对PARP抑制剂耐药的癌症的治疗剂
文献发布时间:2024-04-18 19:52:40
技术领域
本申请要求基于2021年5月18日提交的韩国专利申请No.10-2021-0064278的优先权,其全部内容作为本说明书的一部分并入本文。
本发明涉及一种癌症治疗剂,其可以用于治疗对PARP抑制剂耐药的实体癌患者。
背景技术
DNA突变的积累被认为是癌症的代表性原因之一。哺乳动物从单细胞(受精卵)经过细胞分裂的无限过程生长和发育,并且在这个过程中,在DNA中不可避免地发生突变(以下简称“DNA损伤”)。然而,DNA损伤通过各种DNA修复机制如同源重组(HR)或非同源末端连接(NHEJ)进行修复。各种类型的蛋白质参与每种DNA修复机制,如果这些蛋白质中的一些发生突变,则在DNA修复机制中就会出现问题,从而使癌症的概率增加几倍至几百倍。一般来说,当同源重组功能丧失时,基因组变得不稳定,其诱导各种遗传变化并最终导致肿瘤。
参与受损的DNA修复的BRCA1/2基因是抑制肿瘤发生的基因。众所周知,当BRCA1/2基因中发生突变并且其功能降低时,受损的DNA没有得到适当的修复并且DNA损伤积累,导致癌症。这被称为同源重组缺陷(HRD)。众所周知,与BRCA1/2基因突变相关的乳腺癌和卵巢癌是同源重组缺陷肿瘤。特别是,已知在BRCA1/2基因突变的女性中,患乳腺癌或卵巢癌的概率分别增加高达80%和60%。然而,已知BRCA1/2基因突变不仅与上述乳腺癌和卵巢癌相关,还与胃癌、胰腺癌、前列腺癌、胆囊癌、胆道癌和结肠直肠癌相关。
聚(ADP-核糖)聚合酶(PARP)蛋白质是修复DNA复制过程中不可避免发生的错误所必需的蛋白质,并且是一种通过识别细胞核中受损的DNA而被激活然后通过翻译后过程(多聚ADP-核糖基化修饰(PARrylation))激活DNA修复相关蛋白质的酶。尽管到迄今已知17个PARP家族,但是只有PARP 1/2被确定为具有聚(ADP-核糖基化)活性的DNA修复酶,并且被认为是细胞存活的必需酶。
已知同源重组缺陷肿瘤对由PARP抑制剂引起的DNA损伤表现出敏感响应。因此,PARP抑制剂作为癌症治疗药物在临床实践中具有巨大的潜力。事实上,PARP抑制剂诸如奥拉帕尼(Lynparza
作为PARP抑制剂开发的6-{4-[(5-氧代-1,2,3,4,5,6-六氢苯并[h][1,6]萘啶-8-基)甲基]哌嗪-1-基}烟腈具有下式I的结构。下面式I化合物或其药学上可接受的盐不仅对PARP1/2而且对端锚聚合酶1/2表现出抑制活性。已知端锚聚合酶参与有丝分裂,有丝分裂与Wnt/β-catenin信号通路、DNA修复过程和细胞周期高度相关。另外,端锚聚合酶1/2ADP-核糖基化TRF-1用作端粒长度的正调节因子,使端粒通过端粒酶延长。另外,预期下面式I化合物或其药学上可接受的盐对于对铂基抗癌化疗完全或部分响应的复发性上皮性卵巢癌、高浆液性卵巢癌等具有治疗效果。
<式I>
除了各种PARP抑制剂作为癌症治疗的靶向疗法的潜力或预期之外,包括奥拉帕尼在内的PARP抑制剂像其它抗癌药物一样具有高的先天性/获得性耐药性或难治率。随着同源重组缺陷肿瘤耐药比例的增加,对此的研究正在进行,但迄今尚未取得重大进展。
另外,尚不清楚式I的化合物是否可以治疗对式I以外的其它抗癌药物耐药的实体癌,特别是对现有标准治疗中使用的PARP抑制剂耐药的实体癌。
在此背景下,本发明的发明人从各种角度研究了可以用于治疗对PARP抑制剂表现出耐药性的患者的抗癌剂。结果,通过证实本发明的式I化合物减小了对现有PARP抑制剂(如奥拉帕尼)耐药的患者的肿瘤大小,完成了本发明。
[现有技术文献]
[专利文献]
1、韩国专利注册No.10-1136702(发布日期:2012年4月20日)
2、韩国专利注册No.10-1146806(发布日期:2012年5月22日)
3、韩国专利注册No.10-1837047(发布日期:2018年3月9日)
发明内容
技术问题
本发明的一个目的是提供一种药物组合物,该药物组合物包含6-{4-[(5-氧代-1,2,3,4,5,6-六氢苯并[h][1,6]萘啶-8-基)甲基]哌嗪-1-基}烟腈或其药学上可接受的盐,作为治疗对PARP抑制剂耐药的患者的实体癌的组合物。
本发明的另一目的是提供一种通过向对PARP抑制剂具有耐药性的受试者给药6-{4-[(5-氧代-1,2,3,4,5,6-六氢苯并[h][1,6]萘啶-8-基)甲基]哌嗪-1-基}烟腈或其药学上可接受的盐来治疗受试者的实体癌的方法。
本发明的再一目的是提供6-{4-[(5-氧代-1,2,3,4,5,6-六氢苯并[h][1,6]萘啶-8-基)甲基]哌嗪-1-基}烟腈或其药学上可接受的盐用于治疗对PARP抑制剂耐药的患者的实体癌。
技术方案
为了实现上述目的,本发明提供了一种用于治疗对PARP抑制剂耐药的患者的实体癌的药物组合物,其包含6-{4-[(5-氧代-1,2,3,4,5,6-六氢苯并[h][1,6]萘啶-8-基)甲基]哌嗪-1-基}烟腈或其药学上可接受的盐。
在本发明的一个实施方案中,PARP抑制剂可以是选自奥拉帕尼(olaparib)、鲁卡帕尼(rucaparib)、尼拉帕尼(niraparib)和他拉唑帕尼(talazoparib)中的至少一种,但不限于此。
在本发明的一个实施方案中,患者可能有BRCA1/2突变。
在本发明的一个实施方案中,患者可能有胚系BRCA1/2突变。
在本发明的一个实施方案中,患者可能有体细胞BRCA1/2突变。
在本发明的一个实施方案中,患者可以是有BRCA1/2突变的患者,并且最初对PARP抑制剂响应,但是在治疗期间获得了耐药性并且对PARP抑制剂没有响应或者其癌症复发。
在本发明的一个实施方案中,患者可以是对PARP抑制剂没有响应的有BRCA1/2突变的患者。
在本发明的一个实施方案中,患者可以是没有BRCA1/2突变并且先前没有对PARP抑制剂响应或其癌症复发的患者。
在本发明的一个实施方案中,实体癌可以是已知由BRCA1/2突变引起的卵巢癌、乳腺癌、前列腺癌、胰腺癌、结肠癌、胆囊癌、胆道癌或胃癌,但不限于此。
在本发明的一个实施方案中,实体癌可以是进行性实体癌、复发性实体癌或转移性实体癌的形式。
在本发明的一个实施方案中,如果实体癌是卵巢癌,则它可以是进行性卵巢癌、复发性卵巢癌、高级别浆液性卵巢癌(包括输卵管癌或原发性腹膜癌),并且在原发癌是卵巢癌的转移性癌症的情况下,它可以是乳腺癌、前列腺癌、胰腺癌、结肠癌、胆囊癌、胆道癌、胃癌、肝癌或肺癌,但不限于此。
在本发明的一个实施方案中,6-{4-[(5-氧代-1,2,3,4,5,6-六氢苯并[h][1,6]萘啶-8-基)甲基]哌嗪-1-基}烟腈的药学上可接受的盐可以是柠檬酸盐。
另外,本发明提供了用于治疗对PARP抑制剂耐药的实体癌患者的6-{4-[(5-氧代-1,2,3,4,5,6-六氢苯并[h][1,6]萘啶-8-基)甲基]哌嗪-1-基}烟腈或其药学上可接受的盐。
另外,本发明提供了一种治疗对PARP抑制剂耐药的实体癌患者的方法,该方法通过施用有效量的6-{4-[(5-氧代-1,2,3,4,5,6-六氢苯并[h][1,6]萘啶-8-基)甲基]哌嗪-1-基}烟腈或其药学上可接受的盐。
另外,本发明提供了6-{4-[(5-氧代-1,2,3,4,5,6-六氢苯并[h][1,6]萘啶-8-基)甲基]哌嗪-1-基}烟腈或其药学上可接受的盐在制造用于治疗对PARP抑制剂耐药的实体癌患者的药物中的用途。
有益效果
由于根据本发明的包含6-{4-[(5-氧代-1,2,3,4,5,6-六氢苯并[h][1,6]萘啶-8-基)甲基]哌嗪-1-基}烟腈或其药学上可接受的盐的药物组合物可以有效地减小对PARP抑制剂耐药的实体癌患者的肿瘤大小,因此它可以有效地用于治疗对PARP抑制剂耐药的实体癌患者。
附图说明
图1是示出了分析本发明的式I化合物的柠檬酸盐的抗癌效果的结果的图表(实施例2)。
图2是示出了本发明的式I化合物的柠檬酸盐的抑制Wnt信号通路激活的实验结果的图表(实施例4)。
图3是示出了使用异种移植模型进行评价的本发明的式I化合物的柠檬酸盐的抗癌效果的图(实施例5),以及
图4是示出了使用异种移植模型进行评价的本发明的式I化合物的柠檬酸盐的抗癌效果的图(实施例6)。
具体实施方式
下文中,将详细描述本发明。
在描述和要求本公开的特定特征时,除非另有规定,否则将根据下面阐述的定义使用下面术语。
应当理解的是,尽管本文中的某些方面结合术语“包括”来描述,但是也提供了术语“由...组成”和/或“基本上由...组成”来描述的其它类似方面。
术语“药学上可接受的”是指从药理学/毒理学的角度来看,就组成、配方、安全性等而言,患者可接受的物质,而“药学上可接受的载体”是指不干扰活性成分的生物活性效果且给药时对受试者无毒的介质。
术语“耐药性”是指药物没有预期的响应(抗癌效果)的情况。具体地,在本发明中,它意味着涵盖以下所有情况:药物从一开始就没有响应(难治性),以及在最初对药物响应后从某一点发生复发(癌症病变大小首先减小,然后癌症复发且大小增加的情况;获得性耐药性),尽管有PARP抑制剂标准治疗。这里,“耐药性”和“耐受性”可以互换使用。
术语“患者”或“受试者”或“个体”是指患有病症的生物体,其疾病可以通过给药本发明的药物组合物来治疗,诸如实体癌,并且包括人类和动物。受试者的实例包括但不限于哺乳动物(例如,小鼠、猴、马、牛、猪、狗、猫等),并且优选人类。另外,本发明中的“患者”或“受试者”或“个体”包括对PARP抑制剂耐药的实体癌患者。
术语“BRCA1/2突变”指BRCA1和/或BRCA2的突变,并指在BRCA1基因和BRCA2基因的一个或多个位点处自然发生的突变。因此,突变可以发生在选自BRCA1基因和BRCA2基因中的任一基因中,或者突变可以发生在两个基因中,并且突变可以发生在各个基因的一个位点或两个或更多个位点中。
如上所述,PARP抑制剂在临床实践中作为同源重组缺陷肿瘤的靶向治疗显示出巨大的潜力,但已知具有高的先天性或获得性耐药性的获得率。有多种机制解释其原因如下:i)通过增加ABC转运蛋白增加药物流出,ii)激活PAR链,iii)通过作用于同源重组机制的肿瘤抑制基因诸如p53的突变来重新激活同源重组机制,iv)稳定或保护复制叉的部分,以及v)激活Wnt信号通路。
关于上面iii)的机制,已知多种蛋白质参与同源重组过程,并且在有BRCAl/2突变的癌症患者中,也同时发现参与同源重组过程的其它蛋白质,诸如p53、ATM、ATR和p51的突变。因此,已经解释了它们可能与获得对PARP抑制剂的耐药性有关。
虽然PARP抑制剂作为同源重组缺陷肿瘤患者的特异性靶向治疗,但是它们具有容易获得耐药性的特征。因此,对可以用于治疗对PARP抑制剂耐药的患者的新型抗癌药物的需求日益增加。
多药耐药性(MDR)是抗癌治疗失败的原因之一,近年来已成为抗癌治疗领域中的一个重要问题。这种能力是由于癌细胞中MDR基因的存在。MDR1(ABCB1)基因是一种产生称为P-糖蛋白(下文中简称P-gp)的物质的基因。P-gp是一种帮助各种类型的药物穿过细胞膜的酶,并在从细胞内部排泄药物中发挥作用。当许多抗癌药物持续给药给患者时,就会出现耐药性,P-gp的过度表达是耐药机制之一。因此,当P-gp过表达时,作为P-gp底物的药物被P-gp排泄出细胞,因此不表现出药物功效。在抗癌药物的使用中,这种多药耐药性是一个重要的限制因素,正在进行各种研究来克服这种多药耐药性。因此,P-gp过表达的癌细胞可以通过选择不被用作P-gp底物的抗癌药物来抑制P-gp功能或克服耐药性。
经证实,与常规PARP抑制剂相比,本发明式I化合物的柠檬酸盐具有显著较低的P-gp流出率。因此,它可以有效地用于治疗对PARP抑制剂耐药的患者。
Wnt信号通路与胚胎发育、组织稳态和各种疾病有关。过度活跃的信号传导导致β-连环蛋白的积累,β-连环蛋白转运到细胞核中,促进癌基因的转录和细胞生长。因此,正在努力开发阻断Wnt信号通路的治疗剂。
最近的研究报道,PARP抑制剂的耐药机制与Wnt信号通路有关。即,PARP抑制剂激活Wnt信号通路,并且已知耐药性是通过激活Wnt信号通路而发生的。因此,对PARP抑制剂的耐药性可以通过选择可以阻断对PARP抑制剂获得耐药性的癌细胞中Wnt信号通路的抗癌药物来克服。
发现本发明式I化合物的柠檬酸盐抑制Wnt信号通路。因此,它可以有效地用于治疗对PARP抑制剂耐药的患者。
在本发明中,发现当对PARP抑制剂耐药的实体癌患者采用6-{4-[(5-氧代一1,2,3,4,5,6-六氢苯并[h][1,6]萘啶-8-基)甲基]哌嗪-1-基}烟腈(式I化合物)或其药学上可接受的盐治疗时,实体癌的大小减小。
这一事实可以通过例如以下试验来证实:用式I化合物或其药学上可接受的盐处理对PARP抑制剂(奥拉帕尼、鲁卡帕尼、尼拉帕尼或他拉唑帕尼)耐药的细胞系,或者从对PARP抑制剂耐药的癌症(例如:卵巢癌)患者中分离的原代细胞(CHA-OVA-13细胞)。具体地,在BRCA突变阳性卵巢癌或乳腺癌细胞系诸如HCC1937、SNU-251、BT474和SNU-119中选择对PARP抑制剂耐药的细胞系后(例如:IC
另外,可以通过实验证实与凋亡、同源重组和信号转导相关的蛋白质诸如pATR、pCHK1、pAKT、端锚聚合酶、裂解的半胱天冬酶3、裂解的PARP蛋白质在通过用式I化合物或其药学上可接受的盐处理发生细胞凋亡的PARP抑制剂耐药的细胞系中的表达水平,来证实式I化合物的抗癌活性机制。
这种机制也可以通过针对移植了PARP抑制剂耐药的细胞系的动物模型和实际PARP抑制剂耐药的癌症患者的临床试验结果来证实。
本发明提供了一种药物组合物,包含由下式I表示的化合物“6-{4-[(5-氧代-1,2,3,4,5,6-六氢苯并[h][1,6]萘啶-8-基)甲基]哌嗪-1-基}烟腈”或其药学上可接受的盐,用于治疗对PARP抑制剂耐药的实体癌患者。
<式I>
由于式I化合物示出对PARP1/2的抑制活性,其可以用作同源重组缺陷肿瘤患者的靶向治疗,并且是能够同时抑制端锚聚合酶1/2的抗癌剂。
端锚聚合酶参与端粒稳态、Wnt/β-连环蛋白信号传导、葡萄糖代谢和细胞周期进程。特别地,Wnt/β-连环蛋白参与癌症相关基因的转录过程,Wnt/β-连环蛋白的信号机制在包括胃肠癌在内的多种癌症中被激活。因此,据报道,当端锚聚合酶被抑制时,通过抑制Wnt/β-连环蛋白信号传导获得抗癌作用。事实上,已经尝试开发端锚聚合酶抑制剂作为抗癌剂。
因此,式I化合物或其药学上可接受的盐可以像被用作常规标准治疗的奥拉帕尼一样抑制PARP1/2,并且另外还抑制端锚聚合酶。因此,可以看出它的作用机制不同于奥拉帕尼等。
在本发明中,式I化合物的药学上可接受的盐是由药学上可接受的游离酸形成的有用的酸加成盐。酸加成盐通过常规的方法制备,例如,通过将化合物溶解在过量的酸水溶液中,并使用与水混溶的有机溶剂诸如甲醇、乙醇、丙酮或乙腈来沉淀出盐。即,它可以通过在水中加热等摩尔量的化合物和酸或醇(如乙二醇单甲醚),然后从混合物中蒸发溶剂并干燥,或抽滤沉淀的盐来制备。
此时,有机酸和无机酸可以用作游离酸。无机酸可以包括盐酸、磷酸、硫酸、硝酸等。有机酸可以包括甲磺酸、对甲苯磺酸、乙酸、三氟乙酸、马来酸、琥珀酸、草酸、苯甲酸、酒石酸、富马酸、扁桃酸、丙酸、柠檬酸、乳酸、乙醇酸、葡萄糖酸、半乳糖醛酸、谷氨酸、戊二酸、葡萄糖醛酸、天冬氨酸、抗坏血酸、碳酸、香草酸、氢碘酸等,但不限于此。
特别地,可以优选地使用式I化合物的柠檬酸盐。
在本发明的一个实施方案中,作为式I化合物的药学上可接受的盐,可以使用式I化合物的柠檬酸盐的无水、一水合物或二水合物,并且可以使用结晶或无定形的形式,或者结晶和无定形的形式的混合形式。
式I化合物的各种形式的药学上可接受的盐可以通过本领域中已知的方法制备。
在本发明的一个实施方案中,实体癌患者对其耐药的PARP抑制剂包括奥拉帕尼、鲁卡帕尼、尼拉帕尼、他拉唑帕尼等,它们是用于标准治疗的抗癌药物,但不限于此。
在本发明的一个实施方案中,PARP抑制剂可以是奥拉帕尼。
在本发明的一个实施方案中,患有实体癌的患者可以是患有具有BRCA1/2突变的同源重组缺陷肿瘤的患者。
在本发明的一个实施方案中,BRCA1/2突变可以是胚系突变或体细胞突变。
在本发明的一个实施方案中,患者可以是具有BRCA1/2突变,并且最初对PARP抑制剂响应,然后在治疗过程中获得耐药性并且对PARP抑制剂没有响应的患者,或者其癌症已经复发的患者。
在本发明的一个实施方案中,患者可以是示出具有BRCA1/2突变,但是对PARP抑制剂没有响应的患者。
在本发明的一个实施方案中,患者可以是不具有BRCA1/2突变,并且先前没有对PARP抑制剂响应的患者,或者其癌症已经复发的患者。
在本发明的一个实施方案中,实体癌可以是进行性实体癌、复发性实体癌或转移性实体癌。
在本发明的一个实施方案中,实体癌可以是乳腺癌、前列腺癌、胰腺癌、卵巢癌、进行性卵巢癌、高级别浆液性卵巢癌(包括输卵管癌或原发性腹膜癌);以及从原发性癌症转移的乳腺癌、前列腺癌、胰腺癌、卵巢癌,但不限于此。
在本发明的一个实施方案中,实体癌可以是卵巢癌,以及从原发性卵巢癌扩散的转移性癌。
根据本发明的药物组合物可以进一步包含一种或多种药学上可接受的载体或一种或多种赋形剂和/或稀释剂。
药学上可接受的载体的实例包括但不限于:固体和/或液体诸如乙醇、甘油、水等。基于组合物的总重量,本发明的药物组合物中载体的量可以在约5重量%至约99重量%的范围内。药学上可接受的赋形剂和稀释剂的类型包括无毒的且相容的填料、粘合剂、崩解剂、缓冲剂、防腐剂、润湿剂、填充剂、抗氧化剂、润滑剂、调味剂、增稠剂、着色剂、表面活性剂、乳化剂和悬浮剂等,但不限于此。这种赋形剂和稀释剂包括乳糖、葡萄糖、蔗糖、山梨糖醇、甘露糖醇、木糖醇、赤藓糖醇、麦芽糖醇、淀粉、阿拉伯胶树胶、海藻酸盐、明胶、磷酸钙、硅酸钙、纤维素、甲基纤维素、微晶纤维素、聚乙烯吡咯烷酮、水、羟基苯甲酸甲酯、羟基苯甲酸丙酯、滑石、硬脂酸镁和矿物油,但不限于此。对于技术人员来说显而易见的是,可以使用所有其它药学上可接受的载体、赋形剂和稀释剂。
含有本发明化合物或它的盐的药物组合物可以根据常规的方法配制成口服制剂的形式诸如片剂、粉末、颗粒剂、药丸、胶囊、混悬剂、乳液、内溶液、乳液以及糖浆,外用制剂,栓剂或无菌注射液,并使用。
根据本发明的药物组合物可以是无菌可注射制剂的形式,如无菌可注射水性或油性悬浮液。该悬浮液可以根据本领域中已知的技术,使用合适的分散剂或润湿剂(例如,吐温80)和悬浮剂来配制。无菌可注射制剂可以是在无毒的胃肠外可接受稀释剂或溶剂(例如,在1,3-丁二醇中的溶液)中的无菌可注射溶液或悬浮液。可接受的载体和溶剂包括甘露醇、水、林格氏溶液或等渗氯化钠溶液。另外,无菌固定油可以方便地用作溶剂或悬浮介质。为此,可以使用任一温和的固定油,包括合成的甘油单酯或甘油二酯。脂肪酸诸如油酸及它的甘油酯衍生物可以有效地用于可注射制剂以及药学上可接受的天然油(例如,橄榄油或蓖麻油),尤其是它的聚氧乙烯化的油。
根据本发明的药物组合物可以以任一口服可接受的形式口服给药,包括但不限于胶囊、片剂、和水悬浮液和溶液。
本发明药物组合物的肠胃外给药组合物可以制备成用于直肠给药的栓剂或注射剂的形式。栓剂组合物可以通过将本发明的化合物与合适的无刺激性赋形剂混合来制备,该赋形剂在室温下为固体,但在直肠温度下为液体。这种材料可以包括但不限于可可脂、蜂蜡和聚乙二醇。
在可注射组合物的情况下,本发明的化合物可以作为活性成分包含在常规注射用赋形剂中,给药途径可以是静脉注射、肌肉注射、皮下注射等,但不限于此。
本发明的药物组合物中含有治疗有效量或预防有效量的上述新化合物。根据本发明的化合物的优选剂量根据患者的病症和体重、疾病的严重程度、药物的类型、给药途径和持续时间而变化,但是可以由本领域技术人员适当地选择。然而,为了达到理想的效果,本发明的式I化合物或其药学上可接受的盐以每天0.0001mg至1000mg、0.01mg至500mg、0.1mg至300mg、1mg至200mg或50mg至200mg的量给药。可以单次给药,也可以分几次给药。在本发明的组合物中,基于组合物的总重量,式I的化合物可以以0.0001重量%至50重量%的量配制。
除了式I表示的化合物、它的光学异构体、它的外消旋体或其药学上可接受的盐之外,本发明的药物组合物还可以包含至少一种表现出相同或相似药用效果的活性成分。
另外,本发明提供了式I化合物或其药学上可接受的盐在制备用于预防或治疗对PARP抑制剂耐药的实体癌的药物中的用途。
用于制备药物的式I表示的化合物或其药学上可接受的盐可以与药学上可接受的辅料、稀释剂、载体等混合,并与其它活性剂制备成具有协同作用的复合制剂。
另外,本发明提供了一种通过将有效量的式I化合物或其药学上可接受的盐给药给哺乳动物(包括人类)来预防或治疗对PARP抑制剂耐药的实体癌的方法。
本发明的预防或治疗方法包括通过给药由式I表示的化合物或其药学上可接受的盐不仅在症状出现之前治疗疾病本身,还抑制或避免疾病症状。在疾病管理中,特定活性成分的预防或治疗剂量将根据疾病或病症的性质和严重程度以及活性成分的给药途径而变化。给药的剂量和频率将根据个体患者的年龄、体重和响应而变化。考虑到这些因素,本领域技术人员可以容易地选择合适的剂量方案。另外,本发明的预防或治疗方法可以进一步包括将治疗有效量的用于疾病治疗的附加活性剂与由式I表示的化合物一起给药。附加活性剂可以表现出与式I化合物或其药学上可接受的盐协同或叠加作用。
本发明的药物组合物、用途和治疗方法中提到的事项同等适用,除非它们相互矛盾。
本发明的药物组合物可以以包括说明书的试剂盒的形式提供。
除非另有说明,否则说明书和权利要求中使用的所有数字,无论是否提出,在所有情况下都应理解为由术语“约”来修饰。此外,说明书和权利要求中使用的精确数值应理解为形成本公开的额外的实施方案。已努力确保实施例中公开的数字的准确性。然而,任何测量的数字固有地可能包含某些误差值,这些误差值是由在其各自的测量技术中所发现的标准偏差引起的。
下文中,将通过实施例更详细地描述本发明。这些实施例仅用于更详细地解释本发明,并且对于本领域技术人员来说显而易见的是,根据本发明的要点,本发明的范围不受这些实施例的限制。
在本发明中,式I化合物的柠檬酸盐可以通过以下本领域中已知的方法来制备:韩国专利申请No.10-2021-0064416中公开的方法,或与本发明同日提交的申请中公开的方法,本申请要求基于上述申请的优先权。例如,制备式I化合物的柠檬酸盐的方法如下。
制备例1:
制备6-{4-[(5-氧代-1,2,3,4,5,6-六氢苯并[h][1,6]萘啶-8-基)甲基]哌嗪-1-基}烟腈柠檬酸盐(式I化合物的柠檬酸盐)
将甲醇(25.7L)和纯净水(25.7L)加入到6-{4-[(5-氧代-1,2,3,4,5,6-六氢苯并[h][1,6]萘啶-8-基)甲基]哌嗪-1-基}烟腈(式I化合物、7.34kg、18.32mol)中。将柠檬酸(5.28kg、27.49mol)溶解在甲醇和纯净水的1:1混合的溶液(22L)中,然后加入其中。在15℃至25℃下搅拌30分钟之后,将温度升至60℃,并且在60℃至70℃下搅拌2小时。冷却至室温后,过滤混合物以得到式I化合物的柠檬酸盐一水合物(10.7kg、95.8%)。
向式I化合物的柠檬酸盐一水合物(500g、0.82mol)中加入乙醇(2.5L)、丙酮(2.5L)和异丙醇(2.5L),然后向其中加入纯净水(20mL)。将温度升至55℃后,在55℃至75℃下搅拌4小时。冷却至25℃以下后,将混合物搅拌30分钟。过滤所得固体以得到式I化合物的柠檬酸酸酐(470g,收率96.7%)。
实施例1:分析式I化合物的抗癌效果(体外实验)
为了分析式I化合物(6-{4-[(5-氧代-1,2,3,4,5,6-六氢苯并[h][1,6]萘啶-8-基)甲基]哌嗪-1-基}烟腈)的柠檬酸盐在同源重组缺陷型肿瘤中的抗癌作用,使用各种细胞系进行细胞分裂分析。
在该测定中,HCC1937、SNU-251、BT474和SNU-119细胞系用作BRCA突变阳性卵巢癌或乳腺癌细胞系,并且作为BRCA突变阳性卵巢癌的原代细胞,使用从实际BRCA1突变阳性卵巢癌患者中分离的CHA-OVA-13细胞(显示出对奥拉帕尼的获得性耐药性的卵巢癌原代细胞)。
首先,通过测量IC
在对奥拉帕尼耐药的细胞系上使用各种PARP抑制剂来进行下面实验。具体地,将每个细胞悬浮在培养基中,分配到96孔板中,并且在5%CO2和37℃下培养24小时。然后,以剂量依赖性方式处理奥拉帕尼、尼拉帕尼、他拉唑帕尼和式I化合物的柠檬酸盐,72小时后加入MTT试剂,3小时后加入停止缓冲液(10%的SDS)。反应2至4小时后,在595nm处测量吸光度,并在每种药物抑制细胞生长50%的浓度下计算IC
同时,为了在分子水平上请确认用每种药物处理的细胞凋亡机制和抗癌活性机制,通过免疫印迹法分析了与细胞凋亡相关的蛋白质诸如pATR、pCHK1、pAKT、端锚聚合酶、裂解的半胱天冬酶3和裂解的PARP蛋白质的量。
实施例2:分析抗癌效果(体外实验)
为了分析式I化合物(6-{4-[(5-氧代-1,2,3,4,5,6-六氢苯并[h][1,6]萘啶-8-基)甲基]哌嗪-1-基}烟腈)的柠檬酸盐对BRCA野生型细胞和突变型细胞的抗癌作用,使用各种细胞系进行细胞分裂分析。
(1)实验方法
在该测定中,使用野生型BRCA卵巢癌细胞系OVCAR-3、OVCAR-5、SKOV3、NCI/ADR-RES、A2780、A2780 CR(对卡铂耐药的诱导细胞系)和OVCA433R(对奥拉帕尼耐药的细胞系)以及突变型BRCA1卵巢癌细胞系SNU-251。在37℃、5%CO
(2)实验结果
实验结果如下表1和图1中所示。
[表1]
如表1和图1中的图表所示,可以看出,无论BRCA突变如何,式I化合物的柠檬酸盐即使在显著低于奥拉帕尼或尼拉帕尼的浓度下也抑制癌细胞的生长。特别地,,在由于药物流出泵的过表达而显示耐药性的NCI/ADR-RES细胞系中,或者在诱导卡铂耐药性的A2780-CR细胞系中,癌症生长在显著低于其它PARP抑制剂的浓度下得到有效抑制。
实施例3:通过P-gp评价式I化合物的柠檬酸盐的流出率
P-糖蛋白(P-gp)是决定各种药物的吸收和流出的药物转运蛋白之一。药物的这些吸收和流出的过程影响药物在血浆和组织中的浓度,并最终影响药物的最终效果。
已知具有高P-gp底物特异性的化合物致使对多药耐药的细胞中药物积累减少,并且经常介导对抗癌药物的耐药性的发展。PARP抑制剂诸如奥拉帕尼、鲁卡帕尼、尼拉帕尼和他拉唑帕尼都是已知具有高P-gp底物特异性的化合物。
在该实验中,发明人比较和评价了在P-gp中式I化合物的柠檬酸盐和奥拉帕尼(一种代表性的PARP抑制剂)的流出比,以确定式I化合物的柠檬酸盐在治疗对PARP抑制剂耐药的癌症中表现出优异的效果的机制。
(1)实验方法
进行渗透性研究以测量式I化合物的柠檬酸盐和奥拉帕尼(AZD-2281)的流出比。将200μL培养基(其中每个插入物的CaCO
(2)实验结果
实验结果如下表2中所示。
[表2]
注)A:细胞顶端侧,B:细胞基底外侧
如表2中所确认,发现式I化合物的柠檬酸盐的流出比约为奥拉帕尼(一种P-gp底物的PARP抑制剂)的流出比的1/10。
(3)评价实验结果
从上面实验结果可以看出,本发明式I化合物的柠檬酸盐不是P-糖蛋白(P-gp)底物。
利用这种作用机制,式I化合物的柠檬酸盐似乎能够克服多药耐药性,并显示出更好的抗癌效果,这与已知为P-gp底物的PARP抑制剂诸如奥拉帕尼、鲁卡帕尼、尼拉帕尼和他拉唑帕尼不同。
具体地,在实施例2中,式I化合物的柠檬酸盐抑制了各种类型的野生型BRCA卵巢癌细胞系和突变型BRCA卵巢癌细胞系中癌细胞的生长,甚至在显著低于奥拉帕尼或尼拉帕尼的浓度下也是如此。似乎通过P-gp的流出率的作用机制也部分促成了这一优异的效果。特别地,根据实施例2的结果,由于药物流出泵的过表达而表现出耐药性的NCI/ADR-RES细胞系中的式I化合物的柠檬酸盐在显著低于其它PARP抑制剂的浓度下有效地抑制癌症生长。因此,判断这些实验结果是为了更清楚地解释式I化合物的非P-gp底物部分有助于优异的抗癌效果。
此外,已知PARP抑制剂具有高比例的先天性或获得性耐药性获得,并且作为解释这一原因的机制,已知通过增加ABC受体(ABC转运蛋白)来增加药物流出的机制。
因此,与式I化合物的柠檬酸盐的流出比相关的作用机制被认为在理论上支持式I化合物可以有效用于治疗对其它PARP抑制剂诸如奥拉帕尼、鲁卡帕尼、尼拉帕尼和他拉唑帕尼耐药的癌症的事实。并且,该逻辑被下面实施例4(抗癌效果的分析-异种移植模型)、实施例5(抗癌效果的分析-异种移植模型)和实施例6(I期临床试验-NOV140201)的实验结果清楚地支持。
综上所述,本发明式I化合物的柠檬酸盐提供了优异的抗癌效果,其作用机制不同于其它PARP抑制剂药物诸如奥拉帕尼、鲁卡帕尼、尼拉帕尼和他拉唑帕尼的流出机制,并且对这些PARP抑制剂耐药的癌症提供了优异的效果。
实施例4:评价通过抑制与奥拉帕尼耐药性相关的Wnt信号活性的疗效
(1)实验方法
进行TOP/FOP-flash荧光素酶报告基因分析以测量在卵巢癌细胞系PEO1和获得奥拉帕尼耐药性的卵巢癌细胞系PEO1-OR中Wnt信号的激活。对于奥拉帕尼耐药获得的细胞系,从低奥拉帕尼浓度(10nM)到8μM的高浓度依次处理PEO1细胞系,存活的细胞系被称为PEO1-OR。TOP/FOP-flash荧光素酶报告基因分析利用Wnt激活时产生的β-连环蛋白进入细胞核并与TCF/LEF启动子位点结合以转录受Wnt影响的基因的原理。然后用荧光素酶替换该基因,并通过测量非发光底物通过荧光素酶转化的发光来测量Wnt激活的程度。TOP-f1ash是一种实验性质粒,其中发生荧光素酶的转录,因为β-连环蛋白可以结合到TCF启动子的启动子位点。FOP-flash被用作转染对照质粒来测量荧光的基础水平,因为β-连环蛋白不能通过在TCF启动子中引入突变来结合启动子位点。
将TOP-flash或FOP-flash质粒转染到卵巢癌细胞系PEO1和获得奥拉帕尼耐药性的卵巢癌细胞系PEO1-OR细胞系中。将转染的PEO1和PEO1-OR细胞系分别暴露于溶剂对照以及在400nM、10μM和50μM下的式I化合物的柠檬酸盐,转染72小时,然后裂解细胞并加入荧光素酶底物。使用荧光读取器测量并记录来自底物的发光程度。
(2)实验结果
实验结果如图2中所示。
如图2的左图所示,作为卵巢癌细胞系PEO1与已经获得奥拉帕尼耐药性的卵巢癌细胞系PEO1-OR之间的TOP信号转导强度的相对比较的结果,已经获得奥拉帕尼耐药性的PEO1-OR的细胞系示出比其它细胞系高5倍的TOP信号转导强度。这些结果表明,Wnt信号传导是通过在PEO1-OR细胞系中获得奥拉帕尼耐药性而激活的。
另外,从图2的右图可以证实,当用式I化合物的柠檬酸盐处理其中Wnt激活的PEO1-OR细胞系时,与对照组相比,Wnt信号与式I化合物的柠檬酸盐浓度成比例地减少。
综上所述,根据上面实验,当PEO1细胞系获得奥拉帕尼耐药性时,Wnt信号传导增加(见图2左侧的图表),并且由于式I化合物的柠檬酸盐的端锚聚合酶抑制能力,这种增加的信号传导与剂量成比例地减少(见图2右侧的图表)。
(3)评价实验结果
最近有研究报道PARP抑制剂的耐药机制与Wnt信号通路有关。换言之,已知Wnt信号通路被PARP抑制剂激活作为耐药性发展的机制。因此,通过选择能够阻断对PARP抑制剂具有获得耐药性的癌细胞中的Wnt信号通路的抗癌药物,可以克服对PARP抑制剂的耐药性。
与现有的PARP抑制剂诸如奥拉帕尼不同,本发明式I化合物的柠檬酸盐具有对PARP和端锚聚合酶(TNK)的双重抑制作用。即,已知现有的PARP抑制剂在耐药性的发展中激活Wnt信号通路,而本发明式I化合物的柠檬酸盐通过TNK抑制作用有效地抑制Wnt信号传导。
因此,从这一事实可以看出,本发明式I化合物的柠檬酸盐可以有效地用于治疗已经获得对PARP抑制剂耐药性的癌症。
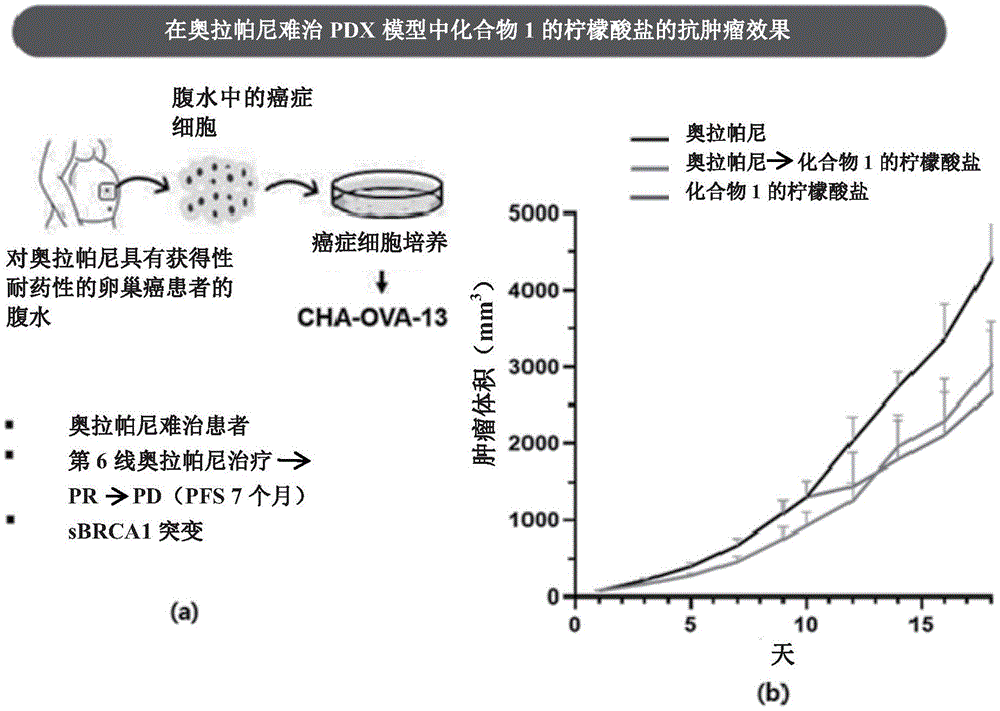
实施例5:分析抗癌效果(异种移植模型)
为了证明式I化合物的柠檬酸盐可以用于治疗实际上对PARP抑制剂耐药的实体癌,创建了使用来自BRCA突变阳性卵巢癌的细胞的异种移植模型,并且比较了PARP抑制剂(奥拉帕尼)和式I化合物的柠檬酸盐的效果。
(1)实验方法
PDX-GFTP 1016是一种来自IIIC期高级别浆液性卵巢癌患者原代组织的细胞,具有TP53突变和BRCA2突变。为了便于跟踪该癌细胞,表达绿色荧光蛋白/荧光素酶(PDX-1016GTFP 1016GFP/luc)并且手术注射到囊内之间的右侧卵巢中,然后在药物治疗前,对癌细胞的定植进行了4周的监测。4周后,将用50mg/kg奥拉帕尼治疗28天的小鼠中癌细胞的再生定义为对奥拉帕尼耐药的PDX-1016GTFP 1016GFP/luc(Ola-R-GTFP-1016)。
将Ola-R-GTFP-1016(一种通过上面方法建立的对奥拉帕尼耐药的PDX)的细胞以1×10
在上述4周的稳定后,基于通量(光子每秒)通过体内通量成像(IVIS光谱体内成像系统,PerkinElmer)收集的图像数据,诸如癌症表达的GFP/荧光素酶的光强度,观察癌症分布和癌症生长。
此时,使用体内通量成像,将具有相似癌症生长率的移植小鼠随机分为对照组(溶剂处理组)和用25mg/kg的式I化合物的柠檬酸盐治疗的组,并且每天口服给药一次。每周记录通量的变化,4周后处死小鼠,通过测量腹水体积、腹水中细胞的体积和癌症转移到淋巴的数量来测量抗癌活性。
(2)实验结果
实验结果如图3(B至E)中所示。
在图3B中,腹部的荧光示出为强光谱颜色的事实表明腹水中正在活跃地发生癌症增殖。即,图3B示出与对照组(溶剂处理组)相比,在用式I化合物的柠檬酸盐治疗的组中癌症生长被有效地抑制。
在图3C的图表中,从药物治疗后2周开始记录的体内荧光用作基础水平的标准,并且通过在移植小鼠的腹部中的体内成像观察相对荧光的变化,直到药物治疗4周。
从图3C的图表可以看出,与对照组(溶剂处理组)相比,用式I化合物的柠檬酸盐治疗的组中的癌细胞生长被显著抑制。
从支持用式I化合物的柠檬酸盐治疗比对照组更有效地抑制癌症生长的数据来看,计数癌症结节的数量和多细胞的数量,以确定癌症的转移潜力。
如图3D中所示,与对照组相比,在用式I化合的柠檬酸盐治疗组中,癌症的结节形成能力显著降低。此外,如图3E中所示,在用式I化合物的柠檬酸盐治疗的组中,腹水中产生的细胞总数比对照组显著减少。
从这些结果可以证实,用式I化合物的柠檬酸盐治疗的组比对照组更有效地抑制了癌症的形成和转移。
实施例6:抗癌效果分析(异种移植模型)
(1)实验方法
CHA-OVA-13是从对奥拉帕尼具有获得性耐药性的卵巢癌患者的腹水细胞建立的原代细胞,并且具有BRCA1突变。将这种CHA-OVA-13注射到NOD/SCID小鼠体内,使原发癌的尺寸生长,当其生长到一定大小时,取出肿瘤,切成小块,然后将肿瘤移植到裸鼠的皮下层中,建立小鼠模型。当肿瘤大小达到80mm
随后,将50mg/kg的奥拉帕尼每日两次口服给药于奥拉帕尼治疗组,并将50mg/kg的式I化合物的柠檬酸盐每日一次口服给药于式I化合物的柠檬酸盐治疗组。每隔两天,通过测径尺测量肿瘤的宽度和高度并记录肿瘤的大小,通过下面等式计算肿瘤体积。
观察奥拉帕尼治疗组的肿瘤大小和式I化合物的柠檬酸盐治疗组的肿瘤大小18天。将奥拉帕尼治疗组在药物治疗10天后随机分为两组,一组继续以相同方式接受奥拉帕尼持续8天(奥拉帕尼治疗组),另一组将药物改为式I化合物的柠檬酸盐并以50mg/kg每天口服给药一次持续8天(奥拉帕尼-式I化合物的柠檬酸盐替代组),观察对肿瘤大小的影响。
(2)实验结果
实验结果如图4中所示。如图4(b)的图表所示,可以证实,在总共18天的观察中,在式I化合物的柠檬酸盐治疗组中的肿瘤生长明显慢于奥拉帕尼治疗组的肿瘤生长。
另外,在奥拉帕尼-式I化合物的柠檬酸盐替代组的情况下,其通过在将奥拉帕尼治疗组分成两组后10天用式I化合物的柠檬酸盐替代奥拉帕尼进行治疗,与单一奥拉帕尼治疗组相比,肿瘤生长速率逐渐减慢,并且从大约第13天开始,示出与式I化合物的柠檬酸盐单一治疗组相似的肿瘤生长抑制作用。
这些结果表明,奥拉帕尼对奥拉帕尼耐药患者的原代细胞异种移植的持续治疗没有抑制肿瘤生长,但是用式I化合物的柠檬酸盐治疗显著减缓了肿瘤生长。
从这些结果中,可以证实式I化合物的柠檬酸盐在抑制对奥拉帕尼耐药的癌症组织的生长方面提供了极好的效果。
实施例7:I期临床试验
进行I期临床试验以评估式I化合物(6-{4-[(5-氧代-1,2,3,4,5,6-六氢苯并[h][1,6]萘啶-8-基)甲基]哌嗪-1-基}烟腈)的柠檬酸盐的稳定性、耐受性、药代动力学和药效学性能及疗效。患者组由年龄为19岁或以上患有组织学或细胞学证实的进行性实体癌的患者组成,对标准治疗难治或不能接受标准治疗。选择预期生存期为12周或更长并且通过一般血液检查等确认血液学和肝功能正常的患者。
根据机构和FDA规定,选定的患者提供了书面同意书。22名患者被纳入剂量递增队列,40名患者被纳入剂量扩大队列(6-{4-[(5-氧代-1,2,3,4,5,6-六氢苯并[h][1,6]萘啶-8-基)甲基]哌嗪-1-基}烟腈的柠檬酸盐)。剂量扩大组接受50mg/天、100mg/天、150mg/天和200mg/天。
根据规定,对所有队列中的患者进行安全性评估、定期采血、通过活检(肿瘤标本)进行生物标志物分析、遗传分析(血液PBMC和肿瘤标本)和肿瘤响应(每6周一次)。另外,即使在给药完成后,仍按照机构和食品药品安全法规进行随访。
在纳入剂量扩大队列(150mg/天)的患者中,一名64岁的女性患者最初被诊断为转移性卵巢癌(3期),并且患有广泛的实体癌组织,包括手术切除的卵巢,并且是一名有胚系BRCA1突变的同源重组缺陷肿瘤患者。
该患者对各种抗癌药物诸如吉西他滨(gemcitabine)、顺铂和紫杉醇几乎没有响应,并且特别地,被判断为对奥拉帕尼具有耐药性,因为在奥拉帕尼给药后立即观察到剩余癌症组织(靶病变)的大小增加和新的病变(进行性疾病)。然后,给药顺铂类药物新铂2个月,但是患者没有响应。之后,患者登记参加该临床试验,并开始以150mg/天单独服用式I化合物的柠檬酸盐,并且在服药期间通过定期CT扫描证实,与基线相比,病变(转移至肝脏的实体癌)的大小减少了超过30%。
实施例8:II期临床试验
进行II期临床试验以评估式I化合物(6-{4-[(5-氧代-1,2,3,4,5,6-六氢苯并[h][1,6]萘啶-8-基)甲基]哌嗪-1-基}烟腈)的柠檬酸盐是否可以用于治疗对PARP抑制剂耐药的患者。患者组由19岁或以上患有组织学或细胞学证实的实体癌的患者组成,患者被证实为HRD(同源重组缺陷)阳性。具体地,患者是患有高级别(2级或3级)浆液性上皮性卵巢癌、输卵管癌或原发性腹膜癌的患者,在参与本临床试验之前,这些患者的癌症在接受超过2线肿瘤抗癌治疗后已经复发或恶化。
抗癌治疗具体是指用吉西他滨(gemcitabine,)、阿霉素、拓扑替康(topotecan)、卡铂、奥沙利铂(oxaliplatin)、顺铂、贝伐珠单抗(bevacizumab)或PARP抑制剂中的一种或组合进行治疗。
作为受试者,选择预期生存期为12周或更长,且通过如一般血液检查等确认了正常血液功能、肾功能和肝功能的患者。
根据机构和FDA规定,选定的患者提供了书面同意书。患者包括那些在既往治疗史中对铂类疗法表现出敏感性的患者,或那些对铂类疗法表现出敏感性但使用现有PARP抑制剂(诸如奥拉帕尼和尼拉帕尼)治疗失败的患者。纳入大约有60名患者,但这是灵活的。
式I化合物的柠檬酸盐以100mg/天给药,必要时可以调整剂量。
根据规定,对所有队列中的患者进行安全性评估、定期采血、遗传分析(血液PBMC和肿瘤标本)和肿瘤响应(每8周一次)。另外,即使在给药完成后,仍按照机构和食品药品安全法规进行随访。
疗效、安全性、耐受性、药代动力学等可以通过该II期临床试验来测量和评价。
功效包括“本发明式I的柠檬酸盐”对“用现有PARP抑制剂治疗失败的HRD突变阳性肿瘤患者等”的治疗效果。更具体地,这项II期临床试验将有效地减少用现有PARP抑制剂治疗失败的患者的实体癌大小。
- 一种基于数字孪生的装配精度仿真分析方法与系统
- 一种基于数字孪生的风机直驱变桨距系统及优化设计方法
- 一种基于数字孪生锂电池系统的故障诊断方法及报警系统
- 一种基于数字孪生的锂电池系统电压传感器故障诊断方法