包括CDC7抑制剂的治疗癌症的方法
文献发布时间:2023-06-19 12:25:57
本申请要求于2018年9月24日提交的美国临时申请第62/735,778号;于2018年11月13日提交的美国临时申请第62/760,638号;和于2019年2月26日提交的PCT申请第PCT/US2019/019676号的权益和优先权。以上引用的申请中的每一个的内容通过引用整体并入本文。
不适用
背景技术
癌症是由活体内细胞的不受控制的、不受限的生长引起的一组疾病。由于癌细胞通常比正常细胞生长得更快,因此癌症能够通过在细胞分裂期间,特别是在染色体分裂期间控制DNA的复制进行治疗。
Cdc7是丝氨酸-苏氨酸蛋白激酶,其对于细胞周期中DNA复制的启动是必需的。具体地,Cdc7与辅因子,如Dbf4(ASK)形成复合物,并且使其底物MCM(微型染色体维持)蛋白磷酸化。据称,这种磷酸化导致Cdc45和DNA聚合酶组装在DNA上以形成MCM复合物,从而启动DNA复制。
Cdc7作为抗癌靶标已经引起人们的极大兴趣,因为Cdc7的表达水平在各种癌细胞系和人肿瘤组织中经常升高。已发现Cdc7不仅在源自人肿瘤的常见建立的细胞系中过表达,而且在取自活组织的细胞中也过表达。
已经证明某些Cdc7抑制剂影响人肿瘤细胞(如海拉(HeLa)和HCT116细胞)的生长,同时对正常细胞仅表现出有限的影响。
目前,不存在可用于使用Cdc7抑制剂治疗癌症的有效治疗性组合物和方法。
发明内容
本文描述了一种治疗癌症的方法,所述方法包括:
向患有所述癌症的受试者施用治疗有效量的由式(I-D)表示的SRA141化合物:
其中所述治疗有效量是介于10-400毫克/天之间或介于10-1000毫克/天之间的绝对剂量。
在一些实施例中,受试者是人。
在一些实施例中,所述治疗有效量为至少10毫克/天、至少20毫克/天、至少40毫克/天、至少80毫克/天、至少160毫克/天或至少320毫克/天。在一些实施例中,所述治疗有效量为至少15毫克/天、至少25毫克/天、至少50毫克/天、至少100毫克/天、至少150毫克/天、至少200毫克/天、至少250毫克/天、至少300毫克/天或至少350毫克/天。
在一些实施例中,口服施用所述SRA141化合物。
在一些实施例中,每天施用所述SRA141化合物。在一些实施例中,施用所述SRA141化合物持续至少连续5天、至少连续7天或至少连续14天。在一些实施例中,其中按照选自由以下组成的组的给药方案施用所述SRA141化合物:每周给药5天,随后非给药2天;每天给药持续1周,随后非给药1周、2周或3周;每天给药持续2周或3周,随后非给药1周或2周;以及在每周周期的第2天和第3天给药。在一些实施例中,单剂量一天一次施用所述治疗有效量。在一些实施例中,一天两次施用所述治疗有效量的一半。
在一些实施例中,癌症选自由以下组成的组:黑素瘤、子宫癌、甲状腺癌、血癌、膀胱癌、乳腺癌、宫颈癌、结肠直肠癌(CRC)、胃癌、子宫内膜癌、肝细胞癌、白血病、淋巴瘤、骨髓瘤、非小细胞肺癌、卵巢癌、前列腺癌、胰腺癌、脑癌、肉瘤、小细胞肺癌、成神经细胞瘤和头颈癌。在一些实施例中,癌症是选自由以下组成的组的血癌:急性髓性白血病(AML)、慢性髓细胞性白血病(CML)、慢性嗜酸性粒细胞白血病和弥漫性大B细胞淋巴瘤(DLBCL)。在一些实施例中,癌症是AML。
在一些实施例中,癌症是转移性结肠直肠癌(mCRC)。在一些实施例中,mCRC未被分类为具有高微卫星不稳定性(MSI-H)状态。在一些实施例中,通过检测选自由以下组成的组的重复DNA序列来确定所述MSI-H状态:单核苷酸重复标志物、二核苷酸重复标志物、准单态标志物和其组合。在一些实施例中,所述检测是通过选自由以下组成的组的方法进行的:PCR分析、多重PCR分析、毛细管电泳、DNA测序和其组合。
在一些实施例中,与所述癌症相关的肿瘤包括选自由以下组成的组的表型:染色体不稳定性(CIN)、纺锤体检查点组装缺陷、有丝分裂缺陷、G1/S检查点缺陷和其组合。在一些实施例中,与所述癌症相关的肿瘤包括Wnt信号传导途径突变。在一些实施例中,所述Wnt信号传导途径突变选自由以下组成的组:腺瘤性结肠息肉病(APC)基因突变、FAT1突变、FAT4突变和其组合。
在一些实施例中,所述方法进一步包括使用存档或新鲜肿瘤活检筛选与所述癌症相关的肿瘤。在一些实施例中,所述筛选包括通过组织化学染色检查染色体分离的模式、通过组织化学染色检查药效学标志物或其组合。在一些实施例中,所述筛选进一步包括确定所述肿瘤是否表现出异常有丝分裂。在一些实施例中,在施用所述SRA141化合物之前进行所述筛选。在一些实施例中,在施用所述SRA141化合物之后进行所述筛选。
在一些实施例中,所述方法导致在施用之后所述受试者体内的所述SRA141化合物的血浆C
在一些实施例中,所述方法导致MCM2磷酸化的体内抑制。在一些实施例中,所述MCM2磷酸化的体内抑制位于氨基酸残基Ser40和/或Ser53处。在一些实施例中,所述MCM2磷酸化的体内抑制位于与所述癌症相关的肿瘤中。在一些实施例中,与所述癌症相关的所述肿瘤中的所述MCM2磷酸化的体内抑制相对于未治疗肿瘤样品为至少50%。在一些实施例中,所述未治疗肿瘤样品是在向所述受试者施用所述SRA141化合物之前获得的活检。在一些实施例中,所述MCM2磷酸化的体内抑制是在单剂量的所述SRA141化合物之后。在一些实施例中,所述MCM2磷酸化的体内抑制是在所述受试者的皮肤中。在一些实施例中,所述MCM2磷酸化的体内抑制通过蛋白质印迹分析、免疫组织化学(IHC)或液相色谱法-质谱法(LC/MS)来测量。在一些实施例中,所述MCM2磷酸化的体内抑制在所述受试者的活检中测量。在一些实施例中,所述MCM2磷酸化的体内抑制在多剂量的所述SRA141化合物值之后测量。在一些实施例中,所述MCM2磷酸化的体内抑制在所述SRA141化合物达到稳态血浆浓度之后测量。
在一些实施例中,所述方法导致与所述癌症相关的肿瘤的生长抑制。在一些实施例中,所述肿瘤的所述生长抑制是相对于未治疗肿瘤至少10%、至少20%、至少30%、至少40%、至少50%、至少60%、至少70%、至少80%或至少90%的最小生长抑制。在一些实施例中,所述肿瘤的所述生长抑制是相对于未治疗肿瘤至少47%的最小生长抑制。在一些实施例中,所述肿瘤的所述生长抑制是相对于未治疗肿瘤至少93%的最小生长抑制。在一些实施例中,所述方法导致与所述癌症相关的肿瘤的消退。在一些实施例中,所述消退是完全消退。在一些实施例中,所述方法导致与所述癌症相关的肿瘤的细胞毒性。在一些实施例中,所述方法导致与所述癌症相关的肿瘤的直径总和减少至少30%。在一些实施例中,所述方法导致所述受试者的部分应答、完全应答或疾病稳定。
在一些实施例中,所述方法进一步包含确定与所述癌症相关的肿瘤或病灶的生长抑制水平。在一些实施例中,所述方法包含将施用SRA141后靶病灶的第二直径与施用SRA141前所述靶病灶的第一直径进行比较来确定靶病灶生长是否被抑制。在某些情况下,确定肿瘤或病灶生长相对于未治疗肿瘤或病灶被抑制至少10%、至少20%、至少30%、至少40%、至少50%、至少60%、至少70%、至少80%或至少90%。
在一些实施例中,所述方法进一步包括向所述受试者施用第二治疗有效量的一种或多种另外的治疗。在一些实施例中,所述一种或多种另外的治疗包括抗肿瘤剂。在一些实施例中,所述抗肿瘤剂选自由以下组成的组:DNA聚合酶抑制剂、受体酪氨酸激酶抑制剂、哺乳动物雷帕霉素靶蛋白(mTOR)途径抑制剂。在一些实施例中,所述抗肿瘤剂包括丝裂原活化蛋白激酶(MAPK)途径抑制剂。在一些实施例中,所述MAPK抑制剂是曲美替尼(Trametinib)。在一些实施例中,所述抗肿瘤剂包括类视黄醇途径调节剂。在一些实施例中,所述类视黄醇途径调节剂是RXR激动剂蓓萨罗丁(Bexarotene)或RAR激动剂维甲酸(Tretinoin)(全反式视黄酸,ATRA)。在一些实施例中,其中所述抗肿瘤剂包括细胞凋亡调节剂。在一些实施例中,所述细胞凋亡调节剂包括细胞凋亡诱导剂。在一些实施例中,所述细胞凋亡诱导剂包括BCL-2抑制剂。在一些实施例中,所述BCL-2抑制剂是ABT-199。在一些实施例中,所述抗肿瘤剂包括磷脂酰肌醇-4,5-二磷酸3激酶(PI3K)途径抑制剂。在一些实施例中,所述PI3K途径抑制剂是库潘尼西(Copanlisib)。在一些实施例中,所述抗肿瘤剂包括PARP抑制剂。在一些实施例中,所述PARP抑制剂是BMN673。在一些实施例中,所述抗肿瘤剂包括极光B激酶抑制剂。在一些实施例中,所述极光B激酶抑制剂是巴拉塞替(Barasertib)。在一些实施例中,每天施用所述一种或多种另外的治疗。在一些实施例中,所述SRA141化合物和所述一种或多种另外的治疗展现出协同效应。
附图说明
参考以下描述和附图,本发明的这些和其它特征、方面和优点将变得更好理解,其中:
图1A是示出在急性髓性白血病的小鼠异种移植模型中用SRA141单一疗法治疗的动物的肿瘤体积的图。
图1B是示出在急性髓性白血病的小鼠异种移植模型中用SRA141单一疗法治疗的动物的肿瘤重量的图。
图1C是示出在急性髓性白血病的小鼠异种移植模型中用SRA141单一疗法治疗的动物的体重变化的图。
图2A是示出在急性髓性白血病的大鼠异种移植模型中用SRA141单一疗法治疗的动物的平均肿瘤体积的图。
图2B是示出在急性髓性白血病的大鼠异种移植模型中用SRA141单一疗法治疗的动物的体重变化的图。
图2C是示出在急性髓性白血病的大鼠异种移植模型中用SRA141单一疗法治疗的个体受试者的肿瘤体积的图。
图3A是示出在一组血液学癌症源性细胞系中SRA141的半最大抑制浓度(IC
图3B示出了根据癌症类型分解的所确定的IC
图3C展示了癌症与正常细胞之间对SRA141的敏感性。
图3D示出了Cdc7抑制剂SRA141、TAK-931和LY-3177833在许多细胞系中的活性。
图3E示出了在72小时(左列)和144小时(右列)后七种细胞系中使用设计用于测量以下的四种正交测定的IC
图3F示出了在对SRA141的不同暴露时间之后,在144小时时通过CTG和CyQuant确定的IC
图4A是示出在结肠直肠癌(CRC)的患者源性异种移植(PDX)模型中用SRA141单一疗法治疗的动物的平均肿瘤体积的图。
图4B是示出在结肠直肠癌(CRC)的患者源性异种移植(PDX)模型中用SRA141单一疗法治疗的动物的体重变化的图。
图5是示出在结肠直肠癌(CRC)的患者源性异种移植(PDX)模型中用SRA141单一疗法治疗的动物的个体受试者的肿瘤体积的图。
图6是示出在结肠直肠癌(CRC)的患者源性异种移植(PDX)模型中,在治疗的第32天(D32)用SRA141单一疗法治疗的动物的平均肿瘤体积的图。
图7A是示出在结肠直肠癌的大鼠异种移植模型中用SRA141治疗的动物的平均肿瘤体积的图。
图7B示出了在结肠直肠癌的大鼠异种移植模型中,体重变化百分比、SRA141的肿瘤浓度和免疫印迹的图,所述免疫印迹示出了用SRA141处理后12小时磷酸化MCM2减少。
图8是示出用SRA141与抗肿瘤剂组合顺序地处理(预处理)的细胞系的细胞生长的抑制(细胞活力与0小时和72小时的未处理对照比较)的图。
图9示出了描绘用SRA141(“塞拉化合物(Sierra compound)1”)与抗肿瘤剂组合共同处理的细胞系的生长抑制的协同作用分数的表。
图10A示出了描绘用SRA141(“塞拉化合物1”)与抗肿瘤剂组合共同处理的细胞系的生长抑制的协同作用分数的表。
图10B是示出用仅吉西他滨或与SRA141组合处理的HT-29细胞的生长抑制百分比的图。
图11A示出了在体外生物化学测定中SRA141对Cdc7的有效抑制。
图11B示出了SRA141与Cdc7结合的停留时间和解离动力学。
图12示出了SRA141和TAK931的激酶组筛选测定的结果。
图13示出了用SRA141在0.033与3.3μM之间的浓度下处理Colo-205细胞持续8小时(图13A)到24小时(图13B),和在0.1和1μM的浓度下持续3、6和24小时(图13C),以及用SRA141在0.5与3.3μM之间的浓度下处理的MV411细胞(图13D),随后评估Cdc7的下游靶标的磷酸化状态。
图14是示出用SRA141在0.1μM和1μM下处理24小时后MCM2的Ser53上的磷酸化的定量的图。
图15示出了在S期(图15A)期间和M期(图15B)开始时来自用SRA141处理的细胞的流式细胞术数据;在用SRA141处理的细胞中的细胞周期和DNA标志物的评估(图15C);和在用Cdc7抑制剂处理的细胞群体中有丝分裂细胞的百分比的评估(图15D)。
图16示出了具有和不具有APC突变的细胞系中SRA141敏感性的汇总。
图17是示出在携带Colo-205肿瘤异种移植物的BALB/c小鼠中用SRA141单一疗法治疗的动物的平均肿瘤体积的图。
图18是示出在携带SW620肿瘤异种移植物的BALB/c小鼠中用SRA141单一疗法治疗的动物的平均肿瘤体积的图。
图19是示出在携带A20肿瘤异种移植物的BALB/c小鼠中用SRA141单一疗法治疗的动物的平均肿瘤体积的图。
图20是示出在携带MDA-MB-486乳腺肿瘤异种移植物的CB-17SCID小鼠中用SRA141单一疗法治疗的动物的平均肿瘤体积的图。
图21示出了在携带皮下SW620肿瘤的BALB/c小鼠中单次SRA141施用后pMCM2水平降低(图21A中示出pMCM2水平,图21B中对抑制%进行定量并相对于肌动蛋白归一化)。
图22示出了携带皮下Colo-205肿瘤的雌性Rowett裸大鼠在SRA141处理后的pMCM2水平。
图23示出了在非荷瘤雌性裸大鼠中通过LC-MS/MS确定的SRA141血浆浓度。
图24A示出了来自用和不用SRA141处理的白血病的大鼠异种移植模型的肿瘤和替代组织中的总MCM2、磷酸化MCM2和gH2AX的免疫组织化学评估。
图24B示出了来自白血病的大鼠异种移植模型的肿瘤和替代组织中的磷酸化MCM2的免疫组织化学评估。
图24C示出了来自用SRA141和媒剂处理的白血病的大鼠异种移植模型的肿瘤的免疫组织化学和H&E评估。
图25A示出了用SRA141以不同剂量处理后异种移植肿瘤中的pMCM2-S40表达的评估结果。
图25B示出了用SRA141以不同剂量处理后,来自大鼠异种移植物的皮肤样品中的pMCM2-S40表达的评估结果。
图26示出了正常人皮肤的总MCM2、pMCM2-S53、pMCM2-S40和gH2AX的评估结果。
图27示出了在Colo-205细胞(图27A,图27G)、SW620细胞(图27B)、A375细胞(图27C)、KG-1细胞(图27D)、MOLM-13细胞(图27E,图27H)和MV411细胞(图27F)中用SRA141与各种另外的抗肿瘤剂组合进行的细胞活力测定的结果。
图28示出了在通过RNAi敲低抑制抗凋亡基因后在细胞中用SRA141处理的结果(图28A)或用ABT-199处理的结果(图28B)。
具体实施方式
本文公开了通过施用有效量的Cdc7抑制剂SRA141,抑制受试者(例如,人)的肿瘤生长的方法。本文还公开了通过施用有效量的Cdc7抑制剂SRA141,抑制受试者(例如,人)的肿瘤生长的方法。本文还公开了通过在组合疗法中施用有效量的Cdc7抑制剂SRA141来抑制受试者(例如,人)的肿瘤生长的方法。
定义
权利要求书和说明书中使用的术语如以下所述的定义,除非另有说明。
本发明的实践包含使用在本领域的技术范围内的有机化学、分子生物学(包含重组技术)、微生物学、细胞生物学、生物化学和免疫学的常规技术。
在本申请中,将参考许多技术名称。所有数字名称,例如pH、温度、时间、浓度和重量,包含其各自的范围,均是近似值,所述近似值通常可以通过例如0.01、0.05、0.1、0.5、1.0、5.0或10.0的增量(视情况而定)变化(+)或(-)。所有数字名称均可以理解为前面是术语“约”。本文所述的试剂是示例性的,并且此类试剂的等效物可以是本领域已知的。
本发明中使用的化合物具有不对称碳原子(光学中心)或双键;外消旋体、非对映异构体、几何异构体、区域异构体和单个异构体(例如,单独的对映异构体)均旨在涵盖在本发明的范围内。本发明的化合物在构成此类化合物的原子中的一个或多个原子处还可以含有非天然比例的原子同位素。例如,化合物可以用放射性同位素放射性标记,例如但不限于氚(
术语“受试者”是指任何哺乳动物,包含人以及哺乳动物,如兽医和研究兴趣的那些动物,包含但不限于:猿猴、牛、马、狗、猫和啮齿类动物。
术语向受试者“施用(administering)”或“施用(administration of)”药物和/或疗法(以及此短语的语法等效物)是指直接施用或间接施用两者,其可以是由医学专业人士向受试者施用,可以是自我施用和/或间接施用,其可以是向受试者开处方或诱导受试者开处方药物和/或疗法的行为。
术语“共同施用(coadministration)”是指以在同一时间段期间发挥其药理作用的方式施用的两种或更多种化合物。这种共同施用可以通过同时、同时期或顺序施用所述两种或更多种化合物来实现。
术语“治疗(treating)”或“治疗(treatment of)”病症或疾病是指采取步骤来减轻病症或疾病(例如,肿瘤生长或癌症)的症状,或以其它方式获得受试者的一些有益或所期望的结果,包含临床结果。任何有益的或所期望的临床结果可以包含但不限于减轻或改善癌症或条件存活的一种或多种症状以及减少肿瘤负荷或肿瘤体积;减轻疾病程度;延迟或减慢肿瘤进展或疾病进展;改善、缓和或稳定肿瘤和/或疾病状态;或其它有益结果。
术语“原位”或“体外”是指在与活生物体分开生长(例如,在组织培养物中生长)的活细胞中发生的过程。
术语“体内”指在活生物体中发生的过程。
术语“Cdc7”是指由CDC7基因编码的细胞分裂周期7丝氨酸/苏氨酸蛋白激酶。
术语“减少”一种或多种症状(以及此短语的语法等效物)是指降低一种或多种症状的严重程度或频率,或消除一种或多种症状。
必须注意,如在说明书和所附权利要求中所使用,单数形式“一个(a)”、“一种(an)”和“所述(the)”包含复数指示物,除非上下文另外清楚地指示。
本发明的方法
本文公开了通过施用Cdc7抑制剂SRA141,抑制受试者(例如,人)的肿瘤生长的方法。对化合物、包括所述化合物的试剂盒及其使用方法的详细描述见下文。
施用
如本文所公开的,本发明的方法包含施用有效量的SRA141。本公开提供了一种治疗方法,其中向受试者施用有效量的SRA141。术语“有效量”或“治疗有效量”是指有效改善疾病症状的量,例如有效抑制肿瘤生长的量。在一些方面,SRA141的有效量小于最大耐受剂量(MTD)。在一些方面,SRA141的有效量小于1000毫克/天、小于500毫克/天或小于400毫克/天。在一些方面,SRA141的有效量小于300毫克/天、小于200毫克/天、小于150毫克/天、小于100毫克/天或小于75毫克/天。在一些方面,SRA141的有效量小于324毫克/天。在一些方面,SRA141的有效量为324毫克/天。在一些方面,SRA141的有效量为至少10毫克/天。在一些方面,SRA141的有效量介于10-400毫克/天之间。在一些方面,SRA141的有效量介于10-324毫克/天之间。在一些方面,SRA141的有效量介于40-400毫克/天之间。在一些方面,SRA141的有效量为至少10毫克/天、至少20毫克/天、至少40毫克/天、至少80毫克/天、至少160毫克/天或至少320毫克/天。在一些方面,SRA141的有效量为至少15毫克/天、至少25毫克/天、至少50毫克/天、至少75毫克/天、至少100毫克/天、至少150毫克/天、至少200毫克/天、至少250毫克/天、至少300毫克/天或至少350毫克/天。在一些方面,SRA141的有效量为10毫克/天、20毫克/天、40毫克/天、80毫克/天、160毫克/天、320毫克/天、325毫克/天、350毫克/天或400毫克/天。在一些方面,SRA141的有效量为15毫克/天、25毫克/天、50毫克/天、75毫克/天、100毫克/天、150毫克/天、200毫克/天、250毫克/天、300毫克/天或350毫克/天。
单一疗法
在一个实施例中,有效量的Cdc7抑制剂作为单一疗法施用。
在一些方面,Cdc7抑制剂单一疗法的有效量小于最大耐受剂量(MTD)。在一些方面,Cdc7抑制剂单一疗法的有效量小于1000毫克/天、小于500毫克/天或小于400毫克/天。在一些方面,Cdc7抑制剂单一疗法的有效量小于300毫克/天、小于200毫克/天、小于150毫克/天、小于100毫克/天或小于75毫克/天。在一些方面,Cdc7抑制剂单一疗法的有效量小于324毫克/天。在一些方面,SRA141单一疗法的有效量为324毫克/天。在一些方面,Cdc7抑制剂单一疗法的有效量为至少10毫克/天。在一些方面,Cdc7抑制剂单一疗法的有效量介于10-400毫克/天之间。在一些方面,Cdc7抑制剂单一疗法的有效量介于10-324毫克/天之间。在一些方面,Cdc7抑制剂单一疗法的有效量介于40-400毫克/天之间。在一些方面,Cdc7抑制剂单一疗法的有效量为至少10毫克/天、至少20毫克/天、至少40毫克/天、至少80毫克/天、至少160毫克/天或至少320毫克/天。在一些方面,Cdc7抑制剂单一疗法的有效量为至少15毫克/天、至少25毫克/天、至少50毫克/天、至少75毫克/天、至少100毫克/天、至少150毫克/天、至少200毫克/天、至少250毫克/天、至少300毫克/天或至少350毫克/天。在一些方面,SRA141单一疗法的有效量为10毫克/天、20毫克/天、40毫克/天、80毫克/天、160毫克/天、320毫克/天、325毫克/天、350毫克/天或400毫克/天。在一些方面,SRA141单一疗法的有效量为15毫克/天、25毫克/天、50毫克/天、75毫克/天、100毫克/天、150毫克/天、200毫克/天、250毫克/天、300毫克/天或350毫克/天。
组合疗法
还如本文所公开的,本发明的方法包含施用有效量的SRA141和共同施用第二有效量的一种或多种另外的治疗的组合疗法。
另外的治疗包含但不限于施用化疗剂、施用抗体或抗体片段(如免疫检查点抑制剂)、施用放射治疗以及施用其组合。另外的治疗还包含但不限于施用抗肿瘤剂,如DNA聚合酶抑制剂、受体酪氨酸激酶抑制剂、哺乳动物雷帕霉素靶蛋白(mTOR)途径抑制剂。抗肿瘤剂还可以包含丝裂原活化蛋白激酶(MAPK)途径抑制剂,例如曲美替尼。抗肿瘤剂还可以包含类视黄醇途径调节剂,如是RXR激动剂蓓萨罗丁和RAR激动剂维甲酸。抗肿瘤剂还可以包含细胞凋亡调节剂,如包括细胞凋亡诱导剂,所述细胞凋亡诱导剂包含但不限于抗Bcl-2剂(例如,ABT-199)。抗肿瘤剂还可以包含磷脂酰肌醇-4,5-二磷酸3激酶(PI3K)途径抑制剂,如库潘尼西。抗肿瘤剂还可以包含PARP抑制剂,如BMN673。抗肿瘤剂还可以包含ATM激酶抑制剂,如KU-60019。抗肿瘤剂还可以包含极光B激酶抑制剂,如巴拉塞替。抗肿瘤剂还可以包含酪氨酸苏氨酸激酶(TTK)抑制剂,如单极纺锤体1激酶(Mps1)的抑制剂(例如,CFI-402257)。抗肿瘤剂还可以包含表皮生长因子(EGF)的抑制剂,如埃罗替尼(Erlotinib)。在一些实施例中,抗肿瘤剂是吉西他滨。在一些实施例中,抗肿瘤剂不是吉西他滨。
另外的治疗还可以包含另外的治疗的组合,如抗肿瘤剂的组合。
在本发明的具体实施例中,将有效量的SRA141作为与第二有效量的另外的治疗的组合疗法向受试者施用。在一些方面,第二有效量是约0.001mg/kg到约15mg/kg的量。在一些实施例中,另外的治疗的第二有效量为0.001、0.005、0.010、0.020、0.050、0.1、0.2、0.5、1.0、2.0、5.0、10.0或15.0mg/kg。在一些实施例中,另外的治疗的第二有效量介于10-2000mg/m
共同施用涵盖以下方法:其中同时给予SRA141和另外的治疗,其中顺序地给予SRA141和另外的治疗,并且其中间歇地或连续地给予SRA141和另外的治疗中的任一者或两者,或以下中的任何组合:同时、顺序地、间歇地和/或连续地。技术人员将认识到,间歇施用不一定与顺序施用相同,因为间歇施用还包含药剂的第一次施用,并且然后同一试剂在时间上稍后的另一次施用。此外,技术人员理解,间歇施用在一些方面也涵盖顺序施用,因为间歇施用确实包含在再次施用第一药剂之前中断药剂的第一次施用和不同药剂的施用。进一步地,技术人员还将知道,连续施用可以通过许多途径(包含静脉内滴注或饲管等)来完成。
此外,并且以更一般的方式,术语“共同施用”涵盖其中向受试者单独施用Cdc7抑制剂和单独施用另外的治疗在任何时间帧期间重叠的任何和所有方法。
给药途径
在一些方面,本公开提供了通过选自由以下组成的组的途径施用SRA141和/或另外的治疗中的任一种或两种或其任何组合的方法:静脉内、皮下、皮肤、口服、肌内和腹膜内。在一些方面,本公开提供了静脉内施用SRA141和/或另外的治疗中的任一种或两种或其任何组合的方法。在一些方面,本公开提供了口服施用SRA141和/或另外的治疗中的任一种或两种或其任何组合的方法。
技术人员应理解,本公开的单位剂型可以按相同或不同的物理形式施用,即通过胶囊或片剂口服施用和/或通过液体口服施用或通过静脉内输注施用等。此外,每次施用的单位剂型可以因特定施用途径方面而不同。对于Cdc7抑制剂和另外的治疗中的任一种或两种,可能存在若干种不同的剂型。因为不同的医学病状可以保证不同的施用途径,因此本文所述的Cdc7抑制剂和另外的治疗的组合的相同组分在组成和物理形式上可能完全相同,但仍可能需要以不同方式给予并且可能在不同时间给予以减轻病状。例如,病状(如持续性恶心,尤其是呕吐)可能使得难以使用口服剂型,并且在这种情况下,可能需要施用另一种单位剂型,可能甚至是与先前或后续使用的其它剂型相同的剂型,代替或还有吸入、口腔、舌下或栓剂途径。具体剂型可以是SRA141和另外的治疗的某些组合的要求,因为可能存在各种因素,如化学稳定性或药物动力学的问题。
给药方案
一方面,向受试者施用SRA141或另外的治疗的频率包含但不限于Q1d、Q2d、Q3d、Q4d、Q5d、Q6d、Q7d、Q8d、Q9d、Q10d、Q14d、Q21d、Q28d、Q30d、Q90d、Q120d、Q240d或Q365d。术语“QnD或qnd”是指每“n”天一次药物施用。例如,QD(或qd)是指每天一次或每天一次给药,Q2D(或q2d)是指每两天一次给药,Q7D是指每7天一次或一周一次给药,Q5D是指每5天一次给药,等等。一方面,SRA141和另外的治疗是按照不同的方案施用的。
在另一方面,对受试者施用SRA141或另外的治疗的频率包含但不限于:每周给药5天,随后非给药2天;每天给药持续1周,随后非给药1周、2周或3周;每天给药持续2周或3周,随后非给药1周或2周;每天两次给药;或在每周周期的第2天和第3天给药。一方面,SRA141和另外的治疗是按照不同的方案施用的。
一方面,本公开提供了间歇地施用SRA141和/或另外的治疗中的任一种或两种或其任何组合的方法。一方面,本公开提供了包括以下的方法:向受试者施用SRA141或另外的治疗中的任一种或两种或其任何组合,其中施用之间具有至少十(10)分钟、十五(15)分钟、二十(20)分钟、三十(30)分钟、四十(40)分钟、六十(60)分钟、两(2)小时、三(3)小时、四(4)小时、六(6)小时、八(8)小时、十(10)小时、十二(12)小时、十四(14)小时、十八(18)小时、二十四(24)小时、三十六(36)小时、四十八(48)小时、三(3)天、四(4)天、五(5)天、六(6)天、七(7)天、八(8)天、九(9)天、十(10)天、十一(11)天、十二(12)天、十三(13)天、十四(14)天、三(3)周或四(4)周延迟。在此类方面,延迟施用遵循这样的模式,其中SRA141和/或另外的治疗中的一种或两种或其任何组合连续施用持续约十(10)分钟到约三百六十五(365)天的给定时间段,并且然后不施用持续约十(10)分钟到约三十(30)天的给定时间段。一方面,本公开提供了间歇地施用SRA141和/或另外的治疗中的任一种或任何组合而连续地给予另一种的方法。
一方面,本公开提供了将有效量的SRA141的组合与第二有效量的另外的治疗顺序地施用的方法。
一方面,本公开提供了同时施用SRA141和另外的治疗的方法。一方面,本公开提供了将有效量的SRA141的组合与第二有效量的另外的治疗顺序地施用的方法。在此类方面,组合还被称为“共同施用”,因为所述术语包含其中受试者暴露于组合中的两种组分的任何和所有方法。然而,此类方面不限于仅在一种调配物或组合物中给出的组合。可能某些浓度的SRA141和另外的治疗更有利于以某些间隔进行递送,并且如此,SRA141的有效量和另外的治疗的第二有效量可以根据所施用的调配物而变化。
在一些方面,本公开提供了其中同时或顺序地施用SRA141和另外的治疗的方法。在一些方面,本公开提供了在第二有效量的另外的治疗之后顺序地施用有效量的SRA141的方法。在一些方面,本公开提供了在有效量的SRA141之后顺序地施用第二有效量的另外的治疗的方法。
在一些方面,本公开提供了以一种调配物施用组合的方法。在一些方面,本公开提供了以两种(2)组合物施用组合,其中以与第二有效量的另外的治疗的调配物分开的调配物施用有效量的SRA141的方法。
在一些方面,本公开提供了在第二有效量的另外的治疗之后顺序地施用有效量的SRA141的方法。在一些方面,本公开提供了在有效量的SRA141之后顺序地施用第二有效量的另外的治疗的方法。在一些方面,施用SRA141和另外的治疗;并且随后间歇地施用SRA141和另外的治疗两者持续至少二十四(24)小时。在一些方面,SRA141和另外的治疗是按照非重叠的每隔一天的方案施用的。在一些方面,在每周方案的第1天施用另外的治疗,并且在第2天和第3天施用SRA141。
在一些方面,本公开提供了在第二有效量的另外的治疗之后不少于四(4)小时施用有效量的SRA141的方法。一方面,本公开提供了在第二有效量的另外的治疗之后不少于十(10)分钟、不少于十五(15)分钟、不少于二十(20)分钟、不少于三十(30)分钟、不少于四十(40)分钟、不少于六十(60)分钟、不少于一(1)小时、不少于两(2)小时、不少于四(4)小时、不少于六(6)小时、不少于八(8)小时、不少于十(10)小时、不少于十二(12)小时、不少于二十四(24)小时、不少于两(2)天、不少于四(4)天、不少于六(6)天、不少于八(8)天、不少于十(10)天、不少于十二(12)天、不少于十四(14)天、不少于二十一(21)天或不少于三十(30)天施用有效量的SRA141的方法。一方面,本公开提供了在有效量的SRA141之后不少于十(10)分钟、不少于十五(15)分钟、不少于二十(20)分钟、不少于三十(30)分钟、不少于四十(40)分钟、不少于六十(60)分钟、不少于一(1)小时、不少于两(2)小时、不少于四(4)小时、不少于六(6)小时、不少于八(8)小时、不少于十(10)小时、不少于十二(12)小时、不少于二十四(24)小时、不少于两(2)天、不少于四(4)天、不少于六(6)天、不少于八(8)天、不少于十(10)天、不少于十二(12)天、不少于十四(14)天、不少于二十一(21)天或不少于三十(30)天施用第二有效量的另外的治疗的方法。
癌症和肿瘤
公开了用于治疗癌症的方法。因此,本公开提供了治疗有需要的受试者(即,患有癌症的受试者或患有癌症的受试者)的癌症的方法,所述方法包括向患有癌症的受试者施用有效量的SRA141,其中所述癌症是黑素瘤、子宫癌、甲状腺癌、血癌、膀胱癌、乳腺癌、宫颈癌、结肠直肠癌(CRC)、胃癌、子宫内膜癌、胆管癌、肝细胞癌、白血病、淋巴瘤、骨髓瘤、非小细胞肺癌、卵巢癌、前列腺癌、胰腺癌、脑癌、肉瘤、小细胞肺癌、成神经细胞瘤或头颈癌。在一些方面,癌症是血癌,如急性髓性白血病(AML)、慢性髓细胞性白血病(CML)、慢性嗜酸性粒细胞白血病和弥漫性大B细胞淋巴瘤(DLBCL)。在一些方面,癌症是AML。
在一些方面,癌症可以分类为不具有高微卫星不稳定性(MSI-H)状态。术语“微卫星不稳定性”是指特征在于在特定重复DNA序列(例如,短串联重复序列或简单序列重复序列)中具有基因组不稳定性的肿瘤。用于检测微卫星不稳定性的方法包含本领域已知的任何方法,所述方法包含但不限于在以下文献中描述的那些方法:Wang M.等人,在结肠直肠癌和林奇综合征中筛选微卫星不稳定性—微型综述(Screening for MicrosatelliteInstability in Colorectal Cancer and Lynch Syndrome–A Mini Review);《美医学与科学杂志(N A J Med Sci.)》2016;9(1):5–11。在某些实施例中,微卫星不稳定性的特征在于检测单核苷酸重复标志物(例如,BAT-25、BAT-26、NR-21、NR-24和MONO-27,也被称为普洛麦格MSI多重系统(Promega MSI Multiplex System))。在某些实施例中,微卫星不稳定性的特征在于检测准单态标志物和二核苷酸重复标志物(例如,BAT25、BAT26、D2S123、D5S346和D17S250,也被称为Bathesda面板)。MSI可以分类为MSI-高(MSI-H)、MSI-低(MSI-L)和MS-稳定(MSS),其中在两个或更多个标志物中的不稳定性被分类为MSI-H,在一个标志物中的不稳定性仅被分类为MSI-L,并且在五个标志物中未观察到的不稳定性被分类为MSS。如果大于30%的所测试的标志物表现出不稳定性(MSI-H),则MSI还可以分类为MSI-H,如果10-30%的所测试的标志物表现出不稳定性,则被分类为MSI-L,并且如果小于10%的所测试的标志物表现出不稳定性,则被分类为MSS。可以使用5个、6个、7个、8个、9个、10个或大于10个标志物测试MSI状态。在一些实施例中,癌症被分类为MSS。癌症还可以具有未知的MSI状态,即不知道肿瘤具有MSI-H状态。
在某些实施例中,术语“微卫星不稳定性”还指特征在于具有与至少一个参考样品相比与错配修复的损失(MMR)相关的本领域已知的一个或多个重复DNA序列的肿瘤。
在一些方面,本公开提供了抑制肿瘤生长的方法,其中所述肿瘤来自为以下的癌症:黑素瘤、子宫癌、甲状腺癌、血癌、膀胱癌、乳腺癌、宫颈癌、结肠直肠癌(CRC)、胃癌、子宫内膜癌、胆管癌、肝细胞癌、白血病、淋巴瘤、骨髓瘤、非小细胞肺癌、卵巢癌、前列腺癌、胰腺癌、脑癌、肉瘤、小细胞肺癌、成神经细胞瘤或头颈癌。肿瘤可以来自血癌,如急性髓性白血病(AML)、慢性髓细胞性白血病(CML)、慢性嗜酸性粒细胞白血病和弥漫性大B细胞淋巴瘤(DLBCL)。肿瘤可以来自AML癌症。肿瘤可以来自转移性结肠直肠癌(mCRC)。肿瘤可能来自未归类为具有MSI-H状态的转移性结肠直肠癌(mCRC)。肿瘤可以具有Wnt信号传导途径突变,如腺瘤性结肠息肉病(APC)基因突变、FAT1突变、FAT4突变或其组合。肿瘤可以具有FBXW7突变。
在一些方面,与癌症相关的肿瘤包括表型,所述表型是通过使用存档或新鲜肿瘤活检并且通过采用用苏木精和曙红(或类似染色)对肿瘤切片的组织化学染色来检查染色体分离的模式并且确定肿瘤是否表现出异常有丝分裂(例如,滞后染色体、后期桥、多极纺锤体)来预期筛选的,如在显微镜下手动地或通过自动化方法检查时。在一些方面,在施用SRA141之后通过使用治疗后肿瘤活检进行组织化学染色来确定异常有丝分裂的药效学标志物。治疗后活检可以在SRA141施用之后若干天获得,如获得同一天的活检以进行pMCM2监测(参见下文)。
受试者
本公开提供了向有需要的受试者施用有效量的SRA141。本公开提供了在组合疗法中向有需要的受试者施用有效量的SRA141和另外的治疗。在一些方面,在施用之前通过基因测试和/测序筛选来自受试者的肿瘤。在一些方面,在施用之后通过基因测试和/测序筛选来自受试者的肿瘤。在一些方面,在施用之后和之前均对来自受试者的肿瘤进行筛选。在一些方面,在施用之前、施用之后或两者,通过基因测试和/或测序筛选来自受试者的健康细胞。在一些方面,通过其它生物测试或测定筛选来自受试者的肿瘤,以确定某些生物标志物(如微卫星或重复DNA)的表达水平。在一些方面,通过基因测试和/测序以及其它生物标志物测试或测定来筛选来自受试者的肿瘤。
在一些方面,本公开提供了其中受试者是哺乳动物的方法。在一些方面,本公开提供了其中受试者是灵长类动物的方法。
在一些方面,本公开提供了其中受试者是小鼠的方法。
在一些方面,本公开提供了其中受试者是人的方法。
在一些方面,本公开提供了其中肿瘤位于患有选自由以下组成的组的癌症的人中的方法:黑素瘤、子宫癌、甲状腺癌、血癌、膀胱癌、乳腺癌、宫颈癌、结肠直肠癌(CRC)、胃癌、子宫内膜癌、胆管癌、肝细胞癌、白血病、淋巴瘤、骨髓瘤、非小细胞肺癌、卵巢癌、前列腺癌、胰腺癌、脑癌、肉瘤、小细胞肺癌、成神经细胞瘤或头颈癌。在一些方面,癌症是血癌,如急性髓性白血病(AML)、慢性髓细胞性白血病(CML)、慢性嗜酸性粒细胞白血病和弥漫性大B细胞淋巴瘤(DLBCL)。在一些方面,癌症是AML。在一些方面,癌症是转移性结肠直肠癌(mCRC)。mCRC可以归类为不具有MSI-H状态。
在一些方面,受试者患有肿瘤,所述肿瘤含有预期赋予对Cdc7抑制的敏感性的基因组改变,如不具有MSI-H状态、具有染色体不稳定性(CIN)表型(例如,CRC CIN表型通常与微卫星不稳定(MSI)CRC肿瘤相互排斥)、纺锤体检查点组装缺陷、有丝分裂缺陷、G1/S检查点缺陷、具有Wnt信号传导途径突变(例如,腺瘤性结肠息肉病(APC)基因突变、FAT1突变、FAT4突变或其组合)或其组合。
药代动力学
本文所述的用于治疗癌症的方法可以导致SRA141化合物的血浆C
本文所述的用于治疗癌症的方法可以导致SRA141化合物的AUC
本文所述的用于治疗癌症的方法可以导致SRA141化合物的肿瘤内浓度大于500ng/mL、大于600ng/mL、大于900ng/mL或大于1300ng/mL。本文所述的用于治疗癌症的方法可以导致SRA141化合物的肿瘤内浓度为至少500ng/mL、至少600ng/mL、至少700ng/mL、至少800ng/mL、至少900ng/mL、至少1000ng/mL、至少1100ng/mL、至少1100ng/mL、至少1200ng/mL、至少1300ng/mL、至少1400ng/mL或至少1500ng/mL。
激酶抑制
在一些实施例中,本文所述的方法导致体内激酶抑制,例如MCM2磷酸化的抑制。如本领域已知的,在进入S期和随后的DNA复制之前,跨基因组建立多个复制起点。这些复制起点由包含六个微型染色体维持蛋白(MCM2-7)(统称为前复制复合物(pre-RC))的多个组分构成。一旦建立前RC,Cdc7就使MCM2磷酸化,从而启动S期进入和DNA复制。随着S期的进展,Cdc7在另外的复制起点处使MCM2磷酸化,直到在进入细胞周期的下一个阶段之前完成DNA复制。因此,MCM2磷酸化可以用作Cdc7活性的替代标志物。
本文公开了用于治疗癌症的方法,所述方法导致MCM2磷酸化的体内抑制,这种磷酸化位于氨基酸残基Ser40或Ser53处。MCM2磷酸化的体内抑制可以位于与癌症相关的肿瘤中。肿瘤中MCM2磷酸化的体内抑制相对于未治疗肿瘤样品可以为至少50%。肿瘤中MCM2磷酸化的体内抑制相对于未治疗肿瘤样品可以为至少10%、20%、30%、40%、50%、60%、70%、80%或90%。MCM2磷酸化的体内抑制可以位于受试者的皮肤中。MCM2磷酸化的体内抑制可以通过本领域的技术人员已知的测定进行测量,包含但不限于蛋白质印迹分析、免疫组织化学(IHC)、液相色谱法-质谱法(LC/MS)或LC/MS/MS。MCM2磷酸化的体内抑制可以在受试者的活检中测量。
本文所述的用于治疗癌症的方法可以不导致(即,可以避免)受试者的脱靶激酶的特异性抑制,其中所述脱靶激酶选自由以下组成的组:WEE1、CDK7、CDK8、CDK9和LATS2。本文所述的用于治疗癌症的方法可以导致脱靶激酶的小于90%的抑制。
肿瘤抑制
本公开涉及使用有效量的化合物(例如,SRA141)来抑制肿瘤的进展、减小所述肿瘤的聚集的聚集体的大小、减小所述肿瘤的体积、减小所述肿瘤的直径和/或以其它方式抑制所述肿瘤的生长的方法。因此,本文所述的用于治疗癌症的方法可以导致与癌症相关的肿瘤的生长抑制。本文所述的用于治疗癌症的方法可以导致与癌症相关的肿瘤的细胞毒性。本文所述的用于治疗癌症的方法可以导致与癌症相关的肿瘤的生长抑制和细胞毒性。本文所述的用于治疗癌症的方法可以导致与癌症相关的肿瘤的生长抑制或细胞毒性。本文所述的用于治疗癌症的方法可以导致与癌症相关的肿瘤的消退,包含完全消退或部分消退。本文还提供了治疗基础疾病(例如,癌症)并且延长受试者的存活的方法。
在一个实施例中,提供了一种抑制有需要的受试者的肿瘤生长的方法,所述方法包括向受试者施用有效量的SRA141。在一些方面,本公开提供了一种向受试者施用有效量的SRA141以抑制肿瘤生长的方法,其中肿瘤生长减少至少47%。在一些方面,本公开提供了一种向受试者施用有效量的SRA141以抑制肿瘤生长的方法,其中肿瘤生长减少至少93%。在一些方面,本公开提供了一种向受试者施用有效量的SRA141以抑制肿瘤生长的方法,其中如通过肿瘤体积所测量的,肿瘤生长减少1%、2%、3%、4%、5%、6%、7%、8%、9%、10%、12%、14%、16%、18%、20%、22%、24%、26%、28%、30%、32%、34%、36%、38%、40%、42%、44%、46%、48%、50%、52%、54%、56%、58%、60%、62%、64%、66%、68%、70%、72%、74%、76%、78%、80%、82%、84%、86%、88%、90%、92%、94%、96%、98%或100%。在一些方面,本公开提供了一种向受试者施用有效量的SRA141以抑制肿瘤生长的方法,其中如通过肿瘤的绝对大小所测量的,肿瘤生长减少1%、2%、3%、4%、5%、6%、7%、8%、9%、10%、12%、14%、16%、18%、20%、22%、24%、26%、28%、30%、32%、34%、36%、38%、40%、42%、44%、46%、48%、50%、52%、54%、56%、58%、60%、62%、64%、66%、68%、70%、72%、74%、76%、78%、80%、82%、84%、86%、88%、90%、92%、94%、96%、98%或100%。在一些方面,本公开提供了一种向受试者施用有效量的SRA141以抑制肿瘤生长的方法,其中如通过所述类型的肿瘤的肿瘤标志物的表达水平所测量的,肿瘤生长减少1%、2%、3%、4%、5%、6%、7%、8%、9%、10%、12%、14%、16%、18%、20%、22%、24%、26%、28%、30%、32%、34%、36%、38%、40%、42%、44%、46%、48%、50%、52%、54%、56%、58%、60%、62%、64%、66%、68%、70%、72%、74%、76%、78%、80%、82%、84%、86%、88%、90%、92%、94%、96%、98%或100%。本公开还涉及使用有效量的化合物SRA141和第二有效量的另外的治疗来抑制肿瘤的进展、减小所述肿瘤的聚集大小、减小所述肿瘤的体积、减小所述肿瘤的直径和/或以其它方式抑制所述肿瘤的生长的方法。本文还提供了治疗基础疾病(例如,癌症)并且延长受试者的存活的方法。
在一个实施例中,提供了一种抑制有需要的受试者的肿瘤生长的方法,所述方法包括向受试者施用有效量的SRA141和第二有效量的另外的治疗。在一些方面,本公开提供了一种向受试者施用有效量的SRA141和第二有效量的另外的治疗以抑制肿瘤生长的方法,其中如通过肿瘤体积所测量的,肿瘤生长减少1%、2%、3%、4%、5%、6%、7%、8%、9%、10%、12%、14%、16%、18%、20%、22%、24%、26%、28%、30%、32%、34%、36%、38%、40%、42%、44%、46%、48%、50%、52%、54%、56%、58%、60%、62%、64%、66%、68%、70%、72%、74%、76%、78%、80%、82%、84%、86%、88%、90%、92%、94%、96%、98%或100%。在一些方面,本公开提供了一种向受试者施用有效量的SRA141和第二有效量的另外的治疗以抑制肿瘤生长的方法,其中如通过肿瘤的绝对大小所测量的,肿瘤生长减少1%、2%、3%、4%、5%、6%、7%、8%、9%、10%、12%、14%、16%、18%、20%、22%、24%、26%、28%、30%、32%、34%、36%、38%、40%、42%、44%、46%、48%、50%、52%、54%、56%、58%、60%、62%、64%、66%、68%、70%、72%、74%、76%、78%、80%、82%、84%、86%、88%、90%、92%、94%、96%、98%或100%。在一些方面,本公开提供了一种向受试者施用有效量的SRA141和第二有效量的另外的治疗以抑制肿瘤生长的方法,其中如通过所述类型的肿瘤的肿瘤标志物的表达水平所测量的,肿瘤生长减少1%、2%、3%、4%、5%、6%、7%、8%、9%、10%、12%、14%、16%、18%、20%、22%、24%、26%、28%、30%、32%、34%、36%、38%、40%、42%、44%、46%、48%、50%、52%、54%、56%、58%、60%、62%、64%、66%、68%、70%、72%、74%、76%、78%、80%、82%、84%、86%、88%、90%、92%、94%、96%、98%或100%。
临床终点
本文提供了用于治疗癌症的方法,例如抑制受试者的肿瘤的生长和/或抑制肿瘤细胞的生长,其中所述方法导致临床相关的终点。
与细胞分裂的正常和健康过程相比,当一个或多个生物细胞生长并且分裂快得多时,肿瘤生长发生,从而导致细胞数量增加。这种现象指示细胞处于疾病状态,如癌症或癌前。此外,肿瘤生长时常发生在聚集细胞形成肿瘤之前的离散阶段。
存在若干种方法可供技术人员用于测量细胞复制速率。细胞内部的总体代谢活性可以通过经标记的生物产物来测量。例如,存在若干可商购获得的染料(例如,MTT),所述染料可以穿透细胞并且与某些酶和其它因子相互作用以产生可检测的产物。而且,细胞生物标志物可以在细胞中测量。例如,BrdU测定可以将胸苷衍生物并入到细胞DNA中并且用抗体进行检测。增殖细胞核抗原(PCNA)是用于检测的另一种此类生物标记。除了标记技术之外,技术人员还可以使用例如显微术或流式细胞术来允许细胞计数。
一方面,细胞复制通过临床终点来测量,所述临床终点包含:生活质量(QOL)分数、应答持续时间(DOR、临床受益率(CBR)、患者报告的结果(PRO)、客观应答率(ORR)分数、无疾病存活(DFS)或无进展存活(PFS)、进展时间(TTP)、总体存活、治疗失败时间(TTF)、RECIST标准、部分应答(PR)、疾病稳定(SD)、进展性疾病(PD)和/或完全应答(CR)。临床终点可以使用本领域的技术人员熟知的方法来确定。
在一些方面,CR是所有靶病灶的消失,其中任何病理性淋巴结(靶标或非靶标)在短轴上减少至<10mm。在一些方面,参考基线总直径,PR是靶病灶的直径总和的至少30%减少。在一些方面,PD是靶病灶的直径总和的至少20%增加,取最小总和(如果基线总和是最小的话,则包含基线总和)作为参考。在一些方面,除了20%的相对增加之外,总和还可以表现出至少5mm的绝对增加。在一些方面,SD既不是足以符合PR的收缩,也不是足以符合PD的增加,取最小总和直径作为参考。
在一些方面,本公开提供了其中在施用有效量的SRA141之后,肿瘤生长减少至少约5、10、20、30、40、50、60、80、90、95、97、99或99.9%的方法。
在一些方面,本公开提供了其中基于一个或多个临床终点的一个或多个测量值来计算减少%的方法。
在一些方面,本公开提供了其中在施用有效量的SRA141之后,如通过MTT测定中总细胞计数的增加或减少所测量的,或如通过ctDNA测定所测量的基因谱的变化所测量的,肿瘤生长减少不超过或至少为5、10、20、30、40、50、60、80、90、95、97、99或99.9%的方法。
在一些总体方面,本公开提供了其中在施用有效量的SRA141之后,肿瘤生长减少至少5、10、20、30、40、50、60、80、90、95、97、99或99.9%的方法。在一些方面,本公开提供了其中在施用有效量的SRA141之后,如通过MTT测定中总细胞计数的增加或减少所测量的,或如通过ctDNA测定所测量的基因谱的变化所测量的,肿瘤生长减少至少为5、10、20、30、40、50、60、80、90、95、97、99或99.9%的方法。
在一些方面,本公开提供了其中施用导致IC
在一些方面,本公开提供了其中施用导致AUC为至少1、10、25、50、100、200、400、600、800或1000的方法。
在一些方面,本公开提供了其中施用导致IC
在一些方面,本公开提供了其中施用导致EC
在一些方面,本公开提供了其中施用导致治疗指数(TI)值的范围为约1.001:1到约50:1、约1.1:1到约15:1、约1.2:1到约12:1、约1.2:1到约10:1、约1.2:1到约5:1或约1.2:1到约3:1的方法。
在一些方面,本公开提供了其中施用导致GI
在一些方面,本公开提供了其中施用导致观察到的最大响应(最大响应)值不超过0.1、0.5、1、2μM、2.5μM、3μM、4μM、5μM或10μM的方法。
肿瘤生长可以表示为总肿瘤体积。存在总体而言并且特定于某些肿瘤模型的公式,技术人员可以基于实体瘤或多或少是球形的假设来使用公式计算肿瘤体积。在此方面,技术人员可以使用实验工具,如:超声成像、手动或数字测径器、超声检查、计算机断层摄影(CT)、显微CT、
在一些方面,本公开提供了其中施用导致在施用有效量的SRA141之后肿瘤体积减少至少5、10、20、30、40、50、60、80、90、95、97、99或99.9%的方法。在一些方面,本公开提供了其中施用导致在施用有效量的SRA141之后,如通过医学成像技术所测量的,肿瘤大小减小至少5、10、20、30、40、50、60、80、90、95、97、99或99.9%的方法。
在一些方面,本公开提供了其中施用导致在一(1)周、二(2)周、三(3)周、四(4)周、六(6)周、八(8)周、十二(12)周、十六(16)周、二十(20)周、二十四(24)周、三十六(36)周或五十二(52)周之后肿瘤体积减少至少5%的方法。
为了评估客观应答或肿瘤生长,可以估计基线处的总体肿瘤负荷并且将其用作后续测量值的比较器。可测量疾病可以通过至少一个可测量病灶的存在来定义。
在一些方面,本公开提供了其中当在基线处存在多于一个可测量病灶时,将代表所有涉及器官的所有病灶鉴定为靶病灶并且在基线处记录和测量,所述所有病灶至多为总共五个病灶的最大值(并且每个器官最多两个病灶)的方法。在一些方面,靶病灶是基于其大小(具有最长直径的病灶)来选择的并且代表所有涉及的器官。在一些方面,靶病灶是那些使其自身可再现地重复测量的靶病灶。在一些情况下,最大病灶不适合于使其自身可再现地测量,在这种情况下,选择可以可再现地测量的次最大病灶,如图3例示的,Eisenhauer等人(2009)。
当通过CT扫描节结的短轴≥15mm时,病理性淋巴结可以定义为是可测量的并且在一些情况下鉴定为靶病灶。在一些实施例中,节结的短轴有助于基线总和。在一些实施例中,节结的短轴是放射科医师用于判断节结是否涉及实体瘤的直径。在一些实施例中,节结大小被报道为在获得图像的平面(例如,用于CT扫描的轴向平面;用于MRI扫描的轴向、矢状或冠状平面,这取决于扫描的采集平面)中的两个维度。在一些实施例中,量度中的较小量度是短轴。在一些实施例中,短轴≥10mm但<15mm的病理性节结被认为是非靶病灶。在一些实施例中,短轴<10mm的节结被认为是非病理性的并且不被记录或跟踪。
在一些方面,计算所有靶病灶的直径的总和(非节结病灶为最长,节结病灶为短轴)并且将其报告为基线总和直径。在一些方面,如果淋巴结将被包含在总和中,则仅短轴被添加到总和中。在一些方面,基线总和直径用作表征客观肿瘤生长或消退的参考。
在一些方面,所有其它病灶或疾病位点(包含病理性淋巴结)被鉴定为非靶病灶并且在基线处记录。在一些情况下,不需要测量值,并且病灶遵循“存在(present)”、“不存在(absent)”或“明确进展(unequivocal progression)”。在一些情况下,可以记录涉及同一器官的多个非靶病灶(例如,“多个扩大的骨盆淋巴结”或“多个肝脏转移”)。
治疗有效量和单位剂型
通常,本发明技术的化合物将以治疗有效量通过用于类似用途的药剂的任何接受的施用模式进行施用。本发明技术的化合物(即活性成分)的实际量将取决于许多因素,如待治疗疾病的严重程度、受试者的年龄和相对健康状况、所使用的化合物的效力、施用途径和施用形式以及技术人员熟知的其它因素。药物可以一天至少一次施用,优选地一天一次或两次。
此类药剂的有效量可以通过常规实验容易地确定,如可以是最有效且方便的施用途径以及最适当的调配物。各种调配物和药物递送系统在本领域中是可获得的。参见例如,Gennaro,A.R.编辑(1995)《雷明顿氏药物科学(Remington's PharmaceuticalSciences)》,第18版,马克出版有限公司(Mack Publishing Co.)。
治疗有效剂量可以使用本领域熟知的各种技术进行最初估计。在动物研究中使用的初始剂量可以基于在细胞培养测定中建立的有效浓度。例如,可以使用从动物研究和细胞培养测定中获得的数据来确定适合于人受试者的剂量范围。
药剂(例如用于本文所述方法中的化合物,例如Cdc7抑制剂SRA141)的有效量或治疗有效量或剂量是指导致受试者的症状改善或存活延长的药剂或化合物的量。此类分子的毒性和治疗功效可以通过标准药学程序在细胞培养物或实验动物中确定,例如通过确定LD
有效量或治疗有效量是将研究人员、兽医、医生或其它临床医生正寻求的在组织、系统、动物或人中引发生物应答或医学应答的化合物或药物组合物的量。剂量具体地落入包含ED
可以单独调整剂量和间隔以提供足以实现所期望效果的活性部分的血浆水平;即最小有效浓度(MEC)。对于每种化合物,MEC将会有所不同,但是可以从例如体外数据和动物实验中估计。实现MEC所需的剂量将取决于个体特性和施用途径。在局部施用或选择性摄取的情况下,药物的有效局部浓度可能与血浆浓度无关。
所施用的药剂或组合物的量可以取决于多种因素,包含待治疗的受试者的性别、年龄和体重、痛苦的严重程度、施用方式以及开处方的医师的判断。
治疗有效量可以与SRA141的有效量和另外治疗的第二有效量中的任一种或两种相同或不同。这是因为本公开提供了如本文所述的方法是有效的,甚至在SRA141的有效量和另外的治疗的第二有效量都不必须是仅改善疾病的症状的量(例如,如果作为个体疗法施用,SRA141和/或另外的治疗的量可以被认为是“亚治疗”量)。然而,本公开确实提供了必须提供治疗有效量的组合,即组合确实至少影响疾病的症状的治疗。
单位剂型是技术人员通常理解的术语。单位剂型是销售用于特定用途的药物产品。药物产品包含一种或多种活性成分以及任何非活性组分,最经常呈药学上可接受的载体或赋形剂的形式。应理解,多个单位剂型是不同的药物产品。因此,一个单位剂型可以是例如SRA141与250mg的另外治疗以每种组分的某一比率的组合,而另一个完全不同的单位剂型是例如SRA141与750mg的另外治疗以上文提及的每种组分的同一某一比率的组合。因此,从一个单位剂型到另一个单位剂型,SRA141的有效量和另外的治疗的第二有效量两者可以保持相同。当然,当SRA141的有效量或另外的治疗的第二有效量之一改变时,单位剂型是不同的。
在一些方面,有效量对于SRA141化合物是独特的,即所述有效量不同于另外的治疗的第二有效量。在一些方面,SRA141的有效量是等效于“治疗有效量”或引起治疗效果和/或有益效果的量的量。在一些方面,SRA141的有效量是“治疗有效量”。在一些方面,另外的治疗的第二有效量是“治疗有效量”。在一些方面,SRA141的有效量和另外的治疗的第二有效量两者都不是“治疗有效量”。在一些方面,第二有效量对于另外的治疗是独特的,即第二有效量对于不同的另外的治疗是不同量。
在一些方面,SRA141和另外的治疗组合被调配成一个(1)单位剂型。在一些方面,同一单位剂型被施用持续至少四(4)小时、六(6)小时、八(8)小时、十二(12)小时、二十四(24)小时、一(1)天、二(2)天、三(3)天、七(7)天、十(10)天、十四(14)天、二十一(21)天或三十(30)天。
在一些方面,SRA141和另外的治疗组合被调配成至少两种(2)分别不同的单位剂型。在一些方面,第一有效量在第一单位剂型中与在第二单位剂型中不同。在一些方面,SRA141的有效量在第一单位剂型中与其在第二单位剂型中相同。
在一些方面,第一单位剂型与第二单位剂型相同。在一些方面,第一单位剂型与第二单位剂型和第三单位剂型相同。在一些方面,第一单位剂型与第二单位剂型、第三单位剂型和第四单位剂型相同。
本发明的化合物
一方面,本公开提供了化合物SRA141的使用方法。
SRA141
化合物SRA141还由以下化学名称鉴定:乙基5-(1H-吡咯并[2,3-b]吡啶-3-基)亚甲基4-氧代-2-{[4-(2,2,2-三氟乙基)哌嗪基]氨基-4,5-二氢呋喃-3-甲酸酯。
SRA141是在国际专利公开号WO 2012133802中公开的化合物,所述国际专利教导的全部内容通过引用并入本文。技术人员将在国际专利公开号WO 2012133802中找到如何合成SRA141。
SRA141结构如下所示:
药物组合物
本文描述了用于抑制肿瘤生长、抑制癌症进展或治疗癌症的方法。所述方法包含施用有效量的SRA141和任选地第二有效量的另外的治疗。SRA141和另外的治疗可以各自调配在药物组合物中。除了一种或多种活性化合物之外,这些药物组合物可以包括药学上可接受的赋形剂、载体、缓冲剂、稳定剂或本领域的技术人员熟知的其它材料。此类材料应该是无毒的并且不应该干扰活性成分的功效。载体或其它材料的确切性质可以取决于施用途径,例如口服、静脉内、皮肤或皮下、鼻、肌内、腹膜内途径。
口服施用的药物组合物可以是片剂、胶囊、粉末或液体形式。片剂可以包含固体载体,如明胶。液体药物组合物通常包含液体载体,如水、石油衍生物、动物油或植物油、矿物油或合成油。可以包含生理盐水溶液、葡萄糖或其它糖溶液或二醇,如乙二醇、丙二醇或聚乙二醇。
对于静脉内、皮肤或皮下注射,或在痛苦位点注射,活性成分将是呈肠道外可接受的水溶液的形式,所述水溶液是无热原的并且具有合适的pH、等渗性和稳定性。本领域的相关技术人员完全能够使用例如等渗媒剂(如氯化钠注射液、林格氏注射液(Ringer'sInjection)、乳酸化林格氏注射液(Lactated Ringer's Injection))制备合适的溶液。根据需要,可以包含防腐剂、稳定剂、缓冲剂、抗氧化剂和/或其它添加剂。
根据待治疗的病状,组合物可以单独地或与其它治疗组合,同时或者顺序地施用。
本发明技术不限于任何特定的组合物或药物载体,因为这些可以变化。通常,本发明技术的化合物将通过以下途径中的任何一种作为药物组合物施用:口服、全身(例如,经皮、鼻内或通过栓剂)或肠胃外(例如,肌内、静脉内或皮下)施用。优选的施用方式是使用可以根据痛苦程度进行调整的方便的每天剂量方案口服。组合物可以采取片剂、丸剂、胶囊、半固体、粉末、缓释调配物、溶液、悬浮液、酏剂、气雾剂或任何其它适当的组合物的形式。用于施用本发明技术的化合物的另一种优选方式是吸入。
调配物的选择取决于各种因素,如药物施用模式和药物物质的生物利用度。为了通过吸入进行递送,可以将化合物调配为液体溶液、悬浮液、气溶胶推进剂或干粉并且装载到合适的分配器中进行施用。存在若干种类型的药物吸入装置-喷雾器吸入器、计量剂量吸入器(MDI)和干粉吸入器(DPI)。喷雾器装置产生高速空气流,所述高速空气流使治疗剂(其被调配成液体形式)作为被携带到受治疗者的呼吸道中的雾而喷射。MDI通常是用压缩气体包装的调配物。在致动时,装置通过压缩气体排出所测得量的治疗剂,从而提供施用设定量的药剂的可靠方法。DPI以自由流动粉末的形式分配治疗剂,所述流动粉末在通过装置呼吸期间可以分散在受试者的吸入空气流中。为了获得自由流动粉末,治疗剂与赋形剂(如乳糖)一起调配。将所测得量的治疗剂以胶囊形式储存,并且在每次致动时分配。
本发明技术的化合物的药物剂型可以通过本领域熟知的任何方法制造,例如通过常规混合、筛分、溶解、熔融、制粒、糖衣丸制备、压片、悬浮、挤出、喷雾干燥、磨细、乳化、(纳米/微)包封、包埋或冻干方法。如上所述,本发明技术的组合物可以包含一种或多种生理学上可接受的非活性成分,其促进将活性分子加工成用于药物用途的制剂。
最近,基于生物利用度可以通过增加表面积(即,减小粒度)而增加的原理,已经开发了尤其是用于显示不良生物利用度的药物的药物调配物。例如,美国专利第4,107,288号描述了颗粒的大小范围为10到1,000nm的药物调配物,其中活性物质负载在大分子的交联基质上。美国专利第5,145,684号描述了药物调配物的生产,其中在表面改性剂的存在下将药物物质粉碎成纳米颗粒(平均粒度为400nm)并且然后分散在液体介质中以给出表现出显著高的生物利用度的药物调配物。
组合物通常由本发明技术的化合物与至少一种药学上可接受的赋形剂组合构成。可接受的赋形剂是无毒的,有助于施用,并且不会不利地影响所要求保护的化合物的治疗益处。此类赋形剂可以是任何固体、液体、半固体,或者在气溶胶组合物的情况下,是本领域的技术人员通常可获得的气态赋形剂。
固体药物赋形剂包含淀粉、纤维素、滑石粉、葡萄糖、乳糖、蔗糖、明胶、麦芽、大米、面粉、白垩、硅胶、硬脂酸镁、硬脂酸钠、单硬脂酸甘油酯、氯化钠、脱脂奶粉等。液体和半固体赋形剂可以选自甘油、丙二醇、水、乙醇和各种油,包含石油、动物、植物或合成来源的那些,例如花生油、大豆油、矿物油、芝麻油等。优选的液体载体,特别是用于可注射溶液的液体载体包含水、盐水、葡萄糖水溶液和二醇。
压缩气体可以用于以气溶胶形式分散本发明技术的化合物。适用于此目的的惰性气体是氮气、二氧化碳等。其它合适的药物赋形剂和其调配物描述于由E.W.Martin(马克出版有限公司,第18版,1990)编辑的《雷明顿氏药物科学》中。
在一些实施例中,药物组合物包含药学上可接受的盐。术语“药学上可接受的盐”是指衍生自本领域熟知的多种有机抗衡离子和无机抗衡离子的盐,仅举例来说,包含钠、钾、钙、镁、铵和四烷基铵,并且当分子含有碱性官能团时,有机酸或无机酸的盐,如盐酸盐、氢溴酸盐、酒石酸盐、甲磺酸盐、乙酸盐、马来酸盐和草酸盐。合适的盐包含在Stahl和Wermuth(编辑),《药用盐的性质、选择和用途手册(Handbook of Pharmaceutical SaltsProperties,Selection,and Use)》;2002中描述的那些盐。
如果需要,本发明的组合物可以存在于包装或分配器装置中,所述包装或分配器装置含有活性成分的一个或多个单位剂型。这种包装或装置可以例如包括金属或塑料箔,如泡罩包装或玻璃,以及橡胶塞,如在小瓶中。包装或分配器装置可以附有施用说明书。还可以制备包括在相容药物载体中调配的本发明技术的化合物的组合物,将其置于适当的容器中,并且标记用于治疗指定病状。
调配物中化合物的量可以在本领域的技术人员采用的全部范围内变化。通常,基于总调配物,调配物将在重量百分比(wt%)的基础上含有约0.01-99.99wt%的本发明技术的化合物,其中余量是一种或多种合适的药物赋形剂。优选地,化合物以约1-80wt%的水平存在。以下描述了代表性药物调配物。
调配物实例
以下是单独或组合的含有SRA141和另外的治疗的代表性药物调配物。
根据待治疗的病状,组合物可以单独地或与其它治疗组合,同时或者顺序地施用。
试剂盒
本公开还提供了一种试剂盒,所述试剂盒包括SRA141以及任选地另外的治疗和使用说明书的组合。本公开进一步提供了一种试剂盒,所述试剂盒包括一种或多种药物组合物,其中一种或多种药物组合物包括SRA141和另外的治疗以及使用说明书,任选地组合包含至少一种药学上可接受的载体或赋形剂。
试剂盒的单独组分可以包装在分开的容器中,并且与此类容器相关的可以是由管理药物或生物产品的制造、使用或销售的政府机构规定的形式的公告,所述公告反映所述机构对制造、使用或销售的批准。试剂盒可以任选地含有概述抗原结合构建体的使用方法或施用方案的说明书或指导。
在一些方面,本公开提供了一种试剂盒,所述试剂盒包括SRA141和另外的治疗以及至少一种药学上可接受的载体或赋形剂的组合。
当试剂盒的一种或多种组分作为溶液(例如,水溶液)或无菌水溶液提供时,容器装置本身可以是吸入剂、注射器、移液管、滴眼器或其它此类相似设备,从所述吸入剂、注射器、移液管、滴眼器或其它此类相似设备可以向受试者施用溶液或将溶液应用于试剂盒的其它组分并且与所述其它组分混合。
试剂盒的组分还可以以干燥或冻干的形式提供,并且试剂盒可以另外含有合适的溶剂用于冻干组分的重构。不论容器的数量或类型如何,本文所述的试剂盒还可以包括用于辅助向患者施用组合物的仪器。此类仪器可以是吸入器、鼻喷雾装置、注射器、移液管、镊子、测量勺、滴眼器或类似的医学上批准的递送媒剂。
在本文所述的另一方面,提供了一种制品,所述制品含有可用于治疗、预防和/或诊断本文所述的病症(例如,抑制肿瘤生长)的材料。制品可以包括容器和在容器上或与容器有关的标签或包装插页。合适的容器包含例如瓶、小瓶、注射器、iv.溶液袋等。容器可以由多种材料(如玻璃或塑料)形成。一个或多个容器容纳有通过本身有效用于治疗、预防和/或诊断病症或与有效用于治疗、预防和/或诊断病症的另一种组合物组合的组合物并且可以具有无菌进入孔(例如,容器可以是静脉内溶液袋或具有可被皮下注射针刺穿的塞子的小瓶)。
本文所述的这个实施例中的制品可以进一步包括指示组合物可以用于治疗特定病状的标签或包装插页。可替代地或另外,制品可以进一步包括第二(或第三)容器,所述容器包括药学上可接受的缓冲剂,如注射用抑菌水(BWFI)、磷酸盐缓冲盐水、林格氏溶液和葡萄糖溶液。制品可以进一步包含从商业和使用者立场上期望的其它材料,所述材料包含其它缓冲剂、稀释剂、过滤器、针和注射器。
多肽和核酸
本文描述了可用于本文所述的方法的基因(例如,Cdc7的基因)的多肽和核酸序列。在一些实施例中,可用于本文所述的方法的多肽和核酸序列与本文所述的或在本文中由数据库登录号提及的序列至少95%、96%、97%、98%或99%相同。在一些实施例中,可用于本文所述的方法的多肽和核酸序列与本文所述的或在本文中由数据库登录号提及的序列100%相同。
在两个或更多个核酸或多肽序列的上下文中,术语“同一性百分比”是指当针对最大对应性进行比较和比对时,如使用下述序列比较算法(例如,BLASTP和BLASTN或技术人员可获得的其它算法)中的一种算法或通过视觉检查测量的,具有指定百分比的相同的核苷酸或氨基酸残基的两个或更多个序列或子序列。根据应用,“同一性”百分比可以超过被比较的序列的区存在,例如,高于功能结构域,或可替代地,超过待比较的两个序列的全长存在。为了进行序列比较,通常一个序列充当与测试序列进行比较的参考序列。当使用序列比较算法时,将测试序列和参考序列输入到计算机,指定子序列坐标(如有必要),并且指定序列算法程序参数。然后,序列比较算法基于所指定的程序参数,计算一个或多个测试序列相对于参考序列的序列同一性百分比。用于比较的序列的最佳比对可以例如通过以下进行:Smith和Waterman,《高级应用数学(Adv.Appl.Math.)》2:482(1981)的局部同源算法;通过Needleman和Wunsch,《分子生物学杂志(J.Mol.Biol.)》48:443(1970)的同源比对算法;通过Pearson和Lipman,《美国国家科学院院刊(Proc.Nat'l.Acad.Sci.USA)》85:2444(1988)的相似性搜索法;通过这些算法的计算机化实施方案(威斯康星州麦迪逊科学大道575号遗传学计算机组(Genetics Computer Group)的威斯康星遗传学软件包中的GAP、BESTFIT、FASTA和TFASTA);或通过视觉检查(通常参见Ausubel等人,《当代分子生物学实验指南(Current Protocols in Molecular Biology)》(2003))。适用于确定序列同一性和序列相似性百分比的算法的一个实例是BLAST算法,所述算法描述于Altschul等人,《分子生物学杂志》215:403-410(1990)中。用于执行BLAST分析的软件可通过美国国家生物技术信息中心(National Center for Biotechnology Information)(www.ncbi.nlm.nih.gov)公开获得。
实例
以下是用于实施本发明的具体实施例的实例。提供实例仅出于说明性目的,并且不旨在以任何方式限制本发明的范围。已经做出努力以确保关于所使用的数字(例如,量、温度等)的准确性,但是当然应该允许有一些实验误差和偏差。
除非另有说明,否则本发明的实践将采用本领域的技术之内的蛋白质化学、生物化学、重组DNA技术和药理学的常规方法。此类技术在文献中有充分解释。参见例如,T.E.Creighton,《蛋白质:结构与分子性质(Proteins:Structures and MolecularProperties)》(W.H.弗里曼和公司(W.H.Freeman and Company),1993);A.L.Lehninger,《生物化学(Biochemistry)》(沃思出版社有限公司(Worth Publishers,Inc.),现行版);Sambrook等人,《分子克隆:实验室手册(Molecular Cloning:A Laboratory Manual)》(第2版,1989);《酶学方法(Methods In Enzymology)》(S.Colowick和N.Kaplan编辑,学术出版社有限公司(Academic Press,Inc.));《雷明顿氏药物科学》,第18版(宾夕法尼亚州伊斯顿市:马克出版公司(Easton,Pennsylvania:Mack Publishing Company),1990);Carey和Sundberg,《高等有机化学(Advanced Organic Chemistry)》,第3版(普莱南出版社(PlenumPress)),第A和B卷(1992)。
缩略词
表1-缩略词
实例1:小鼠AML异种移植模型的SRA141治疗
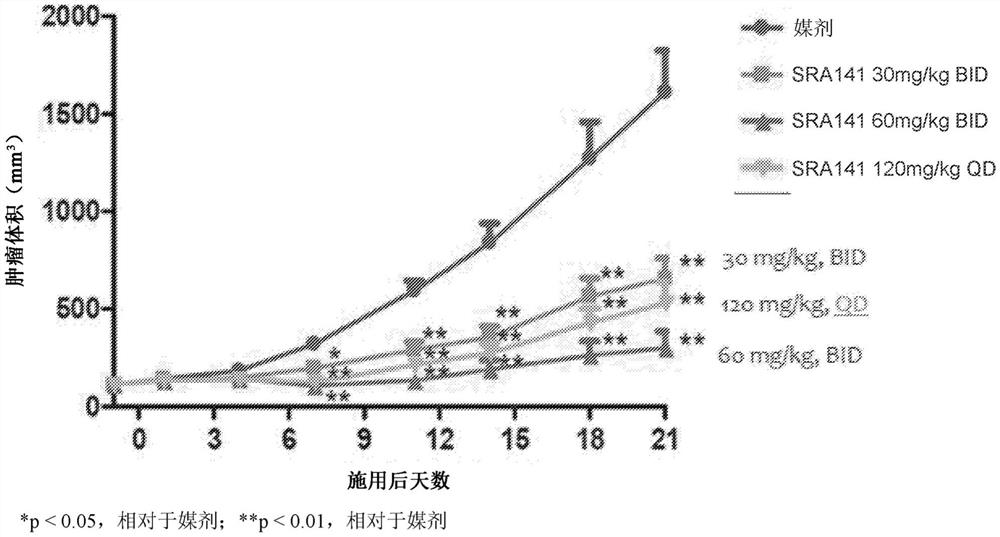
使用急性髓性白血病细胞系MV-4-11在异种移植物中,测试了单独的Cdc7抑制剂(化合物I-D,也被称为SRA141)作为单一疗法的功效。以不同剂量向携带MV-4-11异种移植肿瘤的雌性BALB/c小鼠施用SRA141。60只5-6周龄的雌性BALB/C小鼠皮下接种MV-4-11肿瘤细胞。用5.0×10
如图1A所示,在媒剂和30mg/kg BID、60mg/kg BID和120mg/kg QD mg/kg剂量水平下,四个组在第21天的平均肿瘤体积分别为1612.66mm
所有处理都是耐受的,没有显著的体重减轻(图1C)。在第21天四个组的平均相对体重变化(RCBW)分别是12.11%、16.39%、3.18%和8.47%。
这些结果证明SRA141单独作为单一疗法可以提供用于治疗急性髓性白血病的有效疗法。
实例2:大鼠AML异种移植模型的SRA141治疗
在急性骨髓性白血病(AML)的大鼠异种移植模型中,测试了Cdc7抑制剂(化合物SRA141)作为单一疗法的功效(图2)。雌性rnu/rnu大鼠皮下接种1×10
如图2A所示,在用75mg/kg SRA141(86.1%TGI)和150mg/kg SRA141(86.7%TGI)处理的大鼠中观察到显著的肿瘤生长抑制(TGI)。在用75mg/kg SRA141处理的大鼠中的另外的研究导致在处理的第27天观察到92%的TGI并且在处理的第34天观察到85%的TGI。媒剂对照组的中值肿瘤体积为13247(n=10),经SRA141处理组75mg/kg的中值肿瘤体积为1838(n=8),并且经SRA141处理的组150mg/kg组的中值肿瘤体积为1764(n=9)。在治疗过程中示出每只个体大鼠的每次治疗的结果(图2C)。值得注意的是,以75mg/kg BID用SRA141处理产生了1次完全消退(3次连续测量的肿瘤体积<13.5mm3)和3次部分消退(3次连续测量的肿瘤体积是初始体积的<50%)。完全消退持续到研究终止。以150mg/kg QD用SRA141处理产生了2种部分消退,其中一只动物在研究完成时没有可测量的肿瘤。
通常,所有处理都是耐受良好的,没有显著的体重减轻(图2B)。在研究过程期间发现三只动物死亡(分别在以75mg/kg BID的给药组中,n=2,并且在以150mg/kg BID的给药组中,n=1)。这些死亡的原因被分类为非治疗相关的。
这些结果证明SRA141作为单一疗法可以提供用于治疗AML的有效疗法。
实例3:各种癌细胞系的SRA141治疗
在一组235个癌细胞系中利用CellTiter-Glo
SRA141在一系列实体肿瘤细胞系中表现出有效的抑制活性(IC50<3μM),包含膀胱(5673,RT112/84)、肉瘤(143B,CADO-ES1,RD-ES,A673)、结肠和盲肠(Colo-205,DLD-1,LS1034,SK-CO-1,SNU-C1)、肾(UO.31)、头颈(CNE-2Z,RPMI-2650)、黑素瘤(A2058,A875)和胃(HGC-27)谱系的那些。
在血液源性细胞系中也观察到显著的活性。所测试的59种血液癌细胞系的结果示出于图3A中。这些结果证明SRA141单独作为单一疗法可以提供用于治疗血液癌的有效疗法,如急性髓性白血病(图3A,细胞系表示为“C”)和慢性嗜酸性粒细胞白血病(图3A,细胞系表示为“L”)。还在i ALL系(RS411,SUP-B15,Reh)和其它白血病和淋巴瘤(KHYG-1和JeKo-1)中观察到显著的活性。
在图3B中汇总了所确定的IC50值的汇总,并且根据癌症类型进行分解。总体上,血液癌系示出比实体瘤系更高的敏感性,并且在后者内,结肠直肠癌系是之中最敏感的。
在范围为0.03到33.3μM(48小时温育)的药物浓度下,评估SRA141在Colo-205结肠癌细胞相对于正常人真皮成纤维细胞(NHDF)中的细胞毒性差异活性。处理后,通过流式细胞术分析用碘化丙啶标记的细胞,以确定由于细胞凋亡介导的DNA片段化而在亚G1部分中发现的垂死细胞的百分比。如图3C所示,观察到癌细胞与正常细胞之间对SRA141的敏感性的显著差异。值得注意的是,多达62%的癌细胞是死亡的并且在化合物浓度介于1μM与10μM之间下发现于亚G1部分中。相比之下,如通过亚G1部分确定的,少于10%的正常细胞是凋亡的,这与所公开报告一致,所述所公开报告示在Cdc7抑制之后未经转化的细胞的最小细胞毒性(Montagnoli,2004)。因此,这些发现支持SRA141在肿瘤与未经转化的组织之间的潜在治疗指数。
如图3C,B部分所示,使用两种不同活力测定的实验证明SRA141在许多细胞系中相对于其它Cdc7抑制剂TAK-931和LY-3177833具有相当的或优越的体外活性。总之,SRA141在多种细胞系中跨若干适应症表现出有效的抗增殖活性。
SRA141的抗增殖活性进一步在七种细胞系(Colo-205、SW620、SNU-398、NCI-H716、MDA-MB-231、NCI-H1573和SW1116)中使用设计用于测量以下的四种正交测定进行表征:ATP水平(CTG);代谢活性(CellTiter-Blue(CTB));细胞的DNA含量(CyQuant);和酯酶活性(钙黄绿素AM)。每种测定形式内的细胞系的相对敏感性通常是一致的。如图3D所示,在所有所测试的测定和细胞系中,当与72小时(左列)相比时,在144小时(右列)的较长处理持续时间下观察到SRA141功效的一致增加,这表明最佳抗肿瘤活性将需要连续的靶覆盖。在对SRA141的不同暴露时间之后,在144小时时还通过CTG和CyQuant确定单一疗法SRA141IC50。如图3E所示,与早期评估一致,SRA141功效随着处理持续时间超过24小时而增加(24小时左列、48小时中间列或72小时右列)。
实例4:患者源性CRC的异种移植小鼠模型的SRA141治疗
在结肠直肠癌(CRC)的患者源性异种移植(PDX)模型中测试了Cdc7抑制剂(化合物SRA141)作为单一疗法的功效。向携带CTG-1009患者源性APC-和TP53-突变体结肠直肠肿瘤异种移植物的雌性nu/nu小鼠施用(i)媒剂对照(0.1M HCl/0.5%MC)或(ii)以120mg/kg QD施用SRA141持续28天或(iii)以150mg/kg QD施用SRA141持续7天周期的连续2天,持续28天(n=8每SRA141处理组;n=6媒剂对照组;冠军研究1250-003)。在研究的最后一天第32天计算肿瘤生长抑制。由于体重减轻,在SRA141 120mg/kg QD和150mg/kg QD组中,暂时中止对多只动物的给药(图4B)。在SRA141 120mg/kg QD和150mg/kg QD处理组中,这些给药假期的范围分别是1-3天(n=1和3只动物)、4-8天(n=7和0只动物)。
如图4A所示,SRA141施用在120mg/kg QD和150mg/kg QD剂量水平下分别导致49%和36%的肿瘤生长抑制。
在SRA141(120mg/kg,QD 5D给药,其中剂量假期范围为5-7D,并且随后QD给药)之后观察到的平均肿瘤生长抑制(TGI)为49.4%(P=0.0057)。在实验时间期间SRA141给药4次或6次导致对D32的平均TGI为35.8%(P=0.0385)。SRA141后观察到的平均TGI(120mg/kg,QD 5D给药,其中剂量假期范围为5-7D,并且随后QD给药)为49.4%(P=0.0057)。来自平均媒剂肿瘤体积(TV)的个体TGI为:23.5、31.3、47.8、50.8、59.9、63.6和68.6%。在实验时间期间SRA141给药4次或6次导致对D32的平均TGI为35.8%(P=0.0385)。具有4个剂量的3只小鼠的来自平均媒剂TV的TGI为12.4、27.2和53.2%。接受6个剂量的5只小鼠的来自平均媒剂TV的TGI为15.6、18.2、48.2、65.2和71.2%。在治疗过程中(图5)和第32天(图6)示出每只个体小鼠的每次治疗的结果。因此,SRA141的单一疗法导致显著的肿瘤生长抑制(TGI),如针对研究的D32所确定的。
通常,所有处理都在治疗结束时耐受性良好,相对于媒剂(实心圆)没有显著的体重减轻。在120mg/kg组中初始给药5天后观察到适度的体重减轻(实心正方形),但在若干天药物假期后恢复到媒剂水平(图4B)。
这些结果证明SRA141作为单一疗法可以提供用于在APC和TP53突变的CRC设置中治疗结肠直肠癌的有效疗法。
实例5:患者源性CRC异种移植大鼠模型的SRA141治疗
在结肠直肠癌(CRC)的大鼠异种移植模型中,测试了Cdc7抑制剂(化合物SRA141)作为单一疗法的功效(图7)。雌性Rowett裸大鼠皮下接种2×10
如图7A所示,SRA141的施用在150mg/kg QD和75mg/kg BID剂量水平下分别导致50%和93%的显著肿瘤生长抑制。在4/7大鼠中观察到完全肿瘤消退。此外,在7只动物中的4只中观察到完全肿瘤消退(定义为3个连续测量没有可测量的肿瘤)并且在7只动物中的1只中以75mg/kg BID剂量观察到部分消退(3次连续测量的肿瘤体积是初始体积的<50%)。所有4次完全消退持续到研究完成。
如图7B所示,与用媒剂处理的大鼠相比,大约1.2μM的肿瘤SRA141浓度导致磷酸化MCM2(丝氨酸53)降低大约50%,这表明Cdc7的选择性抑制与肿瘤细胞存活率不相容。用单剂量的SRA141或媒剂处理Colo-205荷瘤大鼠,并且12小时后收集肿瘤。通过LC/MSMS或蛋白质印迹分析从每个处理组三只动物制备的肿瘤匀浆。结果证明SRA141作为单一疗法可以提供用于治疗结肠直肠癌的有效疗法。
实例6:SRA141与另外的抗肿瘤剂的组合
为了确认Cdc7抑制剂与抗肿瘤剂组合对癌细胞致敏的功效,在人癌细胞中进行实验。
用于组合评估的方法:
使用SRA141和配偶体化合物的共同处理给药方案进行组合筛选。在时间零(0小时)时添加增强物(enhancee)(SRA141)和增强子(配偶体化合物)两者。将细胞暴露于SRA141和增强子持续整个72小时处理时间。所有数据点通过自动化过程收集并且经受质量控制并且使用专有软件分析。如果测定板通过以下质量控制标准,则其被接受:在整个实验中相对原始值是一致的,Z因子分数大于0.6并且未处理的/媒剂对照在板上表现一致。
生长抑制(GI)用作细胞生长的量度。GI百分比是通过应用以下测试和等式来计算的:
如果T 如果T≥V 其中T是测试物品在72或96小时的信号量度,V是未处理的/经媒剂处理的对照量度,并且V 作为细胞活力的量度的抑制:0%的抑制水平表示没有通过处理抑制细胞生长。100%的抑制表示在处理窗口期间细胞数量没有加倍。细胞抑制和细胞毒性处理两者均可以产生100%的抑制百分比。抑制百分比如下计算:I=1-T/U,其中T为处理的并且U是未处理的。 协同作用分数分析 为了测量超过Loewe加和性的组合效应,使用标量量度来表征称为协同作用分数的协同相互作用的强度。协同作用分数计算为: 协同作用分数=logf 相对于所有经媒剂处理的对照孔的中值,计算矩阵中每种组分药剂和组合点的分数抑制。协同作用分数等式将实验观察到的超过模型表面的矩阵中每一点处的活性体积进行积分,所述模型表面在数值上源自使用Loewe加和性模型的组分药剂的活性。协同作用分数等式(上文)中的另外术语用于使用于单个药剂的各种稀释因子归一化并且允许在整个实验中比较协同作用分数。包含正抑制门控或Idata倍增器去除了接近零效应水平的噪声,并且偏置了在高活性水平下发生的协同相互作用的结果。具有更高的最大生长抑制(GI)效应的组合或在低浓度下协同的那些组合将具有更高的协同作用分数。具有统计学上取代基线自交值的协同作用分数的那些组合可以被认为是协同的。GI效应的量级可以与细胞的生长速率相关,所述细胞的生长速率因所检查的每种细胞系而变化。 使用等效线图评估效力变动,所述等效线图证明当与达到期望效果所需的单一药剂剂量相比时,组合中需要多少较少药物来实现所述效果水平。通过鉴定对应于越过所指示抑制水平的浓度的基因座绘制等效线图。这通过找到剂量矩阵中每种单一药剂浓度跨其它单一药剂浓度的交叉点来完成。实际上,每个垂直浓度C 协同作用分数分析:Loewe体积分数分析 Loewe体积分数用于评估超过Loewe加和性模型的组合相互作用的总体量值。当区分表型活性(正Loewe体积)相对于协同拮抗作用(负Loewe体积)的协同增加时,Loewe体积是特别有用的。当观察到拮抗作用时,如在当前的数据集中,评估Loewe体积以检查在拮抗作用与特定药物靶活性或细胞基因型之间是否存在任何相关性。这个模型将加和性定义为非协同组合相互作用,其中组合剂量矩阵表面与其自身交叉的任一药物均不可区分。加和性的计算为:满足(X/X 为了确认Cdc7抑制剂与抗肿瘤剂组合杀死癌细胞的功效,在Colo-205结肠直肠腺癌细胞和急性髓性白血病细胞中进行实验,其中采用Cdc7抑制剂SRA141与抗肿瘤剂组合顺序治疗或同时治疗。为了确认当与哺乳动物雷帕霉素靶蛋白(mTOR)途径抑制剂、DNA聚合酶抑制剂、受体酪氨酸激酶(RTK)抑制剂、丝裂原活化蛋白激酶途径(MAPK)抑制剂或磷脂酰肌醇-4,5-二磷酸3激酶(PI3K)途径抑制剂组合使用时Cdc7抑制剂对癌细胞致敏的功效,将人Colo-205结肠直肠腺癌细胞顺序地用SRA141处理,然后用作为单一药剂的依维莫司(everolimus)、阿非迪霉素(aphidicolin)或米哚妥林(midostaurin)处理(图8)。将Colo-205细胞的烧瓶用SRA141(1μM和200nM)和DMSO对照进行预处理持续48小时;将细胞洗涤并接种到384孔板中,并且使板温育6小时,随后在铺板后六小时用作为单一药剂的依维莫司、阿非迪霉素或米哚妥林进行处理。72小时后,使用ATPlite测量ATP水平。用SRA141预处理COLO-205细胞证明了增强子效力向米哚妥林敏感性增加转变,并且与DMSO预处理相比,用200nM SRA141预处理观察到3.19EC 为了确认Cdc7抑制剂与MAPK途径抑制剂或PI3K途径抑制剂组合用于杀死癌细胞和用于治疗癌症的功效,将Colo-205细胞用MAPK途径抑制剂、曲美替尼、PI3K途径抑制剂、库潘尼西和抗代谢物(抗叶酸盐)、甲氨蝶呤同时处理(图9和10A)。在第0天将细胞接种到384孔板中;使板温育24小时;将曲美替尼和库潘尼西与SRA141组合给予细胞,并且在72小时后,使用ATPlite测量ATP水平。在COLO-205细胞中观察到强协同作用,同时用MAPK途径抑制剂和PI3K途径抑制剂和抗叶酸抗代谢药、甲氨蝶呤进行组合处理。 为了确认当与丝裂原活化蛋白激酶途径(MAPK)抑制剂、类视黄醇途径调节剂、细胞凋亡调节剂、PARP抑制剂或哺乳动物雷帕霉素靶蛋白(mTOR)途径抑制剂组合使用时Cdc7抑制剂对癌细胞致敏的功效,人急性髓性白血病细胞MOLM-13细胞顺序地用SRA141处理,随后用作为单一药剂的曲美替尼或蓓萨罗丁处理(图8)。将MOLM-13细胞的烧瓶用SRA141(1μM和200nM)和DMSO对照进行预处理持续48小时;将细胞洗涤并接种到384孔板中;使板温育6小时,随后在铺板后六小时用作为单一药剂的曲美替尼或蓓萨罗丁进行处理。72小时后,使用ATPlite测量ATP水平。对于曲美替尼,与DMSO预处理相比,用200nM SRA141预处理观察到2.63EC 为了确认Cdc7抑制剂与类视黄醇途径调节剂、细胞凋亡调节剂或PARP抑制剂组合用于杀死癌细胞并用作治疗癌症的组合疗法的功效,人MOLM-13和KG-1细胞用PARP抑制剂BMN673、细胞凋亡调节剂ABT-199、类视黄醇途径调节剂、蓓萨罗丁或类视黄醇途径调节剂、维甲酸同时处理(图9和10A)。在第0天将细胞接种到384孔板中;使板温育24小时;将BMN673、ABT-199、蓓萨罗丁或维甲酸与SRA141组合给予细胞,并且在72小时后,使用ATPlite测量ATP水平。MOLM-13细胞证明对BMN673(PARP抑制剂)、ABT-199(BCL-2抑制剂)、蓓萨罗丁和维甲酸(类视黄醇途径调节剂)的强协同作用。这些结果证实了,包括利用顺序施用或同时施用Cdc7抑制剂将Cdc7抑制剂与mTOR途径抑制剂、DNA聚合酶抑制剂和受体酪氨酸激酶抑制剂以及MAPK途径抑制剂、类视黄醇途径调节剂、细胞凋亡调节剂或PARP抑制剂组合使用的组合疗法对于杀死癌细胞是有效的,并且证明,将Cdc7抑制剂与mTOR途径抑制剂、DNA聚合酶抑制剂和受体酪氨酸激酶抑制剂以及MAPK途径抑制剂、类视黄醇途径调节剂、细胞凋亡调节剂或PARP抑制剂组合施用是类视黄醇途径调节剂、细胞凋亡调节剂或PARP抑制剂是有效的癌症治疗方案。 为了确认Cdc7抑制剂与复制应激诱导性抗肿瘤剂吉西他滨组合的功效,将人HT-29结肠直肠腺癌细胞用递增浓度的吉西他滨(0.037到1μM)进行处理96小时。图10B中所示的数据证明,大于100nm的吉西他滨剂量导致处理细胞的生长抑制接近100%。进一步地,添加浓度为1.1μM或3.3μM的SRA141显著降低了吉西他滨的抑制作用。 实例7:SRA141在生物化学测定中的效力和选择性 使用体外激酶测定测量SRA141对Cdc7的抑制。 如图11A所示,SRA141在体外生化测定中证明对Cdc7的有效抑制(IC50=4nM)。在测定之前将SRA141与Cdc7酶预温育1小时适度地改善化合物效力(IC50=1.4nM),从而证实了用潜在有利的抑制剂结合动力学对Cdc7的有效抑制。这些发现与快速稀释研究一致,所述快速稀释研究表明SRA141与Cdc7结合的停留时间长(t1/2=215分钟)并且解离动力学慢(在5nM下,Koff=0.0032),如图11B中所示。 使用含有大约430种天然激酶和突变激酶的DiscoverX激酶组筛选测定评估SRA141对Cdc7的选择性。在500nM的化合物浓度下检测到最小脱靶激酶活性。将SRA141对Cdc7的选择性与TAK-931进行比较。激酶组筛选测定的结果(图12)证明,在化合物浓度为500nM下,与TAK-931相比,SRA141具有更少的脱靶活性。 实例8:SRA141对若干种细胞周期激酶的细胞底物的影响 将Colo-205细胞用SRA141(在浓度介于0.033与3.3μM之间下)进行处理持续8到24小时,随后评估Cdc7(MCM2,Ser40/41和Ser53)和推定的脱靶激酶CDK8(STAT1、Ser727)的下游靶标的磷酸化状态;CDK7和9(RNA pol II,Ser2);LATS2(YAP1,Ser127)以及已知由CDK9间接控制的MCL-1的水平(Gregory,2015)。 将MV411细胞还以剂量依赖性方式用SRA141(在浓度介于0.5与3.3μM之间下)进行处理,并且随后评估MCM2在Ser40/41和Ser53处的磷酸化状态。 如图13所示,Cdc7水平在这些研究中没有变化,而其靶MCM2在Ser40/41和Ser53处的磷酸化以0.033μM(Colo-205)和0.5μM(MV411)开始的浓度依赖性方式降低,从而证明在细胞中稳健的中靶抑制。 在SRA141浓度高达1μM下,任何细胞底物的磷酸化状态(或MCL-1的蛋白水平)均没有显著变化。在3.3μM下观察到STAT1、YAP1和MCL-1的最小抑制,但是总蛋白水平的共事件降低表明这些发现可能是由于SRA141介导的细胞死亡。 总之,这些数据证实了SRA141在细胞环境中的效力和选择性。 实例9:SRA141在AML三维(3D)培养物中的活性 分别在由三种AML细胞系(EOL-1、Molm-16和MV-4-11)和三种AML PDX模型(AM5512、AM7440和AM7577)制备的体外和离体三维(3D)培养物中研究SRA141的活性。将肿瘤培养物用SRA141(0.08到33μM)进行处理持续5天。SRA141在六个模型中的五个模型中证明了有效的抗细胞活性(IC50范围为0.15到1.6μM),而在AM5512制剂中注意到活性降低(IC50=3.2μM;表1)。后一种模型还对阿糖胞苷和顺铂具有相对抗性(IC50>8.9μM)。 表1:AML 3D培养物中的SRA141活性
注意:最大抑制表示为紧接着IC50的百分比 SRA141(0.3-5μM)的抗增殖活性还在另外的3D生长研究中进行评估,所述生长研究是在甲基纤维素生长基质中使用正常人髓样(n=3)和温育14天的AML-母细胞祖细胞进行的。 正常骨髓制剂的平均SRA141 IC50值为0.25μM,而AML样品的平均IC50值为大约为0.16μM(其中对抗性祖细胞样品AML810进行了审查;表2)。阳性对照胞嘧啶阿拉伯糖苷(AraC)对于正常骨髓和AML-母细胞祖细胞样品两者均具有均等的IC50值(大约0.004μM)。还计算了每个样品的治疗指数[(原发性AML IC 总体来说,这些数据证明SRA141在AML 3D培养物中具有有效的细胞毒性活性,其中潜在治疗指数超过正常骨髓祖细胞。 表2:在3D培养物中生长的正常骨髓和AML-母细胞祖细胞中的SRA141活性
**所审查的样品 实例10:SRA141在培养细胞中抑制Cdc7的耐久性 在用两种浓度的SRA141(0.1μM和1μM)处理48小时,随后立即收获细胞,或去除药物温育物,随后在收获细胞之前另外温育24小时的Colo-205细胞中研究药物诱导的Cdc7抑制的耐久性。在两种条件之间将MCM2在Ser53(Cdc7活性的靶位点)处的磷酸化进行比较。 如图14所示,来自这项研究的数据表明,在0.1μM药物浓度下化合物清除后24小时Cdc7活性恢复,而在较高温育浓度(1μM)清除后24小时观察到pMCM2抑制延长。这些结果与先前的生物化学测定一致,其指示缓慢的靶解离速率和随后延长的酶抑制。 实例11:SRA141细胞周期敏感性分析 在与双胸苷阻断同步的Colo-205细胞中确定在细胞周期的各个阶段对SRA141处理的敏感性。在阻断释放时,在细胞达到S期或M期时添加SRA141(1μM)。 如图15所示,用碘化丙啶标记的细胞的流式细胞术分析证明,指示凋亡细胞的亚G1群体在细胞进展通过M期时积聚,条件是在先前的S期期间用SRA141处理其(图15A),但是如果在S期完成(M期开始)后开始处理,则不积聚(图15B)。如果在M期首先添加SRA141,则亚G1积聚被延迟并且要求细胞在显示细胞凋亡的迹象之前在SRA141的存在下进展通过随后的S期。 这些数据支持以下假设:抑制Cdc7在S期中起作用,最可能的是由MCM2/MCM4磷酸化介导的复制起源激发,导致细胞与复制不足的DNA进入有丝分裂,从而引起凋亡细胞死亡的诱导。 如图15C所示,通过蛋白质印迹评估用0.1μM SRA141处理48小时的Colo-205细胞的细胞周期和DNA损伤标志物。结果证明在用SRA141处理后存在G2/M期的有丝分裂标志物。这些结果与通过使用高含量成像获得的数据一致,所述高含量成像证明用SRA141处理48小时的Colo-205和SW620细胞群体在有丝分裂中具有细胞积聚(图15D)。与其它Cdc7抑制剂相比,用SRA141处理的群体中有丝分裂中细胞的百分比更大。 实例12:体外细胞遗传敏感性分析 对235-细胞系组的SRA141敏感性的分析证明,结肠直肠癌属于最敏感的实体瘤系之一(图16)。此外,235个细胞系的基因组学和甲基化模式的生物信息学分析示出FAT1的突变与SRA141敏感性相关。考虑到FAT1在结肠直肠癌中通常改变的Wnt信号传导途径中的作用,进一步检查了16个结肠直肠癌系的敏感性与2个其它Wnt途径基因APC和FAT4中突变的存在之间的相关性。 结果示出,通常在染色体不稳定(CIN)结肠直肠肿瘤中发现的APC突变与对SRA141的增加的敏感性相关(p=0.04,图16)。尽管未确定FAT4突变与SRA141敏感性在统计学上相关,但是发现最敏感系中的三个具有FAT4突变。这些初步数据支持SRA141在CIN结肠直肠癌中的用途。 实例13:Colo-205结肠直肠异种移植研究(小鼠) 携带Colo-205肿瘤异种移植物的雌性BALB/c小鼠口服施用(i)媒剂(0.2M HCl/0.5%MC400)或(ii)以30或60mg/kg BID施用SRA141持续17天或(iii)以120mg/kg QD施用SRA141持续17天(n=8每处理组)。在研究的最后一天第16天计算肿瘤生长抑制。 所有处理都是耐受的;然而,由于体重过度减轻(>15%损失),对于来自SRA14160mg/kg BID的两只小鼠将剂量暂时中止两天,并且对于来自120mg/kg QD处理组的两只小鼠将剂量暂时中止1天和5天。 如图17中所示,SRA141施用在30mg/kg BID、60mg/kg BID和120mg/kg QD剂量水平下分别导致15%、58%和37%的肿瘤生长抑制。这些初步数据支持SRA141在结肠直肠癌中的用途。 实例14:SW620结肠直肠异种移植研究(小鼠) 携带SW620肿瘤异种移植物的雌性BALB/c小鼠口服施用(i)媒剂(0.1M HCl/0.5%MC400)或(ii)以60mg/kg每天三次(TID)施用SRA141连续4天,随后是2天给药假期,持续21天,或(iii)以120mg/kg QD施用SRA141持续21天(n=10每处理组)。在研究的最后一天第21天计算肿瘤生长抑制。所有媒剂对照动物在第21天保留在研究中。 以60mg/kg TID用SRA141处理的10只小鼠中的四只经历大于20%的最大体重减轻。中止对这些动物的给药直到体重减轻恢复到小于20%。以120mg/kg QD用SRA141处理的小鼠中没有一个满足20%体重减轻阈值。 如图18所示,SRA141施用在60mg/kg TID和120mg/kg QD下分别导致78%和73%的肿瘤生长抑制。这些初步数据支持SRA141在结肠直肠癌中的用途。 实例15:MV-4-11人白血病系统存活率研究 静脉内接种MV-4-11癌细胞的雌性NOD SCID小鼠施用(i)媒剂对照(0.1M HCl/0.5%MC400,口服)或(ii)最初以60mg/kg BID施用SRA141持续从第0天到第21天的7天周期的连续5天,并且然后由于显著的体重减轻,以40mg/kg BID,按照统一方案从第22天到第73天(n=10每处理组)。鉴于癌症模型的全身性质,存活率是这项研究中的唯一功效终点。 通常,基于体重减轻数据,SRA141处理是耐受的。十只经SRA141处理的小鼠中有一只经历大于20%的体重减轻。 在研究的第17和18天发现SRA141处理组中的五只动物死亡,这可能是由于技术误差或耐受性而不是疾病进展。对于媒剂和SRA141处理组,包含早熟死亡的所有动物的中值存活率分别为66和35天,并且对于SRA141处理组(其中早熟死亡已被审查)的中值存活率大于99天。因此,经审查的处理组的存活率增加支持SRA141在白血病治疗中的用途。 实例16:A20免疫活性淋巴瘤异种移植模型(小鼠) 携带A20肿瘤异种移植物的免疫活性雌性BALB/c小鼠口服施用(i)对照(PBS,腹膜内)或(ii)以120mg/kg施用SRA141;n=8只动物每组。在第0天到第4天、第7天到第11天以及第14天到第18天进行给药。在研究的最后一天第23天计算肿瘤生长抑制。 通常,SRA141的处理是耐受的。一只动物经历大于20%的体重减轻并且随后将其安乐死。 如图19所示,SRA141施用导致59%的肿瘤生长抑制。这些初步数据支持SRA141在淋巴瘤中的用途。 实例17:MDA-MB-486乳腺异种移植研究(小鼠) 携带MDA-MB-486乳腺肿瘤异种移植物的雌性CB-17SCID小鼠口服施用(i)媒剂对照(0.1M HCl/0.5%MC400)或(ii)以30或60mg/kg BID施用SRA141持续5周(n=10每处理组)。在研究的最后一天第35天计算肿瘤生长抑制。 在30mg/kg BID处理下的媒剂对照和SRA141耐受性良好。以60mg/kg BID用SRA141处理的10只小鼠中有六只经历大于20%的最大体重减轻。在第18天和第19天,暂时中止对这些动物的给药,其中由于体重减轻延长,将一只动物安乐死。 如图20所示,SRA141施用在30mg/kg BID和60mg/kg BID下分别导致26%和53%的肿瘤生长抑制。这些初步数据支持SRA141在乳腺癌中的用途。 实例18:小鼠SW620结肠直肠异种移植模型PK/PD评估 在携带SW620异种移植物的小鼠中确定SRA141处理对肿瘤磷酸-MCM2(pMCM2)(Cdc7的直接底物和Cdc7抑制的下游标志物)的影响。在药物施用以构建基本PK/PD模型后48小时期间,pMCM2的变化与肿瘤和血浆中的SRA141浓度相关。 具体地,向携带皮下SW620肿瘤的雌性BALB/c小鼠以30mg/kg、60mg/kg或120mg/kg(n=15每组)施用单次口服剂量的SRA141。向对照动物施用媒剂(0.1M HCl/0.5%MC400;n=10)。在处理后2、4、8、24和48小时,终止来自每组的三只动物(对于对照,n=2),并且对血浆和肿瘤取样进行分析。 如图21所示,在单次SRA141施用后pMCM2水平降低,对于30mg/kg和60mg/kg剂量在2-4小时之间达到峰值,并且对于120mg/kg剂量在2-8小时之间达到峰值(图21A中示出pMCM2水平,图21B中对抑制%进行定量并相对于肌动蛋白归一化)。在以30mg/kg、60mg/kg和120mg/kg剂量施用后2到4小时分别还观察到接近于1μg/mL、1.5μg/mL和2μg/mL的最大SRA141血浆浓度。药物的肿瘤浓度类似。来自这项研究的PK和PD数据的相关性表明,需要大约1.1μM的循环血浆浓度和肿瘤内组织浓度来将pMCM2抑制50%。 实例19:大鼠Colo-205结肠直肠异种移植模型PK/PD评估 在携带皮下Colo-205肿瘤的雌性Rowett裸大鼠中还确定了SRA141处理后pMCM2的肿瘤抑制。在单次口服剂量的SRA141(75或150mg/kg)后十二小时,动物终止给药,并且对肿瘤取样进行分析(n=3每组)。向对照动物施用与Colo-205大鼠模型中所使用的同一媒剂(参见实例5;n=3每组)。在施用75和150mg/kg剂量后12小时,分别观察到接近于0.5和0.9μg/mL的SRA141肿瘤浓度。在单次剂量的SRA141后十二小时,pMCM2被抑制大约50%到60%的对照(图22)。这些结果表明,1μM的肿瘤内组织浓度足以抑制MCM2磷酸化(pMCM2)50%。 实例20:体内非临床药代动力学汇总 体外分布研究示出,SRA141与人和大鼠血浆蛋白(>90%)高度结合,而在小鼠和狗基质中观察到中等结合(58%到77%)。另外,SRA141不会优先地分配到任何物种基质中的红细胞中。 在临床前和人肝细胞中进行的体外代谢研究表明在大鼠、狗和人制剂中SRA141的有利代谢稳定性。跨物种鉴定出十一种代谢物,其中6种发生在人基质中。鉴定出一种人特异性代谢物,但仅处于痕量水平。小鼠、大鼠和狗肝细胞代谢物谱含有所有其它人代谢物。 在临床相关浓度下,SRA141示出CYP 3A4/5的体外时间和代谢依赖性抑制(IC50=6.2到6.7μM)以及人有机阴离子转运蛋白OATP1B1、OATP1B3和OAT3的体外抑制(在10μM下,>50%抑制)。总的来说,这些体外CYP和转运蛋白抑制数据表明,SRA141可以潜在地改变已知是这种CYP同种型或这些转运蛋白的底物的共同施用的药物的代谢和/或分布。 SRA141(呈现为游离碱悬浮液)在禁食的小鼠、大鼠和狗中证明了中等的绝对口服生物利用度(%F),范围为31%到54%,其中在猴子中口服生物利用度明显较低(%F=1%到33%)。作为悬浮液中的双盐酸盐的呈现似乎并未显著增加任何物种中的暴露。对狗的进一步研究表明,在餐后施用之后,悬浮液中的双盐酸盐的绝对口服生物利用度可以得到改善。然而,在对狗的单独研究中,在施用两种提出的临床药物产品胶囊呈现后注意到中等的绝对口服生物利用度(%F=41%到62%),并且餐食状态似乎对口服生物利用度没有明显影响。口服给药后的全身暴露(Cmax、AUC)通常随着剂量增加而增加,但是以低于剂量比例的方式增加。 以50mg/kg(QD×12天)或100或150mg/kg(给药5天/停药2天/给药5天)用SRA141(n=3每种途径)口服处理非荷瘤雌性裸大鼠。媒剂是含0.5%CMC-Na/1%Lutrol的水。在最后一次给药后,通过LC-MS/MS确定SRA141血浆浓度。如图23所示,全身暴露随着口服剂量从50mg/kg增加到150mg/kg而增加。 实例21:体内非临床毒理学汇总 在大鼠和狗中进行评估SRA141的毒理学研究,然后进行单次和重复口服给药持续至多4个周期(每个周期给药5天/停药2天)。另外,在体外评估SRA141的潜在遗传毒性。表3中示出了发现的汇总。 表3:在大鼠和狗的4周期研究中NOAEL、MTD和HNSTD以及相关的血浆暴露
M=雄性;F=雌性 来自大鼠和狗重复剂量毒性研究的发现表明,SRA141相关毒性的潜在靶器官是胆囊/胆管系统、淋巴组织(脾脏、胸腺、淋巴结和GALT)、骨髓、肝脏、肾脏、雄性和雌性生殖道、外周白细胞和唾液腺,其中狗似乎是最敏感的物种。 在高发生率下并且在大鼠和狗两者中一致地观察到对胆管系统的影响。然而,这些胆管变化通常最小到轻度以及部分(两个物种的胆管增生)或完全可逆(狗的胆囊;大鼠中不存在组织)。此外,在动物中药物诱导的胆管发现尚未可靠地预测人中的类似发现(Hailey,2013;MacDonald,2004)。 胆管和库普弗细胞(Kupffer cell)变化是在向大鼠28天重复剂量施用SRA141之后观察到的仅有的发现,其中偶然的血液学和临床化学发生变化。然而,在大鼠的剂量范围研究中,鉴定出其它器官和组织的变化,包含肝脏(肝细胞变性)、脾脏(减少的红髓细胞性)和胃(炎症和上皮增生)。尽管尚不清楚为什么在大鼠的28天研究中没有观察到可比较的发现,但是在关键研究中药物的周期性施用可能促成不同的毒理学结果。在7天大鼠研究和28天狗研究中,胆管系统之外的一些组织变化的相似性,尽管调配物、剂量和给药持续时间有所不同,这表明狗可能比大鼠对SRA141更敏感。 尽管颧骨唾液腺变化将被认为对受影响的狗不利,但是人的相当的唾液腺的低发生率和缺乏使得这种潜在靶标与人的临床相关性不确定。类似地,淋巴组织变化的低量值和可恢复性,连同与间歇剂量周期相关的外周白细胞的周期性变化未被判断为是不利的;这些变化可以相互关联并且可以表示白细胞运输中的瞬时组织和外周血改变。骨髓细胞性的潜在不利的降低还可能与这种提出的白细胞运输变化相关。胸腺减少比其它淋巴组织变化更严重,但是这种组织变化可以部分地或完全地继发于应激并且不反映直接的SR141相关毒性。 在28天的狗研究中,似乎与更受胆囊、肾小管和骨髓的变化影响的雄性存在潜在的性别差异。然而,在每个测试组中少量的动物以及性别之间的低量值的差异不允许确定组织应答中的特异性性别差异。 在体外细菌反向突变和人淋巴细胞染色体畸变测定中,SRA141的致突变性和类遗传性(clastogenicity)均为阴性。 总之,这些研究的结果表明,施用SRA141后的毒性发现在临床环境中通常是可监测的,支持性疗法在晚期肿瘤学研究群体中是可用的和/或相关性较低。具体地,在大鼠和狗中,在给药5天/停药2天的给药方案持续4周之后MTD和最高非严重毒性剂量(HNSTD)分别被认为是100mg/kg/天(600mg/m2/天)和10mg/kg/天(200mg/m2/天)。 实例22:SRA141 CRC临床试验 进行临床试验以确认SRA141单一疗法在治疗患有CIN表型的转移性CRC的患者的癌症中的功效,所述表型通过排除确实具有MSI-H状态的患者而富集,而在一些情况下,将来探索具有高染色体不稳定性率的其它癌症。 用于肿瘤学临床试验的人类等效起始剂量通常是在10%的啮齿类动物中严重毒性剂量的1/10(STD10)或在非啮齿类动物中最高非严重毒性剂量(HNSTD)的1/6(ICH S9,2009)中的较小者。来自GLP关键毒理学研究的数据(参见实例21)表明,在大鼠中最大耐受剂量(MTD作为STD10的替代物)为100mg/kg/天(600mg/m2/天;所测试的最高剂量)以及在狗中10mg/kg/天(200mg/m2/天)的HNSTD作为更敏感的物种。因此,使用等于54mg的绝对剂量(对于60kg的患者而言)的33.3mg/m2/天(0.90mg/kg/天)的人等效剂量(HED)。为了额外保证受试者安全性,使用口服施用的较低的10mg的SRA141起始剂量。 临床给药方案符合已经证明抗肿瘤活性的临床前给药方案,即5天给药/2天停药。SRA141以羟丙基甲基纤维素(HPMC)胶囊提供。除非赞助者基于另外的数据指导,否则SRA141胶囊应空腹服用(受试者在施用前和施用后1小时禁食持续至少2小时)。 剂量递增阶段 进行剂量递增阶段。征募介于15与50之间个患有转移性CRC的受试者。使用加速滴定设计。最初由单个受试者组成的群组接受递增剂量的SRA141。初始剂量递增以如基于新出现的安全性概况被视为临床上适当的100%一样大的增量进行。一旦SRA141相关的国家癌症研究所—不良事件的通用术语标准(NCI-CTCAE)在第1周期期间在特定群组中观察到2级或更大毒性,所述群组被扩展到3到6名受试者,并且随后剂量水平群组遵循滚动6设计。此后SRA141的剂量以增量递增,以在检查所有可用安全性数据后确定。SRA141的剂量递增,直到已鉴定出MTD,除非由赞助者另外确定,例如如果寻求替代性方案。具有替代性方案的剂量递增在任何时间开始并且并行运行或代替原始方案中的持续剂量递增。 DLT评估期是从第1周期第1天的第一剂量到第一治疗周期结束。在一些情况下替换对于DLT评估不可评估的受试者,例如出于SRA141的不耐受性/毒性之外的原因未完成DLT评估期的受试者。 如果DLT被视为极有可能或可能与SRA141相关,则其被定义为以下事件中的任何事件(由NCI-CTCAE v4.03定义): -尽管不给予给药和/或提供支持性护理,但仍持续>7天的4级中性粒细胞减少症、贫血或血小板减少症; -4级发热性中性粒细胞减少症; -具有≥3级出血的≥3级血小板减少症; -≥3级非血液学毒性,除了与3级恶心、呕吐和/或腹泻相关的例外,不存在足够的预防或治疗、疲劳以及短暂和无症状的3级实验室异常;(有关详细信息,请参阅方案)。 -由于不耐受性或毒性,无法接受75%的计划剂量的SRA141。 出于剂量递增决定的目的考虑剂量限制性毒性;然而,如果累积毒性变得明显,则当确定递增中的下一个剂量水平或RP2D时应考虑到这一点。 剂量扩展阶段 还进行剂量扩展阶段。将大约30名肿瘤不是已知高微卫星不稳定性(MSI-H)状态的患有转移性CRC的受试者纳入单个扩展群组,以进一步表征安全性概况并评估初步功效。在一些情况下,如果存在抗肿瘤活性的证据或如果SRA141血浆浓度达到基于新出现的非临床数据的最小有效阈值,则在确定MTD或RP2D之前开始征募扩展群组。SRA141在所提出的RP2D和基于试验的剂量递增阶段结果的方案下进行施用。在一些情况下,SRA141的RP2D等于或低于MTD。可替代地,如果在确定RP2D之前开始征募,则SRA141最初以在每个受试者的征募时的剂量递增阶段期间已经清除的最高剂量施用。一旦确定同一方案的较高剂量水平在递增群组中是安全的,在一些情况下允许受试者递增到较高剂量水平。 方法学 SRA141治疗是在一个或多个初始群组中,在28天周期中根据给药5天/停药2天的方案每天口服施用一次。基于新出现的安全性和耐受性数据,考虑替代性给药方案。 试验由筛选、治疗、安全性随访(SFU)和长期随访组成。在一些情况下,治疗期持续到满足预先指定的标准之一,包含疾病进展、不可接受的毒性和妊娠。 针对WOCBP受试者进行筛选和基线评估:人口统计学、疾病史、基线疾病评估、基线安全性评估和妊娠测试。收集用于PDn和回顾性基因组评估的预处理(基线)样品。 执行安全性评估: -连续不良事件(AE)评估和伴随治疗回顾; -在第1周期期间和在每个后续周期的第1天,在给药前和给药后2到3小时每周一次以及在SFU下的生命迹象; -在第1周期和第2周期期间每周进行临床生物化学,并且在第3周期后每两周进行一次临床生物化学,接着在第7周期向前每月进行一次临床生物化学,并且在SFU下进行临床生物化学; -在第1周期到第3周期期间每周进行一次血液学,并且在随后的周期中每两周进行一次血液学,并且在SFU处进行血液学; -在SFU处的肌钙蛋白;在每个周期的第1天和在SFU处的尿分析; -在第2周期第1天和在SFU处的超声心动图; -在第1期、第2周期的第1天以及随后的每三个周期以及在SFU时的心电图(ECG)(局部读取); -在第1周期第1天和第19天(在给药后的多个时间点)的中央ECG;以及 -如临床上指示的症状定向的身体检查。 另外,在第1周期第1天的第一剂量的SRA141之后进行包含直立性生命迹象在内的强血压监测。 评估功效: -在第1周期第1天之后每8周并且在SFU处通过CT/MRI对胸部、腹部、骨盆以及所有可疑的疾病位点进行肿瘤评估,除了中止记录的疾病进展的受试者之外; -第1周期第1天后每4周并且在SFU处进行临床评估和血清癌胚抗原(CEA)水平;以及 -生存状态 评估药物代谢动力学: -在第1周期第1天和第19天的24小时密集PK取样。 -C3、4、5和8第1天的给药前取样 评估药效学。收集替代组织(皮肤穿刺活检)和肿瘤组织并分析PDn生物标志物pMCM2。在基线处并且在第1周期第18天或第19天在给药后4到8小时收集皮肤穿刺样品(即,SRA141的预期Cmax)。当SRA141的血浆浓度在一些情况下达到低谷水平(Cmin)时,在第1周期或更晚周期中的更晚时间点,从接受SRA141剂量水平的受试者收集第三次皮肤穿刺活检,所述剂量水平高于从第18天或第19天的剂量活检后4到8小时已经观察到可测量的生物标志物效应的剂量水平。第三次皮肤穿刺活检的定时由赞助者基于新出现的PK和PDn数据进行通信。需要来自参与递增剂量阶段的所有受试者和参加扩展剂量阶段的最少10名受试者的皮肤活检样品。在基线处并且在第1周期第4天或第5天、给药后4到8小时,需要来自扩展剂量阶段的最少6名受试者的肿瘤组织。来自扩展剂量阶段的另外受试者和来自递增剂量阶段的受试者的肿瘤组织是任选的。 评估基因组学。在基线收集存档或新鲜肿瘤组织(原发性或转移性)和血液样品用于回顾性基因组分析,以探索与SRA141治疗应答相关的基因组改变。还在SFU处收集血液样品。 纳入标准 基于以下标准选择用于临床试验的受试者: -在进行任何试验特异性程序、取样和分析之前,需签署书面知情同意书 -给予同意时,年满18岁 -组织学和/或细胞学证实的转移性CRC -用含有氟尿嘧啶或卡培他滨、奥沙利铂和伊立替康的方案进行的先前治疗 -预期寿命为至少12周 -在接受其第一剂量的IMP之前的1周内测量的血液学和生物化学指数在表4中所示的范围内 表4:纳入的血液学和生物化学指数
-世界卫生组织(WHO)体力状态0-1 -按照RECIST标准的可放射照相测量的疾病 -在第一剂量的IMP之前18个月内收集的或由赞助者批准的归档肿瘤组织,并且可用于回顾性肿瘤概况分析或可接近的肿瘤以及意愿同意收集肿瘤组织的活检 -对于参与扩展阶段的最少6名受试者,可接近的肿瘤和意愿同意接受高达2次肿瘤活检以收集肿瘤组织用于PDn测定 排除标准 基于以下标准将受试者从临床试验中排除: -使用Cdc7抑制剂的任何先前治疗 -具有已知高微卫星不稳定性的结肠直肠癌,即MSI-H -已经接受以下先前或当前抗癌疗法: a.在第一剂量的SRA141之前的4周内任何靶病灶(用作可测量疾病的病灶)的放射疗法(除了症状控制之外并且其中病灶将不用作可测量疾病) b.在第一剂量的SRA141之前的3周内的化疗 c.在第一剂量的SRA141之前,如果先前免疫疗法较短并且由赞助者批准的话,则在4周内或5个半衰期内的先前免疫疗法 d.在第一剂量的SRA141之前的6周内的亚硝基脲或丝裂霉素C e.在第一剂量的SRA141之前,如果疗法较短并且由赞助者批准的话,则在4周内或5个半衰期内的其它IMP或靶向疗法 -同时施用自然疗法药物、草药补充剂或其它替代性疗法,就研究者而言,已知所述疗法影响CYP450酶 -从研究者的角度来看,目前非处方药物或酒精依赖严重到足以影响研究依从性或安全性 -在过去2年内的其它恶性肿瘤,除了与大约95%或更高的预期5年无疾病存活率相关的经充分治疗的肿瘤之外,除非得到赞助者批准 -先前治疗的持续毒性表现大于NCI-CTCAE 1级。对此的例外是脱发或某些毒性,研究者和赞助者或赞助者的指定监护人认为这不应排除受试者 -新的或进展的脑转移。在过去6个月内中枢神经系统转移史(仅扩展群组)。具有脑转移的受试者包含在递增群组中,所述脑转移在一些情况下在8周时间段内是放射性稳定的 -已经怀孕或正进行哺乳的女性。具有生育能力的女性(WOCBP),除非其血清或尿妊娠测试筛查结果为阴性(即,在第一次给药前7天内),并且同意从第一次施用IMP起、贯穿整个试验以及在之后6个月遵循有效的避孕要求 -具有生育能力的伴侣的男性,除非其同意通过在整个试验以及在之后6个月遵循避孕要求而采取不是亲生孩子的措施。必须建议怀孕或正进行哺乳的伴侣的受试者使用避孕套加上杀精子凝胶以防止在整个试验中以及在最后剂量的SRA141后6个月胎儿或新生儿暴露 -受试者尚未恢复的大手术 -由于非恶性全身性疾病(包含活性不受控感染)而处于高医疗风险下 -已知对于乙型肝炎、丙型肝炎或人免疫缺陷病毒(HIV)是血清学阳性的,除非用阴性病毒负荷控制并且得到赞助者批准 -严重的心脏或心血管病状,如并发性充血性心力衰竭,根据纽约心脏协会[NYHA]标准的III/IV类心脏病的既往史、在基线处的左心室射血分数<45%、在过去6个月内的心脏局部缺血史、需要治疗的显著性心律失常的既往史、局部缺血性脑血管疾病或症状性周围血管疾病的既往史,除非得到赞助者批准 -在成年男性中,QTcF>450毫秒,并且在成年女性中,>470毫秒 -在第一剂量的IMP之前的8周内,向大于25%骨髓进行骨髓移植或广泛放射疗法之前 -胃肠(GI)功能受损或在一些情况下显著改变IMP吸收的GI疾病(例如,溃疡性疾病、不受控的恶心、呕吐、腹泻或吸收不良综合征) -在没有咀嚼或压碎的情况下不能吞咽胶囊 -已知的对SRA141或其赋形剂的过敏、超敏反应或不耐受 -是参与或计划参与另一介入临床试验,同时参与这项试验。参与观察试验或介入临床试验将是可接受的,所述观察试验或介入临床试验不涉及IMP的施用并且在研究者和赞助者或赞助者的指定人员的观点中不会对受试者造成不可接受的负担 -在研究者或赞助者观点中的任何其它病状将不会使受试者成为临床试验的良好候选者。 RECIST标准 根据修订的RECIST标准v1.1对这项研究中的疾病应答进行评估。RECIST标准更详细地描述于Eisenhauer等人(实体瘤中的新应答评价标准:修订的RECIST指南(Newresponse evaluation criteria in solid tumours:Revised RECIST guideline)(1.1版).《欧洲癌症杂志(Eur J Cancer)》[互联网].2009),所述文献教导的全部内容通过引用并入本文。 在基线处,肿瘤病灶/淋巴结通常如下分类为可测量的或不可测量的: 可测量的 肿瘤病灶:通常在至少一个维度(待记录在测量平面中的最长直径)中以如下最小大小准确地测量: -CT扫描10mm(CT扫描切片厚度不大于5mm;关于成像指导,参见Eisenhauer等人,[Eisenhauer,2009]中的附录II) -通过临床检查进行10mm卡尺测量(不能用卡尺准确测量的病灶应记录为不可测量的) -胸部X射线20mm 恶性淋巴结:被认为是病理学上扩大的且可测量的,当通过CT扫描(CT扫描切片厚度建议为不大于5mm)评估时,淋巴结在短轴上通常为15mm。在基线和随访中,通常仅测量和随访短轴。 不可测量的 所有其它病灶,包含小病灶(最长直径<10mm或短轴≥10到<15mm的病理性淋巴结)以及真正不可测量的病灶。被认为是真正不可测量的病灶通常包含:柔脑膜疾病、腹水、胸膜或心包积液、炎性乳腺疾病、皮肤或肺的淋巴管介入、通过身体检查鉴定的不可通过可再现的成像技术测量的腹部肿块/腹部器官肿大。 骨病灶、囊性病灶和先前用局部疗法治疗的病灶需要特定评述: 骨病灶: -骨扫描、PET扫描或平片通常被视为不足以测量骨病灶的成像技术。然而,这些技术可以用于确认骨病灶的存在或消失。 -如果软组织组分满足上述可测量性的定义,则可以通过横截面成像技术(如CT或MRI)进行评估的溶骨性病灶或混合的溶解性-成纤维细胞病灶通常被视为是可测量的病灶。 -成纤维细胞骨病灶通常是不可测量的。 囊性病灶: -满足放射照相定义的简单囊肿的标准的病灶通常被视为不是恶性病灶(既不是可测量也不是不可测量的),因为所述病灶通过定义是简单囊肿。 -被认为表示囊性转移的“囊性病灶”通常被视为是可测量的病灶,如果所述病灶满足上述可测量性的定义的话。然而,如果在同一受试者体内存在非囊性病灶,则这些病灶对于作为靶病灶的选择是优选的。 具有先前局部治疗的病灶: -位于先前辐照区域或经受其它局部区域疗法的区域中的肿瘤病灶通常被视为是不可测量的,除非证明在病灶中存在进展。研究方案总体上详述了此类病灶通常被视为是可测量的条件。 评估方法 所有测量值通常以度量符号记录,如果临床评估的话,使用卡尺。所有基线评估通常尽可能接近治疗开始进行并且在治疗开始之前从不超过4周进行。 通常使用同一评估方法和同一技术来表征基线处和随访期间每个经鉴定和经报告的病灶。通常总是进行基于成像的评估,而不是进行临床检查,除非所跟踪的一个或多个病灶无法成像,但可通过临床检查进行评估。 在使用卡尺(例如,皮肤结节)进行评估时,当临床病灶是浅表并且直径≥10mm时,所述临床病灶通常被视为是可测量的。对于皮肤病灶的情况,建议通过彩色摄影进行记录,包含用于估计病灶大小的标尺。如上所述,当病灶可以通过临床检查和成像来评估时,通常会进行成像评估,因为其更客观并且在一些情况下还会在研究结束时进行回顾。 胸部CT通常优于胸部X射线,特别是当进展是重要的终点时,因为CT比X射线更敏感,特别是在鉴定新病灶时。然而,在一些情况下,如果胸部X射线上的病灶被充气肺清楚地限定和围绕,则其被视为是可测量的。 CT通常是用于测量选择用于应答评估的病灶的目前最佳可获得且可再现的方法。这项指南已经基于CT切片厚度为5mm或更小的假设限定了病灶在CT扫描上的可测量性。当CT扫描的切片厚度大于5mm时,可测量病灶的最小大小是切片厚度的两倍。MRI在某些情况下(例如,对于身体扫描)也是可接受的。Eisenhauer等人的出版物中提供了关于使用CT和MRI两者来评估客观肿瘤应答评估的更多详细信息。 超声通常不可用于评估病灶大小,并且通常不用作测量方法。超声检查通常不能完整地再现以供稍后的独立回顾,并且因为超声检查依赖于操作者,因此通常不能保证将从一个评估到下一个评估采取相同的技术和测量(在Eisenhauer等人(2009)中更详细地描述)。如果在研究过程中通过超声鉴定新病灶,则通常建议通过CT或MRI进行确认。如果担心在CT处有辐射暴露,则在一些情况下在所选实例中使用MRI代替CT。 通常不建议使用内窥镜检查和腹腔镜检查技术进行客观肿瘤评估。然而,当获得活检时,所述活检通常可用于确认完全病理性应答,或用于在其中完全应答或外科手术切除后的复发是终点的试验中确定复发。 单独的肿瘤标志物通常不用于评估客观肿瘤应答。然而,如果标志物最初高于正常上限,则标志物通常针对完全应答中待考虑的受试者进行归一化。 如果方案需要,细胞学和组织学通常在极少数情况下用于区分PR和CR(例如,肿瘤类型,如生殖细胞肿瘤中的残留病灶,其中已知的残留良性肿瘤可以保留)。当已知积液是治疗的潜在不良反应时(例如用某些紫杉烷化合物或血管生成抑制剂),如果可测量的肿瘤已经满足应答或疾病稳定的标准以便区分应答(或疾病稳定)和进行性疾病,则通常考虑在治疗期间出现或恶化的任何积液的肿瘤起源的细胞学确认。 肿瘤应答评估 为了评估客观应答或将来的进展,通常会估计在基线处的总体肿瘤负荷并且将其用作后续测量的比较器。可测量疾病通常可以通过至少一个可测量病灶的存在来定义。 当在基线处存在多于一个可测量病灶时,代表所有相关器官的高达最多五个病灶的所有病灶(以及每个器官最多两个病灶)通常被鉴定为靶病灶,并且在基线处被记录和测量(这意味着在受试者仅具有分别涉及最多两个和四个病灶的一个或两个器官位点的情况下被记录)。靶病灶通常基于其大小(具有最长直径的病灶)来选择并且通常代表所有相关器官,但另外,通常是使其自身可再现地重复测量的那些。在一些情况下,最大病灶不适合于使其自身可再现地测量,在这种情况下,通常选择可以可再现地测量的次最大病灶,如图3例示的,Eisenhauer等人(2009)。 特别值得一提的是淋巴结,因为淋巴结是正常的解剖结构,其在一些情况下即使不涉及肿瘤也可通过成像可见。被定义为可测量的并且在一些情况下被鉴定为靶病灶的病理性节结通常通过CT扫描满足短轴≥15mm的标准。只有这些节结的短轴通常对基线总和有贡献。节结的短轴通常是放射科医生通常用来判断节结是否涉及实体瘤的直径。节结大小通常被报告为在其中获得图像的平面中的两个维度(对于CT扫描,这几乎总是轴向平面;对于MRI,在一些情况下采集平面是轴向、矢状或冠状)。这些量度中的较小量度是短轴。例如,被报告为20mm×30mm的腹部节结的短轴为20mm并且有资格作为恶性可测量的节结。在此实例中,20mm应记录为节结测量值。所有其它病理性节结(短轴≥10mm但<15mm的那些病理性节结)通常被视为是非靶病灶。短轴<10mm的节结通常被视为是非病理性的并且通常不被记录或跟踪。 通常计算所有靶病灶的直径的总和(非节结病灶为最长,节结病灶为短轴)并且将其报告为基线总和直径。如果淋巴结将被包含在总和中,则如上所述,仅短轴被添加到总和中。基线总和直径通常用作参考,以进一步表征疾病的可测量维度上的任何客观肿瘤消退。 所有其它病灶(或疾病位点)(包含病理性淋巴结)通常被鉴定为非靶病灶并且通常在基线处记录。通常不需要测量,并且这些病灶通常遵循为“存在”、“不存在”或在极少数情况下遵循为“明确进展”(更多详细信息如下)。另外,可以将涉及同一器官的多个非靶病灶记录为病例记录表上的单个项目(例如,“多个扩大的骨盆淋巴结”或“多个肝脏转移”)。 应答标准 完全应答(CR):所有靶病灶的消失。任何病理性淋巴结(无论是靶标还是非靶标)在短轴上均减少到<10mm。 部分应答(PR):靶病灶的直径总和减少至少30%,取基线总和直径作为参考。 进行性疾病(PD):靶病灶的直径总和增加至少20%,取最小总和作为参考(如果基线总和是研究中最小的话,则这包含基线总和)。除了20%的相对增加之外,总和通常显示至少5mm的绝对增加。(注意:一个或多个新病灶的出现通常也被视为是进展)。 疾病稳定(SD):既不是足以符合PR的收缩,也不是足以符合PD的增加,取最小总和直径作为参考。 被鉴定为靶病灶的淋巴结通常记录实际的短轴测量值(在与基线检查相同的解剖学平面中测量),通常即使节结消退到低于10mm。这意味着当淋巴结被包含作为靶病灶时,在一些情况下病灶的“总和”不是零,即使满足完全应答标准,因为正常淋巴结通常被定义为短轴<10mm。因此,在一些情况下,病例报告表或其它数据收集方法被设计成将靶节结病灶记录在单独部分中,其中为了符合CR,每个节结通常会达到短轴<10mm。对于PR、SD和PD,节结的实际短轴测量值优选地包含在靶病灶的总和中。 而在研究中,在基线处记录的所有病灶(节结和非节结)通常在每次后续评估时记录其实际测量值,甚至非常小(例如,2mm)也是如此。然而,有时在基线处被记录为靶病灶的病灶或淋巴结在CT扫描中变得如此衰弱,以致放射科医生在一些情况下不愿意分配精确的量度并且在一些情况下将其报告为“太小而无法测量”。当这种情况发生时,通常重要的一点是将值记录在案例报告表上。如果放射科医生认为病灶可能已经消失,则测量值通常被记录为0mm。如果病灶被认为存在并且隐约可见,但是太小而无法测量,则通常将默认值指定为5mm。(注意:一般来说,不太可能将这种规则用于淋巴结,因为淋巴结在正常情况下通常具有可定义的大小并且常常被脂肪包围,例如在腹膜后腔中;然而,如果淋巴结被认为存在并且隐约可见,但是太小而无法测量,在这种情况下通常也将默认值指定为5mm)。此默认值是从5mm CT切片厚度得出的(但通常不会随着CT切片厚度的变化而改变)。这些病灶的测量可能是不可再现的,因此提供此默认值通常防止基于测量误差的错误应答或进展。然而,重申一下,如果放射科医生能够提供通常会记录下来的实际量度,即使其低于5mm。 当非节结病灶“片段化”时,通常将片段化部分的最长直径相加在一起以计算靶病灶总和。类似地,当病灶聚结时,通常维持病灶之间的平面,这将有助于获得每个单独病灶的最大直径测量值。如果病灶已经真正聚结使得其不再可分离,则在这种情况下最长直径的向量通常是“聚结病灶”的最大最长直径。 尽管一些非靶病灶在一些情况下实际上是可测量的,但是通常不对所述一些非靶病灶进行测量,而是通常仅在方案中指定的时间点进行定性评估。 完全应答(CR):所有非靶病灶的消失以及肿瘤标志物水平的归一化。所有淋巴结的大小均是非病理性的(短轴<10mm)。 非CR/非PD:一个或多个非靶病灶的持久性和/或肿瘤标志物水平维持在正常极限以上。 进行性疾病(PD):现有非靶病灶的明确进展(参见以下评述)。(注意:一个或多个新病灶的出现也被视为是进展)。 当受试者也患有可测量疾病时,为了在非靶疾病的基础上实现“明确进展”,通常非靶疾病的总体水平显著恶化,使得即使在靶疾病中存在SD或PR,总体肿瘤负荷也已经增加到足以值得停止治疗的地步。一个或多个非靶病灶的大小的适度“增加”通常不足以符合明确进展状态。因此,通常仅在非靶疾病在面对靶疾病的SD或PR时的变化的基础上的总体进展的指定是极其罕见的。 在一些III期试验中出现仅患有不可测量疾病的受试者,这不是将研究纳入可测量疾病的标准。如上所述,相同的一般概念适用于此,然而,在这种情况下,不存在可测量疾病评估来解释不可测量疾病负荷的增加的原因。因为非靶疾病的恶化通常不容易定量(通过定义:如果所有病灶均是真正不可测量的),当评估受试者明确进展时通常可以应用的有用测试将考虑基于不可测量疾病的变化的总体疾病负荷的增加是否在量值上与宣布可测量疾病的PD所需的增加相当:即肿瘤负荷的增加表示“体积”的另外73%增加(其等效于可测量病灶的20%增加直径)。实例包含胸膜积液从“痕量”增加到“大量”,淋巴管疾病从局部增加到广泛,或在一些情况下在方案中被描述为“足以要求改变疗法”。如果看到“明确进展”,则通常视为受试者在所述点患有总体PD。尽管具有应用于不可测量疾病的客观标准是理想的,但是所述疾病的本质通常使得很难做到这一点;因此,这种增加通常是实质性的。 新恶性病灶的出现通常表示疾病进展;因此,对新病灶的检测的一些评述通常很重要。通常没有用于鉴定新放射照相病灶的具体标准;然而,新病灶的发现通常是明确的:即通常不可归因于扫描技术的差异、成像模态的改变或被认为表示除了肿瘤之外的某物的发现(例如,一些情况下,一些“新”骨病灶仅仅是早先存在的病灶的愈合或爆发)。当受试者的基线病灶示出部分或完全应答时,这是特别重要的。例如,肝脏病灶的坏死经常在CT扫描报告上报告为“新”囊性病灶,其通常不是这样。 在未在基线处扫描的解剖学位置中在随访研究中鉴定的病灶通常被视为是新病灶并且通常指示疾病进展。这样的实例是在基线处患有内脏疾病的受试者,并且在研究中具有CT或MRI脑有序时,揭示转移。受试者的脑转移通常被视为是PD的证据,即使他/她在基线处没有脑成像。 如果新病灶是模棱两可的,例如由于其较小的大小,则继续疗法和随访评估通常会阐明其是否表示真正的新疾病。如果重复扫描证实明确存在新病灶,则通常使用初始扫描的日期来宣布进展。 尽管FDG-PET响应评估需要另外的研究,但是有时合理的是结合FDG-PET扫描的使用以补充进展评估中的CT扫描(特别是可能的“新”疾病)。在FDG-PET成像的基础上的新病灶通常根据以下算法进行鉴定: a.在基线处的阴性FDG-PET,其中在随访时的阳性*FDG-PET通常是基于新病灶的PD的迹象。*“阳性”FDG-PET扫描病灶通常意指在衰减校正图像上FDG病毒摄取大于周围组织摄取两倍的FDG病毒摄取。 b.在基线处没有FDG-PET并且在随访时的阳性FDG-PET: -如果在随访时的阳性FDG-PET对应于通过CT确认的新疾病位点,则这通常是PD。 -如果在随访时的阳性FDG-PET未被确认为CT上的疾病的新位点,则通常进行另外的随访CT扫描以确定在所述位点是否真的发生进展(如果是这样的话,PD的日期将是初始异常FDG-PET扫描的日期)。“阳性”FDG-PET扫描病灶通常意指在衰减校正图像上FDG病毒摄取大于周围组织摄取两倍的FDG病毒摄取。 -如果在随访时的阳性FDG-PET对应于在解剖图像的基础上未进展的CT上的疾病的预先存在的位点,则这通常不是PD。 最佳总体应答的评估 最佳总体应答通常是从研究治疗开始直到治疗结束所记录的最佳应答。如果在此试验中直到治疗结束之后才记录应答,则治疗后评估通常在确定最佳总体应答时考虑,只要没有给出替代性抗癌疗法。受试者的最佳总体应答分配通常取决于靶疾病和非靶疾病的发现,并且通常还考虑到新病灶的出现。 通常假设在每个方案规定的时间点,发生应答评估。表5提供了在基线处患有可测量疾病的受试者在每个时间点的总体应答状态计算的汇总。 当受试者仅患有不可测量(因此非靶)疾病时,通常使用表6。 当在特定时间点根本未进行成像/测量时,受试者在所述时间点通常不可评估(NE)。如果在评估中仅进行病灶测量的子集,则通常情况经常也被视为是在所述时间点的NE,除非作出一个或多个单个缺失病灶的贡献未改变指定时间点应答的令人信服的论证。这在PD的情况下将最可能发生。例如,如果受试者的基线总和为50mm,具有三个所测得病灶,并且在随访时仅评估两个病灶,但是那些病灶给出的总和为80mm,则受试者通常已经达到PD状态,而不管缺失病灶的贡献如何。 一旦已知受试者的所有数据,通常就能确定最佳总体应答。 在其中通常不需要对完全应答或部分应答的确认的试验中的最佳应答确定:这些试验中的最佳应答通常被定义为跨所有时间点的最佳应答(例如,在第一次评估时患有SD,在第二次评估时具有PR并且在最后一次评估时患有PD的受试者具有最佳的PR总体应答)。当SD被认为是最佳应答时,其通常也满足方案规定的从基线开始的最短时间。如果当SD在其它方面是最佳时间点应答时未满足最短时间,则受试者的最佳应答通常取决于随后的评估。例如,在第一次评估时患有SD、在第二次评估时患有PD并且未满足SD的最短持续时间的受试者将具有最佳的PD应答。在第一次SD评估之后失去随访的同一受试者通常被视为是无法评估的。 当节结疾病包含在靶病灶的总和中并且节结减小到“正常”大小(<10mm)时,在一些情况下,其仍具有扫描所报告的测量值。即使节结是正常的,通常也会记录此测量值,以便在基于节结大小的增加的情况下不会夸大进展。如前所述,这意味着在一些情况下具有CR的受试者在病例报告表(CRF)上的总和不是“零”。 具有整体健康状况恶化的受试者(需要停止治疗)而无客观证据表明当时疾病进展的受试者)通常被报告为“症状恶化”。通常会尽一切努力来记录甚至在停止治疗之后的客观进展。症状恶化通常不是客观应答的描述:这是停止研究疗法的原因。此类受试者的客观应答状态通常通过评估靶疾病和非靶疾病来确定,如表5到6中所示。 定义“EP、早期死亡和不可评估性”的条件是研究特异性的,并且通常清楚地描述在每个方案中(取决于治疗持续时间、治疗周期性)。 在一些情况下,难以将残留疾病与正常组织区分开。当对完全应答的评估取决于这一确定时,通常建议在分配完全应答状态之前研究残留病灶(细针抽吸/活检)。在一些情况下,FDG-PET用于以类似于活组织检查的方式升级对CR的响应,其中认为残余放射照相异常代表纤维化或瘢痕形成。 对于进展的模棱两可的发现(例如,非常小且不确定的新病灶;现有病灶的囊性变化或坏死),在一些情况下继续治疗直到下一次预定评估。如果在下一次预定评估时确认进展,则进展日期通常是怀疑进展的较早日期。 表5:时间点应答:患有靶(+/–非靶)疾病的受试者
CR=完全应答,PR=部分应答,SD=疾病稳定,PD=进行性疾病,并且NE=不可评估。 表6:时间点应答:仅患有非靶疾病的受试者
CR=完全应答,PD=进行性疾病,并且NE=不可评估。 (a)对于非靶疾病,“非CR/非PD”优于“疾病稳定”,因为在一些试验中SD越来越多地用作评估功效的终点,从而当无法测量病灶时不建议将其归类。 应答持续时间 总体应答的持续时间通常根据第一次满足CR/PR(以最先记录的为准)的测量标准直到在研究中记录复发性或进行性疾病的第一日期的时间来测量。 总体完全应答的持续时间通常根据第一次满足CR的测量标准直到客观记录复发性疾病的第一日期的时间来测量。 疾病稳定通常根据治疗开始(在随机化试验中,从随机化日期开始)直到满足进展标准来测量,以研究中的最小总和作为参考(如果基线总和最小,则这是用于计算PD的参考)。 实例23:SRA141 CIN癌症临床试验 如以上实例22中所述进行临床试验,以证实SRA141单一疗法在治疗患有具有高染色体不稳定性率的癌症的患者中的功效。 实例24:替代性SRA141给药方案 一旦足够的临床安全性和耐受性、PK和活性生物标志物数据变得可用,就会研究替代性方案。在一些情况下,在赞助者与研究者之间进行讨论之后,在试验期间的任何时间都将考虑替代性给药方案。在一些情况下,对替代性方案的需要由新出现的安全性和耐受性数据、PK和/或PDn数据指示。在一些情况下,替代性方案包含但不限于以下实例:给药7天/停药7天、给药14天/停药14天、每天给药两次或其它变化。在一些情况下,替代性给药方案群组与连续方案群组并行或代替连续方案群组而开始。 替代性给药方案的初始起始剂量将取决于来自先前群组的安全性、耐受性和PK结果。如果在用先前方案鉴定MTD后要测试替代性方案,则新方案的起始每周总剂量将不超过先前方案的MTD的每周总剂量。如果尚未用先前的方案鉴定MTD,则在一些情况下发生剂量递增步骤;然而,基于每周总剂量,剂量递增通常不会超过50%。 实例25:在双表型B髓单核细胞白血病的大鼠MV-4-11异种移植模型中SRA141处理的免疫组织化学评估 以75mg/kg BID用SRA141或媒剂或以100mg/kg QD用SRA141处理携带皮下双表型B髓单核细胞白血病MV-4-11肿瘤的大鼠,持续5天。在施用最后剂量后12小时从动物中收集肿瘤。通过免疫组织化学评估福尔马林固定石蜡包埋的肿瘤和/或皮肤的总MCM2、丝氨酸40处的磷酸化MCM2(pMCM2-S40)和丝氨酸53处的磷酸化MCM2(pMCM2-S53)、丝氨酸41处的磷酸化MCM2(pMCM2-S41)和丝氨酸139处的磷酸化-组蛋白H2A.X在稳态下的作用。 如图24A和图24B所示,肿瘤中S40和S53处的pMCM2的显著降低与皮肤组织的变化相关,这表明皮肤活检可以用于证明SRA141的中靶活性。正如预期的,在SRA141处理之后未观察到pMCM2-S41的作用,因为S41被Cdk磷酸化,而不是被Cdc7磷酸化。因此,数据进一步证明SRA141对Cdc7抑制具有特异性。还通过免疫组织化学分析了肿瘤中有丝分裂细胞的标志物pHH3,或用苏木精和曙红(H&E)染色。图24C中所示的结果表明,在给药5天之后,与媒剂相比,用SRA141处理的肿瘤导致更少的有丝分裂细胞和凋亡细胞的存在增加(参见图24C中的白色箭头),从而表明在经SRA141处理的MV-4-11肿瘤中存在抗肿瘤活性。 异种移植肿瘤的评估 对异种移植肿瘤(但不是间质或周围大鼠组织)中的肿瘤细胞进行评分。使用主要组分半定量地进行评分,以便在差异强度和H分数下以百分比为单位对肿瘤细胞的总MCM2、pMCM2-S53、pMCM2-S40和gH2AX反应性进行评分。百分比和强度量度是由科学家在每个异种移植样品中的同一肿瘤相对区域(距底部边缘一个40X视野)中进行估计的。 用于评估异种移植组织的H分数方法需要在四点半定量标度(0、1+、2+、3+)上以对应差异强度记录具有核染色的肿瘤细胞的百分比。在这个标度上:0=空、阴性或非特异性染色,1+=低或弱染色,2+=中间或中等染色,并且3+=高或强染色。 通过在四点半定量标度(0、1+、2+、3+)上将细胞与表达强度(棕色染色)乘以其对应差异强度的百分比求和来计算H分数。因此,分数范围为0到300。 H分数=[(在<1处的%)×0]+[(在1+处的%)×1]+[(在2+处的%)×2]+[(在3+处的%)×3] 大鼠皮肤的评估 两名科学家联合对大鼠皮肤样品中表皮的上皮细胞进行评分。使用主要组分定量地进行评分,以对表皮细胞的总MCM2、pMCM2-S53、pMCM2-S40和gH2AX反应性进行评分作为阳性细胞的计数。对于每个皮肤样品,标记四个均匀分布的区域进行检查。将所述区域与每个皮肤样品中的同一近似位置对齐。对于pMCM2-S53和pMCM2-S40,对四个代表性区域处的10X视野中的阳性细胞数进行计数。对于gH2Ax,对四个代表性区域处的20X视野中的阳性细胞数进行计数。如果在10X或20X下容易观察到染色,则无论强度如何,对细胞进行计数。仅在平表皮内对细胞进行计数,而不是在内陷区域中进行计数,以便一致地捕获同一总面积。 对于每种生物标志物,对跨针对每个皮肤样品分析的区域计数的阳性皮肤细胞的数量取平均值。然后将pMCM2-S53和pMCM2-S40的阳性皮肤细胞的平均数表示为相对于总MCM2的阳性皮肤细胞的平均数的百分比。将gH2AX的阳性皮肤细胞的平均数乘以2以说明其在20X相对于10X(归一化)的评估,并且然后还表示为相对于总MCM2的平均数的百分比。 通过记录由两个独立科学家在位于每个样品的同一近似位置的多个皮肤区域(两个区域用于总MCM2,四个区域用于pMCM2和gH2AX)中计数的阳性细胞数(在任何强度下)进行皮肤细胞计数。对于每个样品,对每个生物标志物在每个皮肤区域记录的阳性细胞数取平均值。将pMCM2-S53、pMCM2-S40和gH2AX(针对放大率归一化)的平均细胞计数作为总MCM2的百分比进行比较。针对gH2AX以20X进行细胞计数,并且针对所有其它标记物以10X进行细胞计数。 平均计数(pMCM2,gH2AX)=[区域1阳性细胞]+[区域2阳性细胞]+[区域3阳性细胞]+[区域4阳性细胞]/4 平均计数(总MCM2)=[区域1阳性细胞]+[区域4阳性细胞]/2 总(pMCM2)的百分比=[平均计数pMCM2]/[平均计数总MCM2]×100 总(gH2AX)的百分比=[平均计数gH2AX×2(归一化)]/[平均计数总MCM2]×100 图25A和图25B中所示的结果证明,在大鼠MV-4-11异种移植模型中肿瘤和皮肤两者中pMCM2-S40的剂量依赖性降低。 实例26:人肿瘤和皮肤的免疫组织化学评估 通过免疫组织化学评估福尔马林固定石蜡包埋的人肿瘤和人正常皮肤样品的总MCM2、pMCM2-S53、pMCM2-S40和gH2AX。 人肿瘤的评估 对人转移性结肠癌样品(但不是间质或周围非肿瘤组织组分)中的肿瘤细胞进行评分。如先前所述(实例25)计算H分数。 人皮肤的评估 两名科学家联合对人皮肤样品中表皮的上皮细胞进行评分。使用主要组分定量地进行评分,以对表皮细胞的总MCM2、pMCM2-S53、pMCM2-S40和gH2AX反应性进行评分作为阳性细胞的计数。对于每个皮肤样品,标记两个均匀分布的区域进行检查。将所述区域与每个皮肤样品中的同一近似位置对齐并且在10X下评估MCM2和pMCM2。对于gH2AX,对两个区域处的20X视野中的阳性细胞数进行计数。如果在10X或20X下容易观察到染色,则无论强度如何,对细胞进行计数。仅在平表皮内对细胞进行计数,而不是在内陷区域中进行计数,以便一致地捕获同一总面积。 对于每种生物标志物,对跨针对每个皮肤样品分析的区域计数的阳性皮肤细胞的数量取平均值。然后将pMCM2-S53和pMCM2-S40的阳性皮肤细胞的平均数表示为相对于总MCM2的阳性皮肤细胞的平均数的百分比。将gH2AX的阳性皮肤细胞的平均数乘以2以说明其在20X相对于10X(归一化)的评估,并且然后还表示为相对于总MCM2的平均数的百分比。 如实例25中所述,进行皮肤细胞计数。 平均计数(总MCM2,pMCM2,gH2AX)=[区域1阳性细胞]+[区域2阳性细胞]/2 总(pMCM2)的百分比=[平均计数pMCM2]/[平均计数总MCM2]×100 总(gH2AX)的百分比=[平均计数gH2AX×2(归一化)]/[平均计数总MCM2]×100 正常人组织的免疫组织化学评估证明,pMCM-S40的基线水平是可测量的,并且在用媒剂处理后处于与大鼠皮肤样品相当的水平(图26)。因此,人皮肤似乎是评估SRA141中靶药效学的合理替代组织。 实例27:SRA141与另外的抗肿瘤剂在各种癌细胞系中的组合 在实体肿瘤细胞系(Colo-205、SW620、A375(V600E黑素瘤))以及血液癌细胞系(KG-1、MOLM-13、MV411)中评估SRA141与九种靶向抗肿瘤剂的组合活性。使用先前描述的CTG和CTBlue测定(实例3)评估细胞活力。使用星形孢菌素作为内部对照在96孔板中进行测定。将细胞与单一药剂(I期)或SRA141和第二药剂(II期)的组合一起温育72小时。 在I期中,在所有六种细胞系中,确定以下九种药剂中的每种药剂的20%抑制浓度(IC20)和50%抑制浓度(IC50):ABT-199、ATM激酶抑制剂KU-60019、极光B激酶抑制剂巴拉塞替、MEK抑制剂曲美替尼、PI3K途径抑制剂库潘尼西、类视黄醇途径抑制剂蓓萨罗丁和维甲酸、苏氨酸酪氨酸激酶(TTK)抑制剂CFI-402257和表皮生长因子抑制剂埃罗替尼。 在II期中,在固定浓度下的每种药剂(每种药剂的IC20和IC50)和SRA141的组合的存在下重复测定,其中在SRA141的浓度范围内进行这项测定。使用布利斯独立性模型(Bliss Independence model)计算组合指数(CI)值(参见例如,Foucquier和Guedj,《药理学研究展望(Pharmacol.Res.Perspect.)》2015 3(3),所述文献通过引用整体并入本文)。CI值小于1(CI<1)表明组合作用大于预期的加和效果。 Colo-205细胞中的组合处理的结果(图27A和图27G)证明,SRA141和巴拉塞替协同作用,特别是在0.012μM巴拉塞替存在下。相比之下,SRA141与蓓萨罗丁的组合似乎具有拮抗作用。曲美替尼和库潘尼西在Colo-205细胞中似乎与SRA141具有轻微加和效应。 SW620细胞中的组合处理的结果(图27B)示出巴拉塞替、曲美替尼和库潘尼西全部对SRA141具有轻微加和效应。A375(图27C)和KG-1(图27D)细胞中的组合处理的结果示出,巴拉塞替而不是曲美替尼或库潘尼西与SRA141具有轻微加和效应。 MOLM-13细胞中的组合处理的结果(图27E)证明,ABT-199和SRA141协同作用,特别是在0.1μM ABT-199存在下。此外,巴拉塞替(图27H)、曲美替尼、库潘尼西、蓓萨罗丁和维甲酸全部在MOLM-13细胞中示出与SRA141的轻微加和效应。 MV411细胞中的组合处理的结果(图27F)示出,巴拉塞替、库潘尼西和维甲酸全部与SRA141具有轻微加和效应。 实例28:SRA141与抗凋亡基因的抑制协同作用 使用RNAi敲低在HCT116和海拉细胞中抑制抗凋亡基因BCL-XL、BCL-2和MCL-1。将细胞以2000个细胞/孔铺板在96孔板中并且用RNAi/脂质转染胺RNAimax溶液以10nM的终浓度进行转染。将经转染的细胞用SRA141以一系列剂量处理72小时。使用如先前描述的CTBlue测定(实例3)测量用针对CTRL的RNAi(无毒对照RNAi)、BCL-2、BCL-XL或MCL-1转染的处理细胞的细胞活力。图28A中所示的数据证明,通过RNAi抑制的抗凋亡基因与SRA141协同作用。 在Molm-13细胞中通过用如先前所述的0.1μM ABT-199处理抑制抗凋亡基因BCL-2(参见实例6和24)。在0.04μM到3.30μM的SRA141浓度下,将细胞用0.1μM ABT-199和SRA141处理72小时。在以下浓度的SRA141下证明与BCL-2抑制和SRA141的协同作用:0.12μM、0.37μM、1.10μM和3.30μM(图28B),如由小于1的组合指数值所指示的(参见实例24)。 实例29:Cdc7抑制剂的临床试验评估 为了进一步证实Cdc7抑制剂用于治疗人患者的癌症的功效,进行临床试验。合适的患者包含诊断患有癌症(例如,乳腺癌、结肠癌、肺癌;血癌,如白血病、淋巴瘤、骨髓瘤、急性髓性白血病(AML)和慢性髓性白血病(CML)、黑素瘤、子宫癌、甲状腺癌、慢性嗜酸性粒细胞白血病、弥漫性大B细胞淋巴瘤(DLBCL)、膀胱癌、宫颈癌、结肠直肠癌(CRC)、胃癌、子宫内膜癌、肝细胞癌、非小细胞肺癌、卵巢癌、前列腺癌、胰腺癌、脑癌、肉瘤、小细胞肺癌、成神经细胞瘤和头颈癌)的患者。向患者口服施用Cdc7抑制剂SRA-141。以从上述动物研究转化的最佳剂量开始,证实最大有效剂量。使用本领域的普通技术人员已知的技术确定来自动物(即,大鼠和/或小鼠)研究的人等效剂量(Nair,A.和Jacob,S.《基本临床药学杂志(J.BasicClin.Pharm)》;2016年3月,7(2):27-31)。SRA-141的剂量可以为100-5,000mg;100-1,000mg;1,000-2,000mg;2,000-3,000mg;3,000-4,000mg;4,000-5,000mg;1,500-2,500mg;500-1,000mg;1,000-1,500mg;1,500-2,000mg;2,000mg-2,500mg;2,500-3,000mg;3,000-3,500mg;3,500-4,000mg;4,000-4,500mg或4,500-5,000mg。可以每天、每天两次、每天三次、每隔一天、每周一次、每月一次、每天一次给药施用SRA-141持续1-7天,随后是非给药持续1-28天、每天给药持续1-28天,随后非给药持续1-28天。监测患者的疾病进展。 实例30:Cdc7抑制剂与另外的一种或多种抗肿瘤剂的组合的临床试验评估 为了进一步证实Cdc7抑制剂与mTOR途径抑制剂、DNA聚合酶抑制剂和受体酪氨酸激酶抑制剂和MAPK途径抑制剂、类视黄醇途径调节剂、细胞凋亡调节剂或PARP抑制剂组合在人患者中的功效,进行临床试验。合适的患者包含诊断患有癌症(例如,乳腺癌、结肠癌、肺癌;血癌,如白血病、淋巴瘤、骨髓瘤、急性髓性白血病(AML)和慢性髓性白血病(CML)、黑素瘤、子宫癌、甲状腺癌、慢性嗜酸性粒细胞白血病、弥漫性大B细胞淋巴瘤(DLBCL)、膀胱癌、宫颈癌、结肠直肠癌(CRC)、胃癌、子宫内膜癌、肝细胞癌、非小细胞肺癌、卵巢癌、前列腺癌、胰腺癌、脑癌、肉瘤、小细胞肺癌、成神经细胞瘤和头颈癌)的患者。向患者口服施用Cdc7抑制剂SRA-141。在人患者中,向患者同时施用mTOR途径抑制剂、DNA聚合酶抑制剂和受体酪氨酸激酶抑制剂和MAPK途径抑制剂、类视黄醇途径调节剂、细胞凋亡调节剂、PARP抑制剂或抗叶酸抗代谢药。使用本领域的普通技术人员已知的技术确定待施用的最大有效剂量。监测患者的疾病进展。 所引用参考文献 ·T.E.Creighton,《蛋白质:结构与分子性质(Proteins:Structures andMolecular Properties)》(W.H.弗里曼和公司(W.H.Freeman and Company),1993); ·A.L.Lehninger,《生物化学(Biochemistry)》(沃思出版社有限公司(WorthPublishers,Inc.),现行版); ·Sambrook等人,《分子克隆:实验室手册(Molecular Cloning:A LaboratoryManual)》(第2版,1989) ·《酶学方法(Methods In Enzymology)》(S.Colowick和N.Kaplan编辑,学术出版社有限公司(Academic Press,Inc.)); ·《雷明顿氏药物科学(Remington's Pharmaceutical Sciences)》,第18版(宾夕法尼亚州伊斯顿市:马克出版公司(Easton,Pennsylvania:Mack Publishing Company),1990); ·Carey和Sundberg,《高等有机化学(Advanced Organic Chemistry)》,第3版(普莱南出版社(Plenum Press)),第A和B卷(1992); ·美国专利第5,145,684号 ·美国专利第4,107,288号 ·Monga SP、Wadleigh R、Sharma A等人,用于在患有阻塞性食管癌的患者中缓和的顺铂/肾上腺素可注射凝胶的肿瘤内疗法(Intratumoral therapy of cisplatin/epinephrine injectable gel for palliation in patients with obstructiveesophageal cancer)《美国临床肿瘤杂志(Am.J.Clin.Oncol.)》2000;23(4):386–392; ·Mary M.Tomayko C.,Patrick Reynolds,1989.在无胸腺(裸)小鼠中皮下肿瘤大小的确定(Determination of subcutaneous tumor size in athymic(nude)mice)《癌症化疗与药理学(Cancer Chemotherapy and Pharmacology)》,第24卷,第3期,第148-154页; ·E Richtig、G Langmann、K Müllner、G Richtig和J Smolle,2004.计算肿瘤体积作为脉络膜黑素瘤存活的预后参数(Calculated tumour volume as a prognosticparameter for survival in choroidal melanomas)《眼睛(Eye)》(2004)18,619–623; ·Jensen等人,《BMC医学成像(BMC Medical Imaging)》2008.8:16; ·Tomayko等人,《癌症化疗与药理学(Cancer Chemotherapy andPharmacology)》1989年9月,第24卷,第3期,第148-154页; ·Faustino-Rocha等人,《实验室动物(Lab Anim)》(NY).2013年6月;42(6):217-24; ·Bonte D、Lindvall C、Liu H等人,多种癌症和肿瘤细胞系中的Cdc7-Dbf4激酶过表达与p53失活相关(Cdc7-Dbf4 kinase overexpression in multiple cancers andtumor cell lines is correlated with p53 inactivation)《肿瘤形成(Neoplasia.)》2008年9月;10(9):920–31。 ·Cadigan KM.Wnt信号传导:表面处的复杂性(Wnt signaling:complexity atthe surface)《细胞科学杂志(J Cell Sci)》[互联网].2006;119(3):395–402.可从以下网站获得:http://jcs.biologists.org/cgi/doi/10.1242/jcs.02826 ·Cheng AN、Jiang SS、Fan CC等人,增加的Cdc7表达是口腔鳞状细胞癌的标志物,并且Cdc7的过表达有助于对DNA损伤剂的抗性(Increased Cdc7 expression is amarker of oral squamous cell carcinoma and overexpression of Cdc7 contributesto the resistance to DNA-damaging agents)《癌症通讯(Cancer Lett)》[互联网].2013;337(2):218–25.可从以下网站获得:http://dx.doi.org/10.1016/j.canlet.2013.05.008 ·Cremolini C、Schirripa M、Antoniotti C等人,用于mCRC的一线化疗—基于审阅和证据的算法(First-line chemotherapy for mCRC-a review and evidence-basedalgorithm)《自然评论临床肿瘤学(Nat Rev Clin Oncol)》[互联网].2015;12(10):607–19.可从以下网站获得:http://dx.doi.org/10.1038/nrclinonc.2015.129 ·Dienstmann R、Vermeulen L、Guinney J等人,共有分子亚型和精密医学在结肠直肠癌中的进化(Consensus molecular subtypes and the evolution of precisionmedicine in colorectal cancer)《癌症自然评论(Nat Rev Cancer)》[互联网].2017[2017年3月3日引用];17.可从以下网站获得:http://www.nature.com.proxy.lib.umich.edu/nrc/journal/v17/n2/pdf/nrc.2016.126.pdf ·Ferlay J、Soerjomataram I、Ervik M、Dikshit R、Eser S、Mathers C、RebeloM、Parkin DM、Forman D、Bray,F.GLOBOCAN 2012v1.0,全球癌症发生率和死亡率:IARC癌症基底11号(Cancer Incidence and Mortality Worldwide:IARC CancerBase No.11)[互联网].法国里昂:国际癌症研究机构(Lyon,France:International Agency for Researchon Cancer);2013.可从以下网站获得:http://globocan.iarc.fr,于2018年5月14日访问。 ·Fodde R、Kuipers J、Rosenberg C等人,APC肿瘤抑制基因中的突变引起染色体不稳定性(Mutations in the APC tumour suppressor gene cause chromosomalinstability)《自然细胞生物学(Nat Cell Biol)》[互联网].2001;3(4):433–8.可从以下网站获得:http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=11283620&retmode=ref&cm d=prlinks%5Cnpapers2://publication/doi/10.1038/35070129 ·Gregory GP、Hogg SJ、Kats LM等人,通过地那昔利布的CDK9抑制有力地抑制Mcl-1以诱导侵袭性MYC驱动的体内B细胞淋巴瘤中的持久凋亡应答(CDK9 inhibition bydinaciclib potently suppresses Mcl-1to induce durable apoptotic responses inaggressive MYC-driven B-cell lymphoma in vivo)《白血病(Leukemia)》2015;29:1437-1441。 ·Guinney J、Dienstmann R、Wang X等人,结肠直肠癌的共有分子亚型(Theconsensus molecular subtypes of colorectal cancer)《自然医学(Nat Med)》[互联网].2016年10月12日[2017年3月17日引用];21(11):1350–6.可从以下网站获得:http://www.nature.com/doifinder/10.1038/nm.3967 ·Hailey JR、Nold JB、Brown RH等人,斯普拉格-道来氏大鼠不良/非不良的双生增殖病灶(Biliary Proliferation Lesions in the Sprague-Dawley Rat Adverse/Non-adverse)《毒理学病理学(Toxicologic Pathology)》2013;42(5):844-854 ·Hou Y、Wang H-Q、Ba Y.细胞分裂周期7蛋白的高表达与患有弥漫性大B细胞淋巴瘤的患者的不良预后相关(High expression of cell division cycle 7proteincorrelates with poor prognosis in patients with diffuse large B-celllymphoma)《医学肿瘤学(Med Oncol)》[互联网].2012;29(5):3498–503.可从以下网站获得:http://link.springer.com/10.1007/s12032-012-0223-y ·Howlader N、Noone AM、Krapcho M、Miller D、Bishop K、Kosary CL、Yu M、RuhlJ、Tatalovich Z、Mariotto A、Lewis DR、Chen HS、Feuer EJ、Cronin KA(编辑)《SEER癌症统计评论(SEER Cancer Statistics Review)》,1975-2014,美国国家癌症研究所(National Cancer Institute)贝塞斯达,马里兰州,http://seer.cancer.gov/csr/1975_2014/,基于2016年11月的SEER数据提交,已发布到SEER网站,2017年4月 ·Huggett MT、Tudzarova S、Proctor I等人,Cdc7是胰腺癌中的有效抗癌靶标,这是由于消除了DNA来源的活化检查点(Cdc7 is a potent anti-cancer target inpancreatic cancer due to abrogation of the DNA origin activation checkpoint)《肿瘤靶标(Oncotarget)》[互联网].2016年4月;7(14):18495–507.可从以下网站获得:http://www.oncotarget.com/abstract/7611 ·ICH协调三方指南:抗癌药物的非临床评估S9(ICH Harmonised TripartiteGuideline:Nonclinical Evaluation For Anticancer Pharmaceuticals S9)用于人类使用的药物注册的技术要求协调的国际会议(International Conference onHarmonisation of Technical Requirements for Registration of Pharmaceuticalsfor Human Use)2009. ·Iwai K、Gotou M、Yamanaka K、Ohashi A.,用于确定受新型CDC7选择性抑制剂TAK-931影响的信号传导途径的磷酸-蛋白质组学分析(Phospho-proteomics analysis todetermine the signaling pathways affected by a novel CDC7-selective inhibitorTAK-931)《AACR年度会议(AACR Annual Meeting)》2018.摘要2312。 ·Larasati和Duncker,B.P.(2016).管理DDK调节DNA复制起始的机制(Mechanisms Governing DDK Regulation of the Initiation of DNA Replication)《基因(Genes)》,8(1),3.https://doi.org/10.3390/genes8010003 ·Montagnoli A、Tenca P、Sola F等人,Cdc7抑制揭示了在癌细胞中有缺陷的p53依赖性复制检查点(Cdc7 Inhibition Reveals a p53-Dependent ReplicationCheckpoint That Is Defective in Cancer Cells)《癌症研究(Cancer Res.)》2004;64(19):7110–6. ·Rodriguez-Acebes S、Proctor I、Loddo M,在其开始之前靶向DNA复制:Cdc7作为p53突变型乳腺癌中的治疗靶标(Targeting DNA replication before it starts:Cdc7as a therapeutic target in p53-mutant breast cancers)《美国病理学杂志(Am JPathol)》[互联网].2010年10月;177(4):2034–45.可从以下网站获得:http://www.ncbi.nlm.nih.gov/pubmed/20724597 ·Schukken KM、Foijer F.,CIN和非整倍性:不同的概念,不同的结果(CIN andAneuploidy:Different Concepts,Different Consequences)《生物学测定(生物学)》2018;40(1):1–9。 ·Swords R、Mahalingam D、O'Dwyer M等人,Cdc7激酶—用于药物开发的新靶标(Cdc7kinase-A new target for drug development)《欧洲癌症杂志(Eur J Cancer)》[互联网].2010年1月;46(1):33–40.可从以下网站获得:http://dx.doi.org/10.1016/j.ejca.2009.09.020 ·Therkildsen C、Bergmann TK、Henrichsen-Schnack T、Ladelund S、NilbertM.,KRAS、NRAS、BRAF、PIK3CA和PTEN对于转移性结肠直肠癌中的抗EGFR治疗的预测值:系统综述和元分析(The predictive value of KRAS,NRAS,BRAF,PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer:A systematic review and meta-analysis)《肿瘤学报(Acta Oncol)》(Madr).2014;53(7):852–64。 ·Xiang SY、Lilly E.,选择性CDC7抑制剂(LY3177833)影响染色体动力学并且在PDX肿瘤模型中具有稳健且持久的活性(A selective CDC7 inhibitor(LY3177833)impacts chromosome dynamics and has robust and durable activity in PDX tumormodels)《AACR年度会议(AACR Annual Meeting)》2016.最新文摘(Late breakingabstract)。 ·Zehir A、Benayed R、Shah RH等人,转移性癌症的突变景观揭示自10,000名患者的前瞻性临床测序(Mutational landscape of metastatic cancer revealed fromprospective clinical sequencing of 10,000patients)《自然医学(Nat Med)》[互联网].2017;23(6).可从以下网站获得:http://www.nature.com/doifinder/10.1038/nm.4333
- 包括CDC7抑制剂的治疗癌症的方法
- 使用PI3Kβ抑制剂和包括MEK和RAF抑制剂的MAPK通道抑制剂治疗癌症的组合物和方法