发明反应步骤与操作同实施例1,不同之处在于,反应中加入的N-对甲苯磺酰腙苯乙酮衍生物是2,4-二甲基N-对甲苯磺酰腙苯乙酮2e(47.5mg,0.25mmol);停止反应,经后处理得到黄色油状目标产物1e(35.6mg,产率75%),目标产物通过核磁共振谱和高分辨质谱测定得到确认。
实施例21
本发明反应步骤与操作同实施例1,不同之处在于,反应中加入的N-对甲苯磺酰腙苯乙酮衍生物是对叔丁基N-对甲苯磺酰腙苯乙酮2f(54.5mg,0.25mmol);停止反应,经后处理得到白色固体目标产物1f(39.7mg,产率73%),目标产物通过核磁共振谱测定得到确认。
实施例22
本发明反应步骤与操作同实施例1,不同之处在于,反应中加入的N-对甲苯磺酰腙苯乙酮衍生物是异丙基N-对甲苯磺酰腙苯乙酮2g(51.0mg,0.25mmol);停止反应,经后处理得到白色固体目标产物1g(40.0mg,产率78%),目标产物通过核磁共振谱和高分辨质谱测定得到确认。
实施例23
本发明反应步骤与操作同实施例1,不同之处在于,反应中加入的N-对甲苯磺酰腙苯乙酮衍生物是邻甲氧基N-对甲苯磺酰腙苯乙酮2h(48.0mg,0.25mmol);停止反应,经后处理得到黄色液体目标产物1h(41.1mg,产率85%),目标产物通过核磁共振谱测定得到确认。
实施例24
本发明反应步骤与操作同实施例1,不同之处在于,反应中加入的N-对甲苯磺酰腙苯乙酮衍生物是邻羟基N-对甲苯磺酰腙苯乙酮2i(44.5mg,0.25mmol);停止反应,经后处理得到白色固体目标产物1i(28.6mg,产率69%),目标产物通过核磁共振谱测定得到确认。
实施例25
本发明反应步骤与操作同实施例1,不同之处在于,反应中加入的N-对甲苯磺酰腙苯乙酮衍生物是N,N-二甲基N-对甲苯磺酰腙苯乙酮2j(51.25mg,0.25mmol);停止反应,经后处理得到棕色固体目标产物1j(42.1mg,产率82%),目标产物通过核磁共振谱测定和高分辨质谱得到确认。
实施例26
本发明反应步骤与操作同实施例1,不同之处在于,反应中加入的N-对甲苯磺酰腙苯乙酮衍生物是苯并[d][1,3]二氧杂-5-基N-对甲苯磺酰腙苯乙酮2l(59.5mg,0.25mmol);停止反应,经后处理得到黄色固体目标产物1l(40.1mg,产率78%),目标产物通过核磁共振谱测定得到确认。
实施例27
本发明反应步骤与操作同实施例1,不同之处在于,反应中加入的N-对甲苯磺酰腙苯乙酮衍生物是对三氟甲基N-对甲苯磺酰腙苯乙酮2m(57.5mg,0.25mmol);停止反应,经后处理得到白色固体目标产物1m(39.1mg,产率68%),目标产物通过核磁共振谱测定得到确认。
实施例28
本发明反应步骤与操作同实施例1,不同之处在于,反应中加入的N-对甲苯磺酰腙苯乙酮衍生物是对三氟甲氧基N-对甲苯磺酰腙苯乙酮2n(61.5mg,0.25mmol);停止反应,经后处理得到白色固体目标产物1n(48.0mg,产率78%),目标产物通过核磁共振谱和高分辨质谱测定得到确认。
实施例29
本发明反应步骤与操作同实施例1,不同之处在于,反应中加入的N-对甲苯磺酰腙苯乙酮衍生物是对氰基N-对甲苯磺酰腙苯乙酮2o(46.8mg,0.25mmol);停止反应,经后处理得到白色固体目标产物1o(37.4mg,产率80%),目标产物通过核磁共振谱测定得到确认。
实施例30
本发明反应步骤与操作同实施例1,不同之处在于,反应中加入的N-对甲苯磺酰腙苯乙酮衍生物是对氯基N-对甲苯磺酰腙苯乙酮2p(49.2mg,0.25mmol);停止反应,经后处理得到白色固体目标产物1p(37.1mg,产率80%),目标产物通过核磁共振谱测定得到确认。
实施例31
本发明反应步骤与操作同实施例1,不同之处在于,反应中加入的N-对甲苯磺酰腙苯乙酮衍生物是间溴基N-对甲苯磺酰腙苯乙酮2s(60.2mg,0.25mmol);停止反应,经后处理得到白色固体目标产物1s(35.5mg,产率85%),目标产物通过核磁共振谱测定得到确认。
实施例32
本发明反应步骤与操作同实施例1,不同之处在于,反应中加入的N-对甲苯磺酰腙苯乙酮衍生物是对溴基N-对甲苯磺酰腙苯乙酮2t(60.2mg,0.25mmol);停止反应,经后处理得到白色固体目标产物1t(47.0mg,产率82%),目标产物通过核磁共振谱测定得到确认。
实施例33
本发明反应步骤与操作同实施例1,不同之处在于,反应中加入的N-对甲苯磺酰腙苯乙酮衍生物是噻吩基N-对甲苯磺酰腙2v(42.1mg,0.25mmol);停止反应,经后处理得到褐色固体目标产物1v(29.4mg,产率70%),目标产物通过核磁共振谱测定得到确认。
实施例34
本发明反应步骤与操作同实施例1,不同之处在于,反应中加入的N-对甲苯磺酰腙苯乙酮衍生物是呋喃基N-对甲苯磺酰腙2w(38.0mg,0.25mmol);停止反应,经后处理得到褐色液体目标产物1w(22.4mg,产率60%),目标产物通过核磁共振谱测定得到确认。
实施例35
本发明反应步骤与操作同实施例1,不同之处在于,反应中加入的N-对甲苯磺酰腙苯乙酮衍生物是萘基N-对甲苯磺酰腙2x(53.1mg,0.25mmol);停止反应,经后处理得到白色固体目标产物1x(41mg,产率77%),目标产物通过核磁共振谱测定得到确认。
实施例36
本发明反应步骤与操作同实施例1,不同之处在于,反应中加入的N-对甲苯磺酰腙苯乙酮衍生物是(E)-苯乙烯基N-对甲苯磺酰腙2y(78.6mg,0.25mmol);停止反应,经后处理得到白色固体目标产物1x(30.6mg,产率65%),目标产物通过核磁共振谱测定得到确认。
实施例37
本发明在常温、氮气氛围下,往10mL带有搅拌子的石英反应管中分别加入4-(4-溴-苯基)噻二唑2t(91.8mg,0.25mmol),苯乙炔(1.2equiv),二(三苯基膦)氯化钯(5mol%),三苯基膦(5mol%)和碘化亚铜(10mol%)在三乙胺:四氢呋喃(1:1)中加热50℃,搅拌反应12小时;反应结束后,用乙酸乙酯进行萃取(3×10mL),合并有机层,用无水硫酸钠干燥,过滤、减压下除去挥发组份,然后用硅胶柱层析分离(洗脱液为石油醚(60-90℃馏份)/乙酸乙酯,v/v=5:1),得到白色固体目标产物4(46.0mg,产率70%);目标产物通过核磁共振谱和高分辨质谱得到确认。
实施例38
本发明在常温、氮气氛围下,往10mL带有搅拌子的石英反应管中分别加入4-(4-溴-苯基)噻二唑2t(91.8mg,0.25mmol),苯硼酸(1.5equiv),四(三苯基膦)钯(10mol%),三苯基膦(5mol%),碳酸钾(2.5equiv)在甲苯:乙醇:水(20:5:1)中加热80℃,搅拌反应12小时;反应结束后,用乙酸乙酯进行萃取(3×10mL),合并有机层,用无水硫酸钠干燥,过滤、减压下除去挥发组份,然后用硅胶柱层析分离(洗脱液为石油醚(60-90℃馏份)/乙酸乙酯,v/v=5:1),得到白色固体目标产物5(49.4mg,产率83%);目标产物通过核磁共振谱得到确认。
实施例39
典型化合物表征数据
4-phenyl-1,2,3-thiadiazole(1a):产率94%;35.5mg;白色固体;熔点:75-77℃;如图2所示:
4-(p-tolyl)-1,2,3-thiadiazole(1b):产率85%;34.8mg;白色固体;熔点:73-74℃;如图4所示:
4-(o-tolyl)-1,2.3-thiadiazole(1c):产率72%;29.5mg;黄色液体;如图6所示:
4-(3,4-dimethylphenyl)-1,2,3-thiadiazole(1d):产率72%;34.2mg;褐色油状;如图8所示:
4-(2,4-dimethylphenyl)-1,2,3-thiadiazole(1e):产率75%;35.6mg;黄色油状;如图10所示:
4-(4-(tert-butyl)phenyl)-1,2,3-thiadiazole(1f):产率73%;39.7mg;白色固体;熔点:70-71℃;如图12所示:
4-(4-isopropylphenyl)-1,2,3-thiadiazole(1g):产率78%;40mg;白色固体;熔点:67-67℃;如图14所示:
4-(2-methoxyphenyl)-1,2,3-thiadiazole(1h):产率85%;41.1mg;黄色液体;如图16所示:
2-(1,2,3-thiadiazol-4-yl)phenol(1i):产率69%;28.6mg;白色固体;熔点:157-159℃;如图18所示:
N,N-dimethyl-4-(1,2,3-thiadiazol-4-yl)aniline(1j):产率82%;42.1mg;褐色固体;熔点:115-117℃;如图20所示:
4-([1,1'-biphenyl]-4-yl)-1,2,3-thiadiazole(1k):产率86%;46mg;白色固体:183-184℃;如图22所示:
4-(benzo[d][1,3]dioxol-5-yl)-1,2,3-thiadiazole(1l):产率78%;40.1mg;黄色固体;熔点:124-125℃;如图24所示:
4-(4-(trifluoromethyl)phenyl)-1,2,3-thiadiazole(1m):产率68%;39.1mg;白色固体;熔点:70-72℃;如图26所示:
4-(4-(trifluoromethoxy)phenyl)-1,2,3-thiadiazole(1n):产率78%;48.0mg;白色固体;熔点:65-67℃;如图28所示:
4-(1,2,3-thiadiazol-4-yl)benzonitrile(1o):产率80%;37.4mg;白色固体;熔点:109-110℃;如图30所示:
4-(4-chlorophenyl)-1,2,3-thiadiazole(1p):产率80%;37.1mg;白色固体;熔点:136-138℃;如图32所示:
4-(3,4-dichlorophenyl)-1,2,3-thiadiazole(1q):产率53%;28.9mg;白色固体;熔点:139-140℃;如图34所示:
4-(4-fluorophenyl)-1,2,3-thiadiazole(1r):产率77%;32.6mg;白色固体;熔点:97-97℃;如图36所示:
4-(3-bromophenyl)-1,2,3-thiadiazole(1s):产率85%;35.5mg;白色固体;熔点:152-153℃;如图38所示:
4-(4-bromophenyl)-1,2,3-thiadiazole(1t):产率82%;47.0mg;白色固体;熔点:153-154℃;如图40所示:
4-(4-iodophenyl)-1,2,3-thiadiazole(1u):产率56%;38.6mg;白色固体;熔点:165-166℃;如图42所示:
4-(thiophen-2-yl)-1,2,3-thiadiazole(1v):产率70%;29.4mg;褐色固体;熔点:71-72℃;如图44所示:
4-(furan-2-yl)-1,2,3-thiadiazole(3w):产率60%;22.4mg;褐色液体;如图46所示:
4-(naphthalen-1-yl)-1,2,3-thiadiazole(3x):产率77%;41mg;白色固体;熔点:202-204℃;如图48所示:
(E)-4-styryl-1,2,3-thiadiazole(3y):产率65%;30.6mg;白色固体;熔点:81-83℃;如图50所示:
4-(4-(phenylethynyl)phenyl)-1,2,3-thiadiazole(4):产率70%;46mg;白色固体;熔点:126-128℃;如图52所示:
4-([1,1'-biphenyl]-4-yl)-1,2,3-thiadiazole(5):产率83%;49.4mg;白色固体:183-184℃;如图54所示:
以上所述,仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,都应涵盖在本发明的保护范围之内。
- 含4-(1,1,2,2-四氟乙氧基)-3,5-二氯苯胺基的4-甲基-1,2,3-噻二唑的衍生物及其合成方法和用途
- 2-(4-甲基-1,2,3-噻二唑)-5-(取代基)-1,3,4-噁(噻)二唑类衍生物及其应用