一种西替利嗪口服液体制剂相关杂质的检测方法
文献发布时间:2024-04-18 19:52:40
技术领域
本发明涉及药品检测技术领域,尤其涉及一种西替利嗪口服液体制剂相关杂质的检测方法。
背景技术
盐酸西替利嗪为第二代H
中国专利CN113848271A公开了一种盐酸左西替利嗪口服溶液有关物质方法,采用高效液相色谱法,其中流动相A为25-35mmol/L磷酸二氢钾缓冲液(pH值为2.8~3.2),流动相B为乙腈,梯度洗脱程序;分析了七种杂质:对氯二苯甲醇、对氯二苯甲酮、对氯二苯甲基哌嗪(杂质A)、左西替利嗪酰胺和对羟基苯甲酸。
中国专利CN111821290A公开了一种盐酸西替利嗪有关物质的检测方法,采用磷酸盐缓冲液(pH2.5~3.5)和乙腈为流动相,梯度洗脱,分离了12种杂质:去氯西替利嗪、盐酸西替利嗪乙醇、4-氯苯苯甲基哌嗪、2-氯盐酸西替利嗪、西替利嗪甲酯、3-氯西替利嗪、西替利嗪乙酸、西替利嗪氮氧化物、4-氯苯苯甲醇、4-氯苯苯甲酮、西替利嗪二聚物、乙氧基盐酸西替利嗪。
中国专利CN103263394A公开了一种盐酸西替利嗪片及其有关物质质量控制方法,采用乙腈-0.02%磷酸二氢钾溶液(pH3.0)为流动相A,乙腈-0.05%磷酸溶液为流动相B,研究了杂质A、B、G的洗脱和分离。
发明内容
针对现有技术存在的不足,本发明提出一种西替利嗪口服液体制剂相关杂质的检测方法,能够准确测定西替利嗪杂质A、B、G和西替利嗪甘油酯、西替利嗪丙二醇酯1、西替利嗪丙二醇酯2,同时还能成功将主峰、空白辅料与这些杂质分离。
本发明提供一种西替利嗪口服液体制剂相关杂质的检测方法,采用液相色谱法进行检测,色谱条件包括:
流动相,流动相A为体积浓度为1.05~1.2%的磷酸溶液,流动相B为乙腈;洗脱程序为梯度洗脱;
所述相关杂质包含杂质A、杂质B、杂质G、西替利嗪甘油酯、西替利嗪丙二醇酯1和西替利嗪丙二醇酯2中的一种或多种。
USP分离多个杂质的方法为原料方法,关注杂质基本均为原料的工艺杂质,但本发明旨在实现制剂中有关杂质的检测分离,因此本发明所称“相关杂质”包括合成工艺过程中引入的杂质,及在储存、运输、使用过程中产生的相关杂质。杂质A、G、B为英国药典西替利嗪口服溶液的特定杂质,A、G是原料工艺和原料与制剂的光降解杂质,西替利嗪甘油酯、西替利嗪丙二醇酯1和西替利嗪丙二醇酯2为明确的制剂降解杂质(当制剂为西替利嗪口服溶液时,辅料不含有甘油,故无需检测杂质西替利嗪甘油酯;当制剂为左西替利嗪口服溶液时,辅料不含有丙二醇,故无需检测杂质西替利嗪丙二醇酯),因此,同时检测分离西替利嗪口服液体制剂中已知6个杂质(杂质A、杂质B、杂质G、西替利嗪甘油酯、西替利嗪丙二醇酯1和西替利嗪丙二醇酯2),可以对杂质谱进行控制,准确控制西替利嗪口服溶液的质量,进而反映药品安全的重要指标,必须严格监控,有必要为此探索并建立方便快捷的检测方法。研究发现,当采用浓度为1.05~1.2%的磷酸溶液为流动相A,乙腈为流动相B,进行梯度洗脱,可以同时检测杂质A、杂质B、杂质G、西替利嗪甘油酯、西替利嗪丙二醇酯1和西替利嗪丙二醇酯2。可以理解的是,本发明提供的检测方法也可用于检测仅含有上述6种杂质中部分杂质(如1种、2种、3种、4种、5种)的情形。
根据本发明提供的西替利嗪口服液体制剂相关杂质的检测方法,所述梯度洗脱程序如下表:
表1
。
控制梯度洗脱程序如上,才能配合流动相更好地将6种杂质区分开,包括各自之间的区分以及与主峰、空白辅料的分离。
进一步地,所述梯度洗脱程序如下表:
表2
。
根据本发明提供的西替利嗪口服液体制剂相关杂质的检测方法,柱温为28-32℃,优选为30℃。
根据本发明提供的西替利嗪口服液体制剂相关杂质的检测方法,所述流动相的流速为0.65-0.9ml/min,优选为0.8ml/min。
根据本发明提供的西替利嗪口服液体制剂相关杂质的检测方法,采用紫外检测器进行检测,检测波长为228-232nm,优选为230nm。
根据本发明提供的西替利嗪口服液体制剂相关杂质的检测方法,色谱柱的填料为十八烷基硅烷键合表面多孔硅胶,规格为4.6mm×150mm,2.6μm~3.0μm,优选为2.6μm。
根据本发明提供的西替利嗪口服液体制剂相关杂质的检测方法,所述检测方法包括以下步骤:
步骤1)、制备供试品溶液和对照溶液;
步骤2)、对供试品溶液和对照溶液通过液相色谱进行检测;
步骤3)、对检测结果进行分析。
根据本发明提供的西替利嗪口服液体制剂相关杂质的检测方法,所述西替利嗪口服液体制剂包括盐酸西替利嗪口服溶液、盐酸西替利嗪滴剂、盐酸左西替利嗪口服溶液和盐酸左西替利嗪滴剂。需要说明的是,上述液体制剂需与参比制剂或者原研处方通过一致性实验,也就是说,若与参比制剂或者原研处方相差较大,则本发明的检测方法有可能不适用。
根据本发明提供的西替利嗪口服液体制剂相关杂质的检测方法,当所述西替利嗪口服液体制剂为盐酸西替利嗪滴剂和/或盐酸左西替利嗪滴剂时,所述检测方法可同时检测杂质A、杂质B、杂质G、西替利嗪甘油酯、西替利嗪丙二醇酯1和西替利嗪丙二醇酯2,所述相关杂质与主峰、空白辅料中的羟苯甲酯和/或羟苯丙酯可分离,分离度均大于1.5;
当所述西替利嗪口服液体制剂为盐酸西替利嗪口服溶液时,所述检测方法可同时检测杂质A、杂质B、杂质G、西替利嗪丙二醇酯1和西替利嗪丙二醇酯2,所述相关杂质与主峰、空白辅料中的羟苯甲酯和/或羟苯丙酯可分离,分离度均大于1.5;
当所述西替利嗪口服液体制剂为盐酸左西替利嗪口服溶液时,所述检测方法可同时检测杂质A、杂质B、杂质G和西替利嗪甘油酯,所述相关杂质与主峰、空白辅料中的羟苯甲酯和/或羟苯丙酯可分离,分离度均大于1.5。
本发明提供了一种西替利嗪口服液体制剂相关杂质的检测方法,通过色谱条件的调整,特别是流动相的调整,使得该方法能够有效分离西替利嗪、西替利嗪相关杂质与辅料,特别是辅料中的羟苯甲酯和羟苯丙酯;该方法能够实现在一次检测中同时分离羟苯甲酯、羟苯丙酯、杂质A、杂质B、杂质G、西替利嗪、西替利嗪甘油酯、西替利嗪丙二醇酯1、西替利嗪丙二醇酯2,缩短了分析时间,节约了检测成本;该检测方法专属性强(分离度大于1.5)、耐用性好(每个色谱参数变动的测定结果极差在0.02%范围内);有利于对西替利嗪口服液体制剂的进行精准快捷的质量控制。
附图说明
图1为本发明实施例1检测的色谱图;
图2为本发明实施例1中流动相A磷酸浓度1.05%的色谱图;
图3为本发明实施例1中流动相A磷酸浓度1.2%的色谱图;
图4为本发明实施例1中流动相A比例变化83%~72%的色谱图;
图5为本发明实施例1中流动相A比例变化83%~69%的色谱图;
图6为本发明实施例1中流速0.65ml/min时的色谱图;
图7为本发明实施例1中流速0.9ml/min时的色谱图;
图8为本发明实施例1中柱温28℃时的色谱图;
图9为本发明实施例1中柱温32℃时的色谱图;
图10为本发明实施例1中波长228nm时的色谱图;
图11为本发明实施例1中波长232nm时的色谱图;
图12为本发明对照例1中检测的色谱图;
图13为本发明对照例2中检测的色谱图;
图14为本发明对照例3中检测的色谱图。
具体实施方式
为使本发明的目的、技术方案和优点更加清楚,下面将对本发明中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例1
本实施例提供一种盐酸西替利嗪滴剂(市售制剂)相关杂质的检测方法,具体如下:
专属性试验
1.溶液配制:
1.1稀释剂制备
磷酸盐缓冲液-乙腈(80:20)。
1.2定位溶液
西替利嗪甘油酯:取西替利嗪甘油酯对照品2mg,置100ml量瓶中,加稀释剂稀释至刻度,作为西替利嗪甘油酯贮备液,精密量取5ml,置100ml量瓶中,加稀释剂置刻度,摇匀,制成1μg/ml的溶液。
西替利嗪丙二醇酯1:取西替利嗪丙二醇酯1对照品2mg,置100ml量瓶中,加稀释剂稀释至刻度,作为西替利嗪丙二醇酯1贮备液,精密量取5ml,置100ml量瓶中,加稀释剂置刻度,摇匀,制成1μg/ml的溶液。
西替利嗪丙二醇酯2:取西替利嗪丙二醇酯2对照品2mg,置100ml量瓶中,加稀释剂稀释至刻度,作为西替利嗪丙二醇酯2贮备液,精密量取5ml,置100ml量瓶中,加稀释剂置刻度,摇匀,制成1μg/ml的溶液。
杂质A:取杂质A对照品2mg,置100ml量瓶中,加稀释剂稀释至刻度,作为杂质A贮备液,精密度量取2ml,置100ml量瓶中,加稀释剂置刻度,摇匀,制成0.4μg/ml的溶液。
杂质B:取杂质B对照品2mg,置100ml量瓶中,加稀释剂稀释至刻度,作为杂质B贮备液,精密度量取2ml,置100ml量瓶中,加稀释剂置刻度,摇匀,制成0.4μg/ml的溶液。
杂质G:取杂质G对照品2mg,置100ml量瓶中,加稀释剂稀释至刻度,作为杂质G贮备液,精密度量取2ml,置100ml量瓶中,加稀释剂置刻度,摇匀,制成0.4μg/ml的溶液。
羟苯甲酯:取羟苯甲酯27mg,置100ml量瓶中,加甲醇10ml使溶解,再用稀释剂稀释至刻度,精密量取5ml,置50ml量瓶中,用稀释剂稀释至刻度,制成27μg/ml的溶液。
羟苯丙酯:取羟苯丙酯15mg,置100ml量瓶中,加甲醇10ml使溶解,再用稀释剂稀释至刻度,精密量取5ml,置250ml量瓶中,用稀释剂稀释至刻度,制成3μg/ml的溶液。
1.3系统适用性溶液制备
取本品2ml,置100ml量瓶中,分别加入5ml西替利嗪甘油酯、5ml西替利嗪丙二醇酯1、5ml西替利嗪丙二醇酯2、2ml杂质A、2ml杂质B、2ml杂质G贮备液,用稀释剂稀释至刻度,制成的溶液浓度为盐酸西替利嗪0.2mg/ml、西替利嗪甘油酯1μg/ml、西替利嗪丙二醇酯11μg/ml、西替利嗪丙二醇酯21μg/ml、杂质A0.4μg/ml、杂质B0.4μg/ml、杂质G0.4μg/ml、羟苯甲酯27μg/ml、羟苯丙酯3μg/ml。
1.4供试品溶液制备
取本品2ml,置100ml量瓶中,用稀释剂稀释至刻度。
1.5对照溶液制备
精密量取供试品溶液2ml,置100ml量瓶中,用稀释剂稀释至刻度,精密量取5ml至50ml量瓶中,用稀释剂稀释至刻度,制成盐酸西替利嗪0.4μg/ml的溶液。
1.6空白辅料溶液制备
取根据处方配制的不含西替利嗪的空白辅料,按照供试品溶液稀释方式进行制备。
1.7流动相的配制:
流动相:流动相A为体积浓度1.1%磷酸水溶液,流动相B乙腈。
2.液相色谱条件如下:
色谱柱:Accucore C18 4.6mm×150mm,2.6μm
流速:0.8ml/min
检测波长:230nm
柱温:30℃
梯度设置如下表所示:
表3
3.检测结果
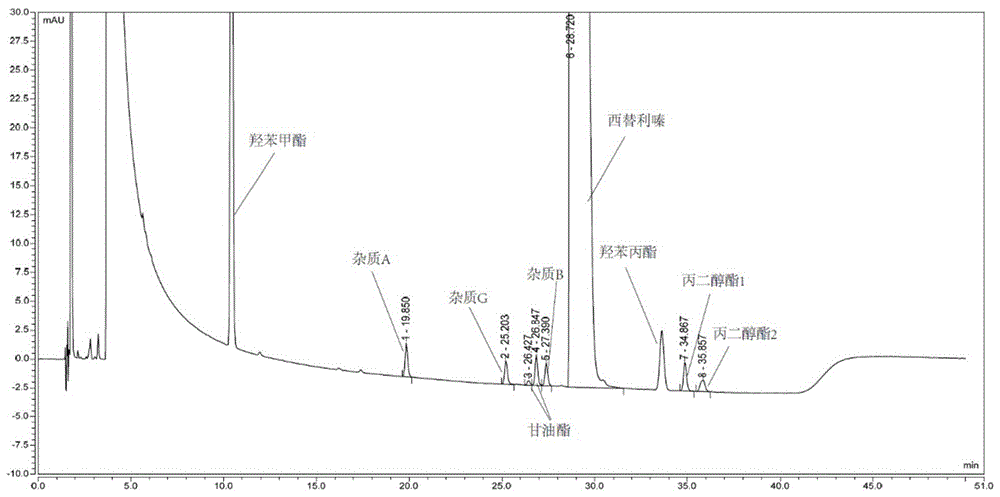
如图1所示,从图1中可以看出,该方法能够实现西替利嗪、杂质A、B、G、西替利嗪甘油酯、西替利嗪丙二醇酯1、西替利嗪丙二醇酯2、羟苯甲酯和羟苯丙酯的检测,能够将杂质峰与主峰、杂质峰之间、杂质与辅料峰之间分离,分离度满足检测要求,很好的满足原料药和制剂工艺过程中的杂质监控及原料药和制剂的成品中的杂质控制要求。
4.实验结论
对空白的稀释剂进行检测,发现稀释剂基线平稳,并且无特征峰,表明空白的稀释剂不干扰供试品溶液中已知杂质的测定;空白辅料中在西替利嗪和杂质出峰位置均无干扰峰;由供试品溶液的谱图(图1)可知,西替利嗪特征峰的保留时间为30.010min。
由6个杂质定位溶液、系统适用性溶液的检测结果可以定位各杂质特征峰的保留时间,杂质A、杂质G、西替利嗪甘油酯(双峰)、杂质B、西替利嗪丙二醇酯1和西替利嗪丙二醇酯2的保留时间分别为:21.075min、26.475min、27.717min(西替利嗪甘油酯峰1)、28.148min(西替利嗪甘油酯峰2)、28.578min、36.255min、37.210min。
空白辅料羟苯甲酯和羟苯丙酯的保留时间分别为:10.552min、34.377min。
采用系统适用性溶液进行测定,发现空白稀释剂和空白辅料不干扰供试品溶液中杂质的检测,系统适用性溶液中西替利嗪与6种杂质的相邻色谱峰之间的分离度均大于1.5,分离度良好。
由上述实验检测结果可知,本发明提供的测定方法,能同时分离西替利嗪与6种已知杂质,并且西替利嗪和6种已知杂质及辅料(羟苯甲酯和羟苯丙酯)的色谱峰表现出显著的分离效果,专属性良好。
耐用性试验
为了考察检测条件对检测效果的影响,采用上述系统适用性溶液进行如下实验:
2-1.磷酸浓度的考察,流动相A设置不同磷酸浓度进行实验(其他条件参考上述实施例设置),具体如下:
2-1-1.磷酸浓度=1.05%(V/V),检测结果如图2所示;
2-1-2.磷酸浓度=1.2%(V/V),检测结果如图3所示;
2-2.流动相比例条件的考察,设置0~35min不同流动相A变化比例条件进行实验,具体如下:
2-2-1.流动相A 83%~72%,检测结果如图4所示;
2-2-2.流动相A 83%~69%,检测结果如图5所示;
2-3.流速条件的考察,设置不同流速条件进行实验,具体如下:
2-3-1.流速0.65ml/min,检测结果如图6所示;
2-3-2.流速0.9ml/min,检测结果如图7所示;
2-4.柱温条件的考察,设置不同柱温条件进行实验,具体如下:
2-4-1.柱温28℃,检测结果如图8所示;
2-4-2.柱温32℃,检测结果如图9所示;
2-5.检测波长条件的考察,设置不同检测波长条件进行实验,具体如下:
2-5-1.检测波长228nm,检测结果如图10所示;
2-5-2.检测波长232nm,检测结果如图11所示;
对图1~11中的分离度及检测结果统计如下表所示:
表4
注释:最小分离度为西替利嗪甘油酯与杂质B的分离度。
由以上结果可知:
磷酸浓度:在1.05%~1.2%范围内调节磷酸浓度,6个杂质检出量的极差0.01%,系统适用性溶液中西替利嗪甘油酯与杂质B分离度最小,其分离度不低于1.53,表明拟定的磷酸浓度适宜,磷酸浓度微小变化,对测定结果无影响。
流动相比例:调节0~35min的流动相比例,流动相A35min比例范围69%~72%,6个杂质检出量的极差0.02%,系统适用性溶液分离度均不低于1.50,表明拟定的流动相梯度适宜,比例微小变化,对测定结果无影响。
流速:在0.65~0.90ml/min范围内调节流速,系统适用性溶液分离度不低于1.47,其中0.90ml/min分离度1.47,虽未达到1.5,但杂质检出结果无影响,6个杂质检出量的极差0.01%,表明拟定的流速适宜,流速的微小变化,对测定结果无影响。
柱温:在28℃~32℃范围内调节柱温,6个杂质检出量的极差0.02%,系统适用性溶液分离度均不低于1.53,表明拟定的柱温适宜,柱温的微小变化,对测定结果无影响。
波长:在228~232nm范围内调节波长,6个杂质检出量的极差0.02%,系统适用性溶液分离度均不低于1.53,表明拟定的波长适宜,波长的微小变化,对测定结果无影响。
综上,色谱条件在磷酸浓度1.05%~1.2%范围内,流动相A相比例在0~35min变化在83%到69%~72%范围内,流速在0.65~0.90ml/min范围内,柱温在28℃~32℃范围内,波长在228nm~232nm范围内,杂质可有效分离,杂质测定结果无影响。拟定方法磷酸浓度1.1%,流动相A相比例在0~35min变化为83%到70%,流速0.8ml/min,柱温30℃,检测波长230nm,可有效分离6个杂质,并准确检测6个杂质含量。
实施例2
本实施例提供一种盐酸西替利嗪口服溶液(市售制剂)相关杂质的检测方法,具体如下:
1.溶液配制:
1.1稀释剂制备
同实施例1。
1.2定位溶液
西替利嗪丙二醇酯1、西替利嗪丙二醇酯2、杂质A、杂质B、杂质G同实施例1。
羟苯甲酯:取羟苯甲酯27mg,置100ml量瓶中,加甲醇10ml使溶解,再用稀释剂稀释至刻度,制成0.27mg/ml的溶液。
羟苯丙酯:取羟苯丙酯15mg,置100ml量瓶中,加甲醇10ml使溶解,再用稀释剂稀释至刻度,精密量取5ml,置25ml量瓶中,用稀释剂稀释至刻度,制成30μg/ml的溶液。
1.3系统适用性溶液制备
取本品20ml,置100ml量瓶中,分别加入5ml西替利嗪丙二醇酯1、5ml西替利嗪丙二醇酯2、2ml杂质A、2ml杂质B、2ml杂质G贮备液,用稀释剂稀释至刻度,制成的溶液浓度为盐酸西替利嗪0.2mg/ml、西替利嗪丙二醇酯11μg/ml、西替利嗪丙二醇酯21μg/ml、杂质A0.4μg/ml、杂质B0.4μg/ml、杂质G0.4μg/ml、羟苯甲酯0.27mg/ml、羟苯丙酯30μg/ml。
1.4供试品溶液制备
取本品20ml,置100ml量瓶中,用稀释剂稀释至刻度。
1.5对照溶液制备
精密量取供试品溶液2ml,置100ml量瓶中,用稀释剂稀释至刻度,精密量取5ml至50ml量瓶中,用稀释剂稀释至刻度,制成盐酸西替利嗪0.4μg/ml的溶液。
1.6空白辅料溶液制备
取根据处方配制的不含西替利嗪的空白辅料,按照供试品溶液稀释方式进行制备。
1.7流动相的配制:
同实施例1。
2.液相色谱检测
色谱条件同实施例1。
3.检测结果
该方法能够实现西替利嗪、杂质A、B、G、西替利嗪丙二醇酯1、西替利嗪丙二醇酯2、羟苯甲酯和羟苯丙酯的检测,能够将杂质峰与主峰、杂质峰之间、杂质与辅料峰之间分离,分离度满足检测要求,很好的满足原料药和制剂工艺过程中的杂质监控及原料药和制剂的成品中的杂质控制要求。
4.实验结论
对空白的稀释剂进行检测,发现稀释剂基线平稳,并且无特征峰,表明空白的稀释剂不干扰供试品溶液中已知杂质的测定;空白辅料中在西替利嗪和杂质出峰位置均无干扰峰。
实施例3
本实施例提供一种盐酸左西替利嗪口服溶液(市售制剂)相关杂质的检测方法,具体如下:
1.溶液配制:
1.1稀释剂制备
同实施例1。
1.2定位溶液
西替利嗪甘油酯、杂质A、杂质B、杂质G同实施例1。
羟苯甲酯:取羟苯甲酯27mg,置100ml量瓶中,加甲醇10ml使溶解,再用稀释剂稀释至刻度,制成0.27mg/ml的溶液。
羟苯丙酯:取羟苯丙酯15mg,置100ml量瓶中,加甲醇10ml使溶解,再用稀释剂稀释至刻度,精密量取5ml,置25ml量瓶中,用稀释剂稀释至刻度,制成30μg/ml的溶液。
1.3系统适用性溶液制备
取本品40ml,置100ml量瓶中,分别加入5ml西替利嗪甘油酯、2ml杂质A、2ml杂质B、2ml杂质G贮备液,用稀释剂稀释至刻度,制成的溶液浓度为盐酸西替利嗪0.2mg/ml、西替利嗪丙二醇酯11μg/ml、西替利嗪丙二醇酯21μg/ml、杂质A0.4μg/ml、杂质B0.4μg/ml、杂质G0.4μg/ml、羟苯甲酯0.27mg/ml、羟苯丙酯30μg/ml。
1.4供试品溶液制备
取本品40ml,置100ml量瓶中,用稀释剂稀释至刻度。
1.5对照溶液制备
精密量取供试品溶液2ml,置100ml量瓶中,用稀释剂稀释至刻度,精密量取5ml至50ml量瓶中,用稀释剂稀释至刻度,制成盐酸西替利嗪0.4μg/ml的溶液。
1.6空白辅料溶液制备
取根据处方配制的不含西替利嗪的空白辅料,按照供试品溶液稀释方式进行制备。
1.7流动相的配制:
同实施例1。
2.液相色谱检测
色谱条件同实施例1。
3.检测结果
该方法能够实现西替利嗪、杂质A、B、G、西替利嗪甘油酯、羟苯甲酯和羟苯丙酯的检测,能够将杂质峰与主峰、杂质峰之间、杂质与辅料峰之间分离,分离度满足检测要求,很好的满足原料药和制剂工艺过程中的杂质监控及原料药和制剂的成品中的杂质控制要求。
4.实验结论
对空白的稀释剂进行检测,发现稀释剂基线平稳,并且无特征峰,表明空白的稀释剂不干扰供试品溶液中已知杂质的测定;空白辅料中在西替利嗪和杂质出峰位置均无干扰峰。
对比例1,英国药典2022西替利嗪口服溶液有关物质方法色谱条件:流动相A为乙腈-磷酸溶液[pH1.5,1.0%磷酸(V/V)](17:83),流动相B为乙腈-磷酸溶液[pH1.5,1.0%磷酸(V/V)](35:65),检测波长230nm,流速1.0ml/min,柱温30℃,色谱柱Acclaim C18 4.6mm×250mm 5μm,其梯度洗脱程序如下:
表5
测试结果如图12所示,从图中可以看出,该方法下,出峰顺序和保留时间分别为羟苯甲酯22.812min、杂质A 36.945min、杂质G 42.260min、西替利嗪甘油酯(峰1)42.673min、西替利嗪甘油酯(峰2)43.247min、杂质B 44.145min、西替利嗪46.448min、西替利嗪丙二醇酯1 52.385min、西替利嗪丙二醇酯2与羟苯丙酯的包合峰53.512min,其中杂质G与甘油酯分离效果差,不能计算分离度,西替利嗪丙二醇酯2与羟苯丙酯完全包合在一起。
另外,将实施例1色谱参数中梯度变更为上述对比例1梯度(按照对比例乙腈与磷酸溶液比例设置梯度),其他参数不变,杂质B与甘油酯不能基线分离,羟苯丙酯与丙二醇酯1不能分离。
对比例2,美国药典43盐酸西替利嗪原料有关物质方法2
色谱条件:流动相A为2g/L四丁基硫酸氢铵和3g/L磷酸二氢钠溶液(1mol/LNaOH调pH至2.8±0.5),B为甲醇,检测波长230nm,柱温40℃,色谱柱ACE super Excel C18 4.6mm×250mm 5μm,其梯度洗脱程序和流速如下:
表6
测试结果如图13所示,从图中可以看出,该方法所测得色谱图中,杂质G与西替利嗪甘油酯不能分离,西替利嗪丙二醇酯1、西替利嗪丙二醇酯2与西替利嗪峰不能分离,并且会有较大梯度峰产生。
对比例3,中国药典2020年版二部盐酸西替利嗪有关物质方法
色谱条件:乙腈-0.1%三乙胺的0.05mol/L磷酸二氢钠溶液(磷酸调pH3.0)(35:65),检测波长230nm,流速1.0ml/min,柱温30℃,色谱柱Acclaim C18 4.6mm×250mm 5μm,
测试结果如图14所示,从图中可以看出,该方法所测得色谱图中,杂质G与主成分分离效果不好,杂质B与西替利嗪甘油酯不能分离,杂质A与西替利嗪不能分离。
对比例1-3的实验总结
参照公开文献的检测条件和方法,对本发明实施例供试品溶液和混合杂质对照品溶液中的6种已知杂质进行分离检测,主要的结果如下所示:
表7
由以上分离结果可知,在文献1、2、3的检测条件下,文献1杂质G与西替利嗪甘油酯双峰中的第一个峰不能分离,羟苯丙酯与西替利嗪丙二醇酯2完全包合,文献2西替利嗪丙二醇酯1、西替利嗪丙二醇酯2与西替利嗪三个峰中邻近双峰均不能分离,西替利嗪甘油酯与杂质G包合,文献3杂质B与西替利嗪甘油酯不能分离,文献1~3这些不能分离的情况,均无法计算分离度,远远低于本发明实施例的各杂质特征分的分离效果1.5,也无法实现对本发明西替利嗪口服溶液的6中杂质和两种辅料的同时分离。
综上所述,采用本发明的检测方法可实现对西替利嗪口服液体制剂中6种已知杂质特征峰和两种辅料(羟苯甲酯和羟苯丙酯)的分离度均大于1.5,各杂质的特征峰之间不存在相互干扰,分离度良好,专属性强。
最后应说明的是:以上实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的精神和范围。
- 一种非布司他及其相关杂质的HPLC检测方法
- 一种用HPLC法检测愈美素口服液体制剂中多种杂质含量的方法
- 一种检测盐酸左西替利嗪口服溶液中有关物质的方法