基于缩醛的化合物、基于缩醛的预聚物、基于缩醛的聚合物、和包括其的光刻胶组合物
文献发布时间:2024-04-18 19:53:33
对相关申请的交叉引用
本申请要求在韩国知识产权局于2021年12月6日提交的韩国专利申请No.10-2021-0173266和2022年10月14日提交的韩国专利申请No.10-2022-0132743的优先权、以及由其产生的所有权益,将其公开内容全部通过引用引入本文中。
技术领域
一种或多种实施方式涉及基于缩醛的化合物、基于缩醛的预聚物、基于缩醛的聚合物、和包括所述基于缩醛的聚合物的光刻胶组合物。
背景技术
精细图案化对于制造高度集成的半导体器件是必需的。近来,由于半导体器件的设计规格正在快速减小,因此正在开发各种用于实现精细图案的光刻技术。特别地,近来存在对于如下的兴趣:作为用于代替通过KrF准分子激光(248nm)和ArF准分子激光(193nm)的光刻工艺的下一代技术的使用通过EUV的曝光(暴露)过程的极紫外(EUV)光刻工艺。然而,在EUV光刻工艺和使用常规的化学放大抗蚀剂(光刻胶)(CAR)的情况中,与ArF光相比,EUV光具有非常高的能量。因此,即使用相同或类似能量的光照射CAR,光子的数量也显著减少,且因此灵敏度降低。因此,为了增加能够产生酸的光子的数量,要求更高的曝光剂量和/或非常长的曝光时间。
发明内容
一种或多种实施方式包括新的基于缩醛的化合物。
一种或多种实施方式包括使用以上描述的基于缩醛的化合物的基于缩醛的预聚物。
一种或多种实施方式包括由以上描述的基于缩醛的预聚物形成的基于缩醛的聚合物。
一种或多种实施方式包括光刻胶组合物,其包括以上描述的基于缩醛的聚合物。
另外的方面将部分地在随后的描述中阐明,且部分地将从所述描述明晰,或者可通过本公开内容的所呈现的实施方式的实践获悉。
根据一种或多种实施方式,提供由式1表示的基于缩醛的化合物。
式1
其中,在式1中,
R
其中Q
除了如下之外:R
a和b各自独立地为0或1-10的整数,
G为由式2或式2a表示的基团,
式2
式2a
其中,在式2和2a中,R
*表示连接位点,和
k为0或1。
根据一种或多种实施方式,提供基于缩醛的预聚物,其为以上描述的基于缩醛的化合物、具有极性基团的第一能聚合的单体、和具有对酸不稳定的基团的第二能聚合的单体的聚合产物。
根据一种或多种实施方式,提供基于缩醛的聚合物,其为以上描述的基于缩醛的预聚物和扩链剂的扩链反应产物。
根据一种或多种实施方式,提供光刻胶组合物,其包括基础聚合物、光致产酸剂、和溶剂,其中所述基础聚合物包括以上描述的基于缩醛的聚合物。
附图说明
由结合附图考虑的以下描述,本公开内容的一些实施方式的以上和其它方面、特征和优点将更加明晰,其中:
图1显示根据实施例1制备的产物(CTA-EGVE)的核磁共振(NMR)谱;
图2显示根据实施例1制备的具有缩醛基团的链转移剂的
图3显示根据实施例2制备的基于缩醛的预聚物的
图4显示根据实施例4制备的基于缩醛的预聚物的
图5显示根据实施例4制备的基于缩醛的聚合物的
图6为用于说明基于缩醛的聚合物的酸处理过程的图解;
图7显示基于缩醛的预聚物、由所述基于缩醛的预聚物获得的基于缩醛的聚合物、和在酸处理之后的产物的凝胶渗透色谱法(GPC)测量的结果;和
图8至12为用于说明使用根据实施方式的光刻胶组合物的图案形成方法的图。
具体实施方式
现在将对实施方式详细地进行介绍,其实例说明于附图中,其中相同的附图标记始终指的是相同的元件。在这点上,本实施方式可具有不同的形式且不应被解释为限于本文中阐明的描述。因此,下面仅通过参考附图描述实施方式以说明方面。
将理解,当一个元件被称为“在”另外的元件“上”时,其可直接在所述另外的元件上或者其间可存在中间元件。相反,当一个元件被称为“直接在”另外的元件“上”时,不存在中间元件。
本文中使用的术语仅用于描述具体实施方式的目的且并不意图为限制性的。如本文中使用的,单数形式“一个(种)(不定冠词)(a,an)”和“所述(该)”意图包括复数形式,包括“至少一个(种)”,除非内容清楚地另外指明。“至少一个(种)”将不被解释为限制“一个(种)”。“或”意味着“和/或”。如本文中使用的,术语“和/或”包括相关列举项目的一个或多个的任何和全部组合。将进一步理解,术语“包含”或“包括”当用在本说明书中时,表明存在所陈述的特征、区域、整体、步骤、操作、元件和/或组分,但是不排除存在或添加一种或多种另外的特征、区域、整体、步骤、操作、元件、组分、和/或其集合。
如本文中使用的“约”或“大约”包括所陈述的值且意味着在如由本领域普通技术人员考虑到所讨论的测量和与具体量的测量有关的误差(即,测量系统的限制)而确定的对于具体值的可接受的偏差范围内。例如,“约”可意味着相对于所陈述的值在一种或多种标准偏差内,或者在±10%或5%内。
除非另外定义,否则在本文中使用的所有术语(包括技术和科学术语)具有与本公开内容所属领域的普通技术人员所通常理解的相同的含义。将进一步理解,术语,例如在常用字典中定义的那些,应被解释为具有与它们在相关领域的背景和本公开内容中的含义一致的含义,并且将不以理想化或过于形式的意义进行解释,除非在本文中清楚地如此定义。
在本文中参照作为理想化实施方式的示意图的横截面图描述示例性实施方式。这样,将预料到作为例如制造技术和/或公差的结果的与图的形状的偏差。因此,本文中描述的实施方式不应解释为限于如本文中所图示的区域的具体形状,而是包括由例如制造导致的形状上的偏差。例如,图示或描述为平坦的区域可典型地具有粗糙的和/或非线性的特征。此外,图示的尖锐的角可为圆化的。因此,图中所示的区域在本质上是示意性的,并且它们的形状不意图说明区域的精确形状且不意图限制本权利要求的范围。
如本文中使用的,对“基于缩醛的”化合物、预聚物、或聚合物的提及将被理解为指的是含有缩醛的化合物、预聚物、或聚合物。
下文中,将详细地描述基于缩醛的化合物、由其制备的基于缩醛的预聚物、基于缩醛的聚合物、其制备方法、和包括所述基于缩醛的聚合物的光刻胶组合物的实施方式。另外,将描述通过使用所述光刻胶组合物形成图案的方法。
由于与ArF光相比,极紫外(EUV)光具有非常高的能量,因此即使当将相同能量的光照射到化学放大抗蚀剂(CAR)上时,光子的数量也是显著小的,且因此,灵敏度降低。因此,为了增加能够产生酸的光子的数量,要求CAR的增加的曝光剂量和非常长的曝光时间。
为了提高CAR型光刻胶的灵敏度,当光刻胶聚合物链的长度通过相对较小量的由光产生的酸而减小时,分子量快速减小,且所述链的溶解度可快速增加。
作为能够减小聚合物链的长度的主链断裂(MCS)聚合物,甲基丙烯酸甲酯聚合物、聚砜、聚碳酸酯等是已知的。然而,当这样的MCS聚合物被使用时,由于显影溶液的必须改变、低的耐蚀刻性、对于显影溶液的差的对比度、缺陷出现、低的玻璃化转变温度、在曝光期间的排气等,所述聚合物的实际应用可更加困难。
因此,本发明人已经对基于缩醛的聚合物进行了研究,所述基于缩醛的聚合物为新的主链断裂(MCS)聚合物,其具有作为对酸不稳定的官能团被引入聚合物主链中的缩醛部分。所述基于缩醛的聚合物可通过由光引起的主链断裂有效地减小聚合物的分子量。使用根据实施方式的基于缩醛的聚合物,聚合物的分子量可通过经由将对酸不稳定的官能团引入聚合物主链中的主链断裂而被有效地减小,且因此,在扩展作为CAR的产酸剂/聚合物的组合的概念的同时改善体系的光敏性。因此,当聚合物的主链断开时,且虽然由于有限的酸产生,侧链的去保护在经曝光的区域中可为不完全的,但是聚合物链的长度减小且分子量降低。这又提供改善的显影,且因此,光敏性可大大增加。
新的基于缩醛的化合物被用在根据一种或多种实施方式的基于缩醛的聚合物的制备中。
在一种或多种实施方式中,提供由式1表示的基于缩醛的化合物。
式1
其中,在式1中,
R
其中Q
除了如下之外:R
a和b各自独立地为0或1-10的整数,
G为由式2或式2a表示的基团,
式2
式2a
其中,在式2和式2a中,R
R
*表示连接位点,和
k为0或1。
在式1中,G表示用于可逆加成断裂链转移(RAFT)聚合的链转移剂(CTA)。
式1的化合物可为,例如,由式1-1表示的化合物。
式1-1
其中,在式1-1中,
R
其中Q
除了如下之外:R
a和b各自独立地为0或1-10的整数,
G为由式2-1或式2a-1表示的基团,
式2-1
式2a-1
其中在式2和2a-1中,R
R
*表示连接位点。
在上式中,各基团的取代基可为至少一个R。R可为氘(-D)、-F、-Cl、-Br、-I、羟基、氰基、或硝基;C
根据一种或多种实施方式的基于缩醛的化合物可为由式3或式3-1表示的化合物。
式3
式3-1
其中,在式3和3-1中,z为C1-C60烷基。
例如,z可为C1-C30烷基、C1-C25烷基、C5-C20烷基、或C10-C15烷基。例如,z可为CH
根据一种或多种实施方式的基于缩醛的化合物可为,例如,由式3-3表示的化合物、或由式3-4表示的化合物。
式3-3
式3-4
在制备根据一种或多种实施方式的基于缩醛的化合物的方法中,所述基于缩醛的化合物可通过如下制备:首先制备具有乙烯基醚末端基团的链转移剂,然后将所述链转移剂与乙二醇和酸一起使用,如稍后将描述的实施例1中所描述地。
根据一种或多种实施方式的基于缩醛的化合物包括缩醛部分,且因此,可使用所述基于缩醛的化合物制备在聚合物主链中具有所述缩醛部分作为对酸不稳定的官能团的基于缩醛的预聚物和基于缩醛的聚合物。
所述基于缩醛的预聚物和所述基于缩醛的聚合物可使用被设计成具有对酸不稳定的官能团的用于RAFT聚合的可逆加成-断裂链转移(RAFT)剂或原子转移自由基聚合(ATRP)引发剂和扩链剂制备。
根据一种或多种实施方式的基于缩醛的预聚物可通过使用其中引入对酸不稳定的基团的RAFT剂制备,以具有缩醛基团位于链中心的结构。所述基于缩醛的预聚物为通过所述基于缩醛的化合物、具有极性基团的第一能聚合的单体、和具有对酸不稳定的基团的第二能聚合的单体的聚合反应获得的产物。
所述第一能聚合的单体和所述第二能聚合的单体的混合摩尔比可为,例如,1:99-99:1、1:9-9:1、2:8-8:2、6:4-4:6、或5:5。
继续地,在通过所述基于缩醛的预聚物的末端基团的取代的改性之后,将经改性的基于缩醛的预聚物与扩链剂混合以引起扩链反应,由此制备基于缩醛的聚合物。
通过所述基于缩醛的预聚物和所述扩链剂的扩链反应获得的基于缩醛的聚合物可具有在所述扩链反应期间根据反应条件可改变的分子量。这些条件可包括扩链剂、催化剂、溶剂等。
当使用光刻胶组合物形成光刻胶膜中的线图案时,由于图案侧壁表面的粗糙度,即,线边缘粗糙度(LER),线宽变化(偏差)可发生。要求线宽变化为尺寸宽度的约10%或更小,并且图案尺寸越小,LER的影响越大。然而,光刻胶组合物中的一般基础聚合物具有如几纳米大的每分子的平均颗粒直径,且因此,难以减小LER。
根据一种或多种实施方式的基于缩醛的聚合物含有作为对酸不稳定的基团的缩醛部分,且因此,可通过减小暴露于光的聚合物的部分中的回转半径而具有改善的LER。另外,通过减小曝光的部分的分子量,在显影溶液中的溶解度增加,由此减少缺陷的出现和改善对比度。另外,灵敏度可改善,使得即使用低剂量的光也可保证优异的对比度。
根据一种或多种实施方式的基于缩醛的聚合物可具有约4,000克/摩尔(g/mol)-约50,000g/mol、约4,000g/mol-约35,000g/mol、约4,100g/mol-约20,000g/mol、约4,300g/mol-约20,000g/mol、约4,500g/mol-约18,000g/mol、或约4,800g/mol-约15,000g/mol的重均分子量(Mw)、以及约1.2-约2.5例如约1.3-2.4的多分散指数(PDI)。在根据一种或多种实施方式的基于缩醛的聚合物中,通过酸处理分解的断裂聚合物链可具有约3,000g/mol或更小、约2,000g/mol或更小、约1,500g/mol或更小、约1,000g/mol或更小、或约500g/mol-约900g/mol的重均分子量(Mw)。通过酸分解的聚合物链可具有约1.05-约1.19的PDI。
如本文中的重均分子量通过凝胶渗透色谱法限定。
根据一种或多种实施方式的基于缩醛的聚合物可用作在制备光刻胶组合物中的基础聚合物。
关于当所述基于缩醛的聚合物用作光刻胶组合物中的基础聚合物时的图案化机理,在曝光的部分中通过光致产酸剂产生酸。作为疏水性基团的所述基于缩醛的聚合物的对酸不稳定的官能团通过所产生的酸分解并且变成亲水性基团。结果,在显影溶液中的溶解度增加,从而增加对比度和保证优异的性能,即使是用小剂量的光。
根据一种或多种实施方式的基于缩醛的预聚物为所述基于缩醛的化合物、具有极性基团的第一能聚合的单体、和具有对酸不稳定的基团的第二能聚合的单体的聚合反应产物。
所述第一能聚合的单体可为由式4表示的化合物。
式4
其中,在式4中,R
R
任选地,至少两个相邻的R
R
例如,所述极性基团可为羟基、缩醛基团、或R'C(=O)O-(其中R'为H或C1-C20烷基)。
所述卤素可为,例如,F、I、Cl、或Br。另外,被卤素取代的C1-C60烷基可为,例如,CF
所述具有对酸不稳定的基团的第二能聚合的单体可为,例如,由式5表示的化合物。
式5
其中,在式5中,R
所述对酸不稳定的基团可为,例如,C4-C12烷基例如叔丁基或叔戊基、或具有脂环族结构的C4-C60烃基团。
所述具有脂环族结构的烃基团可为,例如,具有脂环族基团或具有被脂环族基团取代的烷基的烃基团。
R
/>
所述脂环族基团可包括如下作为单价脂环族基团:金刚烷基、降金刚烷基(noradamantyl)、十氢萘残基、三环癸烷基、四环十二烷基、降莰烷基、柏木醇基团、环己基、环庚基、环辛基、环癸基、和环十二烷基。所述脂环族基团的其它实例可包括金刚烷基、十氢萘残基、降莰烷基、柏木醇基团、环己基、环庚基、环辛基、环癸基、或环十二烷基。所述脂环族结构的取代基的实例包括烷基、卤素、羟基、烷氧基、羧基、或烷氧羰基。所述烷基可为低级烷基,例如甲基、乙基、丙基、异丙基、或丁基。例如,所述烷基可为甲基、乙基、丙基、或异丙基。所述烷氧基可包括C1-C4烷氧基例如甲氧基、乙氧基、丙氧基、或丁氧基。所述烷基和所述烷氧基可进一步具有取代基。所述烷基和所述烷氧基的取代基的实例可为羟基、卤素、或烷氧基。
所述对酸不稳定的基团可为未取代的或取代的基团A,基团A包括金刚烷基、降金刚烷基、十氢萘残基、三环癸烷基、四环十二烷基、降莰烷基、柏木醇基团、环己基、环庚基、环辛基、环癸基、或环十二烷基。
可形成重复单元的第一能聚合的单体的非限制性实例为由下式表示的单体。
/>
/>
/>
在上式中,R
第一单体的另外的非限制性实例为可形成由下式表示的重复单元的单体。
/>
/>
在式中,a为1-6的整数。
所述第二能聚合的单体的另外的非限制性实例为可形成由下式表示的重复单元的单体。
所述第二能聚合的单体的还另外的非限制性实例为可由下式表示的单体。
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
由所述第一能聚合的单体和所述第二能聚合的单体,可分别形成由式4-1表示的第一重复单元和由式5-1表示的第二重复单元。所述第二重复单元为通过酸的作用分解以增加在碱性显影溶液中的溶解度的重复单元。
其中,在式4-1中,R
在式5-1中,R
所述第一重复单元可得自3-羟基苯乙烯、4-羟基苯乙烯、5-羟基-2-乙烯基萘、或6-羟基-2-乙烯基萘。例如,所述第二重复单元可包括基于(甲基)丙烯酸酯的聚合物例如聚(甲基丙烯酸叔丁酯)、聚(甲基丙烯酸降莰烷酯)、或者所述基于(甲基)丙烯酸酯的聚合物的二元或三元共聚物的重复单元。例如,所述第二重复单元可不具有对酸不稳定的基团。没有对酸不稳定的基团的第二重复单元可包括例如聚甲基丙烯酸甲酯和聚(甲基丙烯酸)的重复单元。
所述基于缩醛的预聚物可为,例如,由式6表示的化合物。
式6
E为
D
其中,在式6中,A
Z
R为C1-C30烷基,R
在式6中,例如,A
例如,a1、b1、a2、和b2各自独立地为1-1,000的数、1-100的数、1-50的数、2-50的数、或3-50的数。
在式6中,a1和b1可分别与a2和b2相同。
在式6中,苯乙烯类重复单元和丙烯酸类重复单元的混合摩尔比为,例如,1:99-99:1、1:9-9:1、2:8-8:2、6:4-4:6、或5:5。
例如,所述基于缩醛的预聚物可为由式6-1表示的化合物。
式6-1
其中,在式6-1中,A1和A2各自独立地为H或-C(=O)R
Z
R为C1-C30烷基,和
a1、b1、a2、和b2各自独立地为1-10,000的数、1-1,000的数、1-100的数、或1-50的数。
例如,所述基于缩醛的预聚物可为由式6-2表示的化合物、由式6-3表示的化合物、或由式6-4表示的化合物。
式6-2
式6-3
式6-4
在式6-2至6-4中,a1、b1、a2、和b2各自独立地为1-10,000的数、1-1,000的数、1-100的数、1-50的数、2-50的数、3-50的数、或5-50的数。
所述基于缩醛的预聚物可各自具有约1,500g/mol-约20,000g/mol、约1,800g/mol-约20,000g/mol、约2,000g/mol-约20,000g/mol、或约5,000g/mol-约20,000g/mol的重均分子量。当所述基于缩醛的预聚物具有太高的重均分子量时,这在使光刻胶显影中是不利的,因为即使在通过光产生的酸的分解之后,分子量仍然是太高的。当所述基于缩醛的预聚物具有太低的分子量时,可产生差的机械性质或性能。
化学计量地控制用于制备根据一种或多种实施方式的基于缩醛的预聚物的基于缩醛的化合物的量以获得具有期望的性能的基于缩醛的预聚物。基于1摩尔的基于缩醛的预聚物,所述基于缩醛的化合物的量可为1摩尔或更大、或者2摩尔或更大。当所述基于缩醛的化合物的量在这些范围内时,由所述基于缩醛的预聚物形成的基于缩醛的聚合物的分子量可通过酸处理有效地减小。
所述第一能聚合的单体和所述第二能聚合的单体的混合摩尔比可为1:99-99:1、1:9-9:1、2:8-8:2、6:4-4:6、或5:5。
根据一种或多种实施方式,提供基于缩醛的聚合物,其为以上描述的基于缩醛的预聚物和扩链剂的扩链反应产物。
作为对所述基于缩醛的预聚物的两个末端具有反应性官能团的化合物的扩链剂被用于扩展所述基于缩醛的聚合物的链长度。所述扩链剂的非限制性实例为基于氨基甲酸酯的化合物、由式9表示的化合物、或其组合,所述基于氨基甲酸酯的化合物为由式7表示的二醇和由式8表示的二异氰酸酯的反应产物。
/>
基于1摩尔的所述基于缩醛的预聚物,所述扩链剂的量可为约0.5摩尔-2摩尔。当所述扩链剂的量在该范围内时,可制备其分子量可通过酸处理有效地减小的基于缩醛的聚合物。所述基于缩醛的聚合物可包括如上所述的式4-1的第一重复单元和由式5-1表示的第二重复单元。
所述基于缩醛的聚合物可为,例如,由式10表示的基于缩醛的聚合物。
式10
其中,在式10中,L
L
L
k
a1、b1、a2、b2、和n表示聚合度,且各自独立地为1-10,000的数、1-1,000的数、1-100的数、2-100的数、或3-100的数。
苯乙烯类重复单元和甲基丙烯酸类重复单元的混合摩尔比可为1:99-99:1、1:9-9:1、2:8-8:2、6:4-4:6、或5:5。
所述基于缩醛的聚合物可为由式10-1表示的聚合物或由式10-2表示的聚合物。
式10-1
在式10-1中,a1、b1、a2、b2、和n各自独立地为1-10,000的数、1-100的数、2-100的数、或3-50的数。
在式10-1中,例如,n可为3-20的数。
苯乙烯类重复单元和甲基丙烯酸类重复单元的混合摩尔比可为1:99-99:1、1:9-9:1、2:8-8:2、6:4-4:6、或5:5。
式10-2
在式10-2中,a1、b1、a2、b2、和n表示聚合度,并且各自独立地为1-10,000的数。
a1、b1、a2、b2、和n各自独立地为1-1,000的数、1-100的数、2-100的数、3-50的数、或3-20的数。
苯乙烯类重复单元和甲基丙烯酸类重复单元的混合摩尔比可为1:99-99:1、1:9-9:1、2:8-8:2、6:4-4:6、或5:5。
在一种或多种实施方式中,所述基于缩醛的聚合物可具有约4,000g/mol-约20,000g/mol的重均分子量和约1.2-约2.5的多分散性指数(PDI)。当所述基于缩醛的聚合物的分子量和PDI在这些范围内时,通过酸处理的有效的分子量减小是可能的。酸的非限制性实例为全氟丁烷磺酸、三氟甲磺酸等。
所述基于缩醛的聚合物可为无规共聚物、嵌段共聚物、交替共聚物、接枝共聚物、或其组合,和例如,可为无规共聚物。所述无规共聚物与其它共聚物相比可容易地合成而没有相分离,并且可形成均匀的薄膜。
根据一种或多种实施方式,提供光刻胶组合物,其包括基础聚合物、光致产酸剂、和溶剂,其中所述基础聚合物包括以上描述的基于缩醛的聚合物。
通过包括根据以上描述的一种或多种实施方式的基于缩醛的聚合物,所述光刻胶组合物即使在较小剂量的光下也可提供优异的在曝光灵敏度、分辨率等方面的光刻特性、以及经显影的光刻胶例如经显影的CAR的优异的图案形状。
在100重量份的所述光刻胶组合物的基础上,所述基于缩醛的聚合物的量可为约1重量份-约25重量份、约3重量份-20重量份、或约5重量份-约20重量份。当所述基于缩醛的聚合物的量在这些范围内时,可不仅通过经由光致产酸剂的降解、而且通过分子量降低而另外保证对比度。另外,可引入现有的化学放大抗蚀剂(CAR)结构以采用低剂量的光改善对比度和确保性能。
所述光致产酸剂的实例为三氟甲磺酸三苯基锍、锑酸三苯基锍、三氟甲磺酸二乙基碘
在100重量份的所述基础聚合物的基础上,所述光致产酸剂的量可为约15重量份-约50重量份。当所述光致产酸剂的量在该范围内时,所述基础聚合物的玻璃化转变温度可被保持,而没有由于较少的EUV光子所致的光吸收率(速率)的降低,并且在通过EUV光源的光刻法过程期间没有光致产酸剂剩余,使得由所述光刻胶组合物形成的光刻胶图案可具有改善的分辨率。
所述光刻胶组合物可进一步包括能光分解的猝灭剂(PDQ)。所述能光分解的猝灭剂的非限制性实例可包括由式12表示的猝灭剂。
式12
在100重量份的所述基础聚合物的基础上,所述光刻胶组合物中的能光分解的猝灭剂的量可为约0.1重量份-约20重量份。
在一种或多种实施方式中,所述溶剂可包括丙二醇单乙基醚乙酸酯、聚丙二醇单甲基醚乙酸酯、丙二醇单甲基醚、或其组合。然而,实施方式不限于此,并且可使用本领域中可用的任何溶剂。
另外,为了改善对基板的粘附(例如,为了改善所述光刻胶组合物与基板的粘附),所述光刻胶组合物可另外包括硅烷偶联剂作为粘附增强剂。所述硅烷偶联剂可为,例如,乙烯基三甲氧基硅烷、乙烯基三乙氧基硅烷、乙烯基三氯硅烷、乙烯基三(β-甲氧基乙氧基)硅烷;3-丙烯酰氧基丙基三甲氧基硅烷、3-甲基丙烯酰氧基丙基三甲氧基硅烷、对-苯乙烯基三甲氧基硅烷、3-甲基丙烯酰氧基丙基甲基二甲氧基硅烷、3-甲基丙烯酰氧基丙基甲基二乙氧基硅烷;或包括不饱和的碳-碳键的硅烷化合物例如三甲氧基[3-(苯基氨基)丙基]硅烷。然而,实施方式不限于此。
在酸处理之后的所述基础聚合物的聚合物分解产物可具有3000g/mol或更小、例如2000g/mol或更小、1000g/mol或更小、或约850g/mol-约1000g/mol的重均分子量,并且具有2.0或更小、1.5或更小、或约1.0-约1.5的PDI。
现在将描述用根据一种或多种实施方式的光刻胶组合物形成半导体图案的方法。
所述光刻胶组合物即使在形成具有高的纵横比(高宽比)的图案时也可不经历图案坍塌。因此,为了形成具有例如5nm-100nm、5nm-50nm、或5nm-20nm的宽度的微图案,所述光刻胶组合物可用在使用约5nm-约150nm、约5nm-约80nm、或约5nm-约20nm的波长的光的光刻胶工艺中。因此,使用根据一种或多种实施方式的光刻胶组合物,可使用约13.5nm的波长的EUV光源实施极紫外光刻法。
根据另外的实施方式,提供用根据以上描述的一种或多种实施方式的光刻胶组合物形成图案的方法。例如,所形成的图案可为光刻胶图案。
根据实施方式,图案形成方法可包括:在基底上形成蚀刻目标膜(靶膜);将根据本文中描述的一种或多种实施方式的光刻胶组合物施加到所述蚀刻目标膜上以形成光刻胶膜;通过将所述光刻胶膜图案化而形成光刻胶图案;和用所述光刻胶图案作为蚀刻掩模蚀刻所述蚀刻目标膜。
将参照图8至12描述用根据一种或多种实施方式的光刻胶组合物形成图案的方法。
参考图8,描述待蚀刻的对象(下文中,蚀刻目标)。所述蚀刻目标的实例可为在基板100形成的薄膜102。下文中,以下描述限于其中使用薄膜102作为蚀刻目标的实施方式。为了除去残留在薄膜102上的污染物,清洁薄膜102的表面。薄膜102可为,例如,氮化硅膜、多晶硅膜、或氧化硅膜。随后,在经清洁的薄膜102的表面上涂布用于形成下部抗蚀剂膜104的下部抗蚀剂膜组合物。所述涂布可例如通过如下实施:旋涂,喷涂,浸涂,刀口涂布,或印刷例如喷墨印刷、丝网印刷等。
尽管可省略涂布所述下部抗蚀剂膜,但是下面将描述涂布所述下部抗蚀剂膜的实施方式。之后,进行干燥和烘烤过程以在薄膜102上形成下部抗蚀剂膜104。烘烤处理可在约100℃-约500℃下、例如在约100℃-约300℃下实施。
参考图9,下部抗蚀剂膜104可形成于基板100和光刻胶膜106之间,并且防止在照射辐射从基板100和光刻胶膜106之间的界面反射或者中间层硬掩模被散布到不期望的光刻胶区域中的时候的图案可成形性的恶化和光刻胶线宽的不均匀性。
将以上描述的光刻胶组合物涂布在下部抗蚀剂膜104上以形成光刻胶膜106。通过在将所述光刻胶组合物涂布在基板100上的薄膜102上之后进行的热处理过程,光刻胶膜106可具有固化形式。
更特别地,用根据一种或多种实施方式的光刻胶组合物形成图案可包括:将以上描述的光刻胶组合物通过旋涂、狭缝涂布、喷墨印刷等涂覆于在基板100的表面上具有薄膜102的基板100上;和将经涂布的根据一种或多种实施方式的光刻胶组合物干燥以形成光刻胶膜106。随后,实施加热具有形成于其上的光刻胶膜106的基板100的第一烘烤过程。所述第一烘烤过程可在约80℃-约120℃的温度下实施。
参考图10,使光刻胶膜106经历使用光掩模的选择性辐射曝光。可用在曝光过程中的光的实例可为如下的活化辐射:短波长的光例如i线(365nm的波长)、KrF准分子激光(248nm的波长)、ArF准分子激光(193nm的波长)、高能波长的光例如极紫外(EUV,例如,13.5nm的波长)、E束(电子束)等。在一种或多种实施方式中,用于曝光的光可为在5nm-150nm的波长范围内的短波长的光或者可为高能波长的光例如极紫外(EUV,例如,13.5nm的波长)、E束(电子束)等。
根据通过交联反应的聚合物形成,光刻胶膜106的未经曝光的区域106a可具有与光刻胶膜106的经曝光的区域106b的溶解度不同的溶解度。随后,对基板100实施第二烘烤过程。所述第二烘烤过程可在约90℃-约200℃的温度下实施。通过所述第二烘烤过程,光刻胶膜106的未经曝光的区域106a变为在显影溶液中不可溶的。
图11显示通过如下形成的光刻胶图案108:通过用显影溶液溶解而除去光刻胶膜106的经曝光的区域106b。特别地,在用有机溶剂例如2-庚酮溶解和除去光刻胶膜106的经曝光的区域106b之后,完成对应于正色调图像的光刻胶图案108。
如上所述,在根据一种或多种实施方式的图案形成方法中的显影溶液可为有机溶剂。在根据一种或多种实施方式的图案形成方法中使用的有机溶剂的实例为酮例如甲乙酮、丙酮、环己酮、和2-庚酮;醇例如4-甲基-2-丙醇、1-丁醇、异丙醇、1-丙醇、和甲醇;酯例如丙二醇单甲基醚乙酸酯、乙酸乙酯、乳酸乙酯、乙酸正丁酯、和丁内酯;芳族化合物例如苯、二甲苯、和甲苯;或其组合。
根据一种或多种实施方式,所述光刻胶图案可形成为具有正色调图像。在此情况下,可用于形成正色调图像的显影剂可为氢氧化季铵成分例如氢氧化四乙基铵、氢氧化四丙基铵、氢氧化四丁基铵、或其组合。
在一种或多种实施方式中,通过用短波长的光例如i线(365nm的波长)、KrF准分子激光(248nm的波长)、ArF准分子激光(193nm的波长)、或者还有高能量的光例如极紫外(EUV,13.5nm的波长)或E束(电子束)的曝光形成的光刻胶图案108可具有约5nm-约100nm的宽度。例如,光刻胶图案108可形成为具有约5nm-约90nm、约5nm-约30nm、约10nm-约50nm、或约10nm-约20nm的宽度。
光刻胶图案108可具有约50nm或更小、例如40nm或更小、例如30nm或更小、例如25nm或更小的半间距(节距)、以及约10nm或更小或者约5nm或更小的线宽粗糙度。随后,使用光刻胶图案108作为蚀刻掩模蚀刻下部抗蚀剂膜104。通过如上所述的蚀刻过程,形成有机膜图案112。所形成的有机膜图案112也可具有与光刻胶图案108对应的宽度。
参考图12,用光刻胶图案108作为蚀刻掩模蚀刻薄膜102的暴露的区域。结果,薄膜102形成为薄膜图案114。例如,薄膜102可通过使用蚀刻气体的干法蚀刻进行蚀刻。例如,CHF
在之前进行的曝光过程中,使用通过用EUV光源的曝光获得的光刻胶图案108形成的薄膜图案114可具有与光刻胶图案108对应的宽度。例如,薄膜图案114可具有与光刻胶图案108相同的约5nm-100nm的宽度。例如,通过用EUV光源的曝光过程形成的薄膜图案114可具有像光刻胶图案108一样的约5nm-约90nm、约5nm-约30nm、约10nm-约40nm、或约10nm-约20nm的宽度。例如,薄膜图案114可形成为具有约20nm或更小的宽度。
光刻胶图案可具有离应形成的理想直线形状的线边缘的偏差。所述第一光刻胶图案边缘离应形成的理想直线的偏差程度称为线边缘粗糙度(LER)。线边缘粗糙度的值越大,所述第一光刻胶图案离直线形状的偏差越大。
特别地,当用EUV光形成约20nm-约50nm的非常小的尺寸的图案时,更难以控制粗糙度特性。另外,对于窄线宽的图案,图案线宽可根据线宽粗糙度而具有非常大的均匀性差异,且因此,线宽粗糙度特性可为非常重要的。
在本文中的式中使用的脂族环基团、杂脂族环基团、芳族环基团、和杂芳族环基团如下定义。
脂族环状基团可为单环或多环脂族环状基团。单环脂族环基团的实例可包括环戊基、环己基、或环辛基。多环脂族环基团的实例可包括降莰烷基、降冰片烯基、三环癸烷基、或四环十二烷基。杂脂族环状基团可为如下脂族环状基团:其包括至少一个来自N、O、P或S的杂原子作为形成所述脂族环状基团的环元素,并且包括C作为剩余的环原子。
芳族环基团可为单环或多环芳族环基团。单环芳族环基团的实例可包括苯环。多环芳族环基团的实例可包括萘环、菲环、蒽环、或芴环。杂芳族环基团可为如下芳族环基团:其包括一个或多个作为N、O、P或S的杂原子作为形成所述芳族环基团的环元素,并且包括C作为剩余的环原子。
在整个说明书中除非另外定义,否则式中使用的取代基的定义如下。
本文中的C
本文中的C
本文中的C
本文中的C1-C
本文中的C
本文中的C
本文中的C
本文中的C
本文中的C
本文中的C
本文中的单价非芳族稠合多环基团指的是包括至少两个彼此稠合的环并且仅包括碳作为成环原子的作为整个分子具有非芳香性的(例如,8-60个碳原子的)单价基团。单价非芳族稠合多环基团的具体实例包括茚基、芴基、螺二芴基、苯并芴基、茚并菲基、茚并蒽基等。本文中的二价非芳族稠合多环基团指的是具有与单价非芳族稠合多环基团相同的结构的二价基团。
本文中的单价非芳族稠合杂多环基团指的是包括至少两个彼此稠合的环并且除了碳原子之外进一步包括至少一个杂原子作为成环原子的作为整个分子具有非芳香性的(例如,1-60个碳原子的)单价基团。单价非芳族稠合杂多环基团的具体实例包括吲哚基、苯并吲哚基、萘并吲哚基、异吲哚基、苯并异吲哚基、萘并异吲哚基、苯并噻咯基、苯并噻吩基、苯并呋喃基、咔唑基、二苯并噻咯基、二苯并噻吩基、二苯并呋喃基、氮杂咔唑基、氮杂芴基、氮杂二苯并噻咯基、氮杂二苯并噻吩基、氮杂二苯并呋喃基、苯并吡唑基、苯并咪唑基、苯并
本文中的C
本文中的C
本文中的杂原子指的是除了碳原子之外的任意原子。杂原子的实例包括O、S、N、P、Si、B、Ge、Se、或其任意组合。
现在将参照以下实施例和对比例详细地描述本公开内容的一种或多种实施方式。然而,这些实施例仅用于说明的目的,且不意图限制本公开内容的一种或多种实施方式的范围。
基于缩醛的化合物的制备
实施例1.具有乙烯基醚基团作为末端基团的链转移剂的合成
反应方案1
在方案1中,EDC为1-乙基-3-(3-二甲基氨基丙基)碳二亚胺的缩写。
将作为原材料的5g 4-氰基-4(十二烷基硫基硫代羰基)硫基戊酸(CDSTSP)添加至250ml圆底烧瓶并且用50毫升(ml)二氯甲烷(DCM)完全溶解。将所述烧瓶置于含有冰和丙酮的水浴中,然后搅拌5分钟。
将83.2mg 4-二甲基氨基吡啶(DMAP)添加至所述烧瓶,并且将烧瓶壁上的化合物用20ml二氯甲烷洗掉。进一步向所述烧瓶添加2.6g 1-乙基-3-(3-二甲基氨基丙基)碳二亚胺(EDC)和20ml二氯甲烷并且搅拌10分钟。将1.38g乙二醇乙烯基醚(EGVE)逐滴添加至反应混合物并且搅拌。
将反应混合物在冰浴中搅拌30分钟或更长,然后在室温(25℃)下搅拌。在将反应混合物在室温下搅拌24小时之后,将反应溶液添加至500ml分液漏斗。然后将150ml二氯甲烷添加至所述分液漏斗,并且用碳酸氢钠水溶液(含水碳酸氢钠溶液)萃取有机混合物。将含水碳酸氢钠萃取过程重复4次。
向来自所述萃取的有机层添加硫酸钠以从所述有机层除去剩余的水。用旋转蒸发器除去溶剂(二氯甲烷),并且用真空泵完全除去剩余的溶剂以得到5.6g式11的产物(CTA-EGVE)。使用核磁共振(NMR)波谱仪分析该产物以确认所述产物的结构。
式11
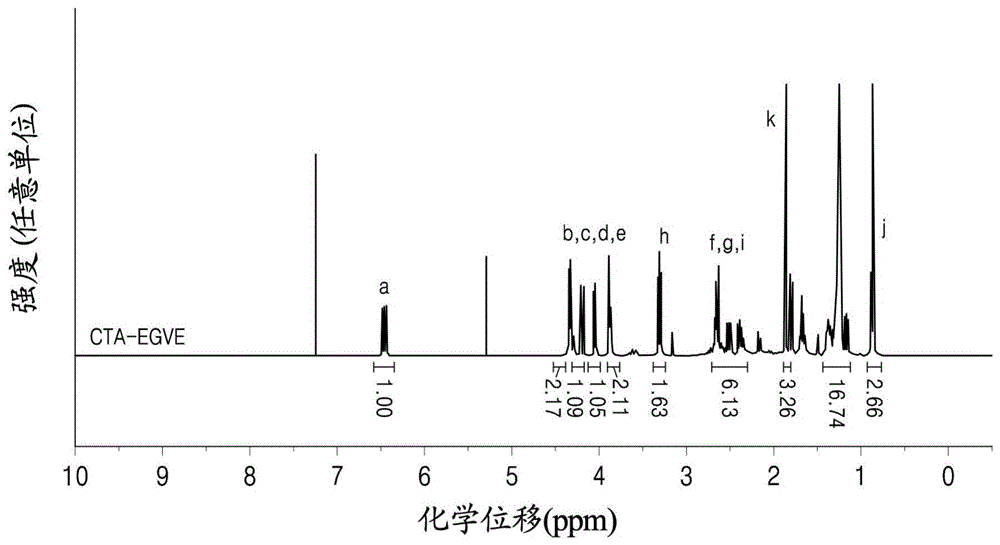
所述产物(CTA-EGVE)的结构可由图1的NMR分析结果证实。图1的
具有缩醛基团的链转移剂的合成
反应方案2
使用式11的化合物(即根据以上描述的过程获得的具有乙烯基醚末端基团的链转移剂)、乙二醇和酸,如下合成式3-3的化合物(具有缩醛基团的链转移剂)(下文中,缩醛链转移剂(ACT-CTA))。
将4.7g式11的化合物(根据以上描述的过程获得的具有乙烯基醚末端基团的链转移剂)添加至25ml圆底烧瓶,且用10ml无水四氢呋喃(THF)完全溶解。将反应烧瓶加至含有冰和丙酮的水浴中,并且将反应混合物搅拌5分钟。然后,将0.31g乙二醇添加至烧瓶并且连续地搅拌以制备所述链转移剂溶解在其中的混合物。
单独地,将0.17g三氟乙酸(TFA)添加至之前在冰浴中冷却的1mL无水四氢呋喃。将三氟乙酸/THF混合物也在冰浴中冷却,然后逐滴添加至所述链转移剂溶解在其中的混合物。将反应混合物在0℃下搅拌30分钟或更多,然后除去冰浴,并且容许反应混合物温热至室温(25℃)且搅拌。
在25℃下搅拌24小时之后,将反应烧瓶置于冰浴中以降低反应混合物的内部温度以使反应终止。在5mL小瓶中将过量的0.5mL至1mL三乙胺溶解在1mL无水四氢呋喃中以获得混合物,并且将所述小瓶置于冰浴中。将冷却的三乙胺溶液在0℃下以10分钟或更多缓慢添加至所述反应烧瓶。使用旋转蒸发器除去溶剂,并且使用真空泵完全除去剩余的溶剂。将所得产物混合物用20mL二氯甲烷溶解并且添加至500mL分液漏斗,并且将另外的150mL二氯甲烷添加至所述漏斗。将有机产物用碳酸氢钠水溶液萃取四次。收集有机层,并且添加硫酸钠以除去剩余的水。然后,用旋转蒸发器除去溶剂,并且用真空泵完全除去剩余的溶剂。固体在室温下缓慢地形成并且重结晶。将所述固体用二乙醚过滤掉,随后使用旋转蒸发器从所获得的溶液除去溶剂以得到5.7g式3-3的化合物。
式3-3
使用核磁共振波谱仪分析式3-3的化合物。分析结果示于图2中,其中在
在图2的
实施例2.采用基于缩醛的化合物的基于缩醛的预聚物的聚合
反应方案3
在式6-2a中,Z为CH
首先,使用氧化铝从4-乙酰氧基苯乙烯除去反应抑制剂以得到经纯化的4-乙酰氧基苯乙烯。然后,将9.5g经纯化的4-乙酰氧基苯乙烯和9.7g 2-乙基-2-甲基丙烯酰氧基金刚烷添加至含有机械搅拌器的50mL Schlenk烧瓶以制备混合物。
将作为缩醛链转移剂的5.5g式3-3的化合物添加至20mL小瓶,并且用7g甲苯完全溶解以制备溶液1。将作为热引发剂的0.45g偶氮二异丁腈(AIBN)添加至5mL小瓶,并且用1g甲苯完全溶解以制备溶液2。然后,在将溶液1和溶液2添加至在反应烧瓶中的混合物之后,通过三次冷冻解冻泵循环(three freeze-pump-thaw cycles)除去气体。随后,在75℃下进行共聚。
在共聚反应完成之后,将反应溶液冷却至室温并且暴露于空气。将所得溶液用四氢呋喃稀释,并且添加甲醇以促进固体的沉淀。将所得黄色固体通过真空过滤收集并且在真空烘箱中干燥以得到18.5g式6-2的基于缩醛的预聚物。
式6-2
在式6-2中,苯乙烯类重复单元和甲基丙烯酸类重复单元的混合摩尔比为1:1,并且聚合度a1、b1、a2、和b2被控制成使得式6-2的聚合物具有1,864g/mol的重均分子量。在此情况下,a1与a2相同,且b1与b2相同。
使用核磁共振波谱法和凝胶渗透色谱法分析所合成的预聚物样品的组成、分子量、和分散度。产物的结构由图3的NMR分析结果证实。在
将式6-2的基于缩醛的预聚物的两个末端基团都用硫醇基团代替以如下制备式6-3的预聚物。
式6-3
在式6-3中,苯乙烯类重复单元和甲基丙烯酸类重复单元的混合摩尔比为1:1,并且聚合度a1、b1、a2、和b2被控制成使得式6-3的基于缩醛的预聚物具有1,810g/mol的重均分子量。在此情况下,a1与a2相同,且b1与b2相同。
将5g式6-2的基于缩醛的预聚物完全溶解在62mL无水四氢呋喃中,然后将0.88g正丁基胺和0.3g三丁基膦添加至烧瓶,并且将混合物在室温下搅拌24小时。将溶液使用旋转蒸发器浓缩,并且在用过量的己烷沉淀之后,将固体通过真空过滤收集并在真空烘箱中干燥以制备式6-3的基于缩醛的预聚物。
使用核磁共振波谱法和凝胶渗透色谱法分析所合成的预聚物样品的组成、分子量、和分散度。式6-2的产物的结构由图3的NMR分析结果证实。在
扩链剂的合成
实施例3.
在烧瓶中将1g烯丙醇和0.041g对甲苯磺酸吡啶
将硫酸钠添加至萃取的产物,并且在除去水之后,使用旋转蒸发器除去二氯甲烷。通过使用己烷和乙酸乙酯作为洗脱剂的柱层析法纯化所得产物。在这时,为了保护缩醛基团,向所述洗脱剂添加三乙胺,由此得到1.5g式9的化合物。使用核磁共振波谱仪分析该化合物的结构。
式9
/>
通过使用缩醛链转移剂和扩链剂的逐步生长聚合的基于缩醛的聚合物的聚合
实施例4.硫醇-乙烯基交联剂的逐步生长聚合
根据反应方案5,将0.2g在实施例2中合成的式6-3的预聚物添加至10mL圆底烧瓶并且用1mL无水四氢呋喃完全溶解。然后,添加作为光引发剂的4.7mg 2,2-二甲氧基-2-苯基乙酰苯,并且将13.1mL根据实施例3制备的作为扩链剂的式9的化合物添加至所述烧瓶。
将含有反应物的烧瓶用365-nm UV灯照射2小时。然后,将所得溶液添加至己烷以促进沉淀。将沉淀的基于缩醛的预聚物在真空烘箱中干燥。所述基于缩醛的预聚物的结构可由图4确认。下面所示的标记用于分配
之后,将0.5g基于缩醛的预聚物和10mL甲醇添加至100ml圆底烧瓶,将28%氨水溶液(每摩尔的4-乙酰氧基苯乙烯70当量)添加至所述烧瓶。在室温下搅拌24小时之后,使用旋转蒸发器除去溶剂。将所得产物用四氢呋喃溶解,然后用水沉淀,在真空下过滤进行收取,随后干燥以得到式10-1的无规共聚物(基于缩醛的聚合物)。通过凝胶渗透色谱法分析产物的分子量和分散性。
反应方案5
/>
在反应方案5中,基于缩醛的聚合物(聚合物A)中的a1、b1、a2、b2、和n被调节成使得聚合物A具有约5,360g/mol的重均分子量,并且基于缩醛的聚合物(聚合物B)中的a1、b1、a2、b2、和n被调节成使得聚合物B具有约5,500g/mol的重均分子量。基于缩醛的聚合物(聚合物A和聚合物B)中的苯乙烯类重复单元和甲基丙烯酸类重复单元的混合摩尔比为1:1。在上式中,a1与a2相同,且b1与b2相同。
实施例5.基于缩醛的聚合物的制备
反应方案6
在式10-1中,b1、a1、b2、a2、和n被调节成使得各聚合物具有5,501g/mol的重均分子量。在获得式10-1的聚合物之前的步骤处的聚合物中,b1、a1、b2、a2、和n可被调节成使得各聚合物具有4,950g/mol的重均分子量。苯乙烯类重复单元和甲基丙烯酸类重复单元的混合摩尔比为1:1。a2和b2分别与a1和b1相同。
根据反应方案6,将0.1g式6-3的预聚物添加至5mL小瓶并且用0.3mL无水四氢呋喃完全溶解。然后,将5.8mg三乙胺添加至所述小瓶并且使用旋涡混合器混合反应溶液。接着,将4.7mg二月桂酸二丁基锡、2.1mg式7的化合物、和22mg式8的六亚甲基二异氰酸酯添加至10mL圆底烧瓶并且搅拌。
式7
在搅拌时的几秒内,反应混合物开始变浑浊,添加缩醛预聚物溶液并且在室温下搅拌19小时。将所得溶液用四氢呋喃稀释,随后添加己烷以获得沉淀物。将沉淀物通过真空过滤收集,然后在真空烘箱中干燥以获得聚合物。
在将0.5g所述聚合物样品和10mL甲醇添加至100mL圆底烧瓶之后,向其添加28%氨水溶液(每摩尔的4-乙酰氧基苯乙烯70当量)。在室温下搅拌24小时之后,使用旋转蒸发器除去溶剂,将所得产物用四氢呋喃溶解,然后用水沉淀,并且在真空下过滤进行收取,随后干燥以得到式10-1的无规共聚物(基于缩醛的聚合物)。
<式10-1>
在式10-1中,a1、b1、a2、b2、和n(聚合度)被调节成使得式10-1的共聚物具有约5,501g/mol的重均分子量。苯乙烯类重复单元和甲基丙烯酸类重复单元的混合摩尔比为1:1。在式10-1中,a2和b2分别具有与a1和b1的那些相同的值。
所述聚合物的结构由图5的
通过凝胶渗透色谱法分析产物的组成、分子量和多分散性。
实施例6.光致产酸剂(PAG)的合成
反应方案4
将2.246g(11.01mmol)碘苯、0.655g(5.51mmol)氯化亚砜、和0.117g(1.10mmol)高氯酸钠与12mL四氢呋喃混合,然后搅拌3小时。之后,在减压下除去反应溶剂,并且用30mL水和30mL二氯甲烷进行萃取以提供有机层。将所述有机层分离并且用Na
在将3.73g(8.20mmol)化合物A-2溶解在15mL苯中之后,将2.778g(9.85mmol)三氟甲磺酸酐在0℃下逐滴添加至溶液并且在室温下搅拌1小时。将反应混合物用20mL水和50mL乙酸乙酯萃取以提供有机层,然后将所述有机层分离,用饱和NaHCO
将4.91g(7.39mmol)化合物A-1和3.146g(7.39mmol)2-(1-金刚烷羰氧基)-1,1-二氟乙烷磺酸三乙基铵与45mL二氯甲烷和5mL水混合,并且搅拌1小时。将所得有机层分离并且用MgSO
实施例7:式9的聚合物的制备
按以下方式合成由式9表示的聚合物。
将0.94g 2,2'-偶氮二(2-甲基丙酸)二甲酯(Waco Chemicals)、3.03g甲基丙烯酸2-乙基-2-金刚烷酯(TCI Chemicals)、和1.98g 4-乙酰氧基苯乙烯(Sigma-Aldrich)溶解在四氢呋喃中,然后在80℃下聚合8小时。之后,在添加甲醇时,形成沉淀物,且将所述沉淀物在40℃真空烘箱中干燥12小时以获得白色聚合物粉末。将所获得的聚合物在甲醇钠和甲醇的混合溶液中用磁力搅拌子搅拌6小时,然后用乙酸酸化以获得经酸化的聚合物。之后,将经酸化的聚合物用蒸馏水沉淀,并且将所得沉淀物在真空烘箱中干燥48小时以获得由式9表示的白色聚合物粉末。
式9
在式9中,x和y各自为约50的数。
实施例8:光刻胶组合物的制备
根据以下过程以根据实施例5获得的基于缩醛的聚合物制备光刻胶组合物,所述基于缩醛的聚合物于是被用作基础聚合物。
添加和混合100重量份作为无规共聚物的基于缩醛的聚合物、20.29重量份式11的光致产酸剂、12重量份式12的能光分解的猝灭剂(Chemieliva Pharmaceutical ProductList)、以及7500重量份其中丙二醇单乙基醚乙酸酯(Aldrich)和丙二醇单甲基醚(Aldrich)以30:70的重量比混合的共溶剂以制备光刻胶组合物。
对比例1:光刻胶组合物的制备
根据以实施例8中相同的方式制备光刻胶组合物,除了如下之外:使用根据实施例7获得的由式9表示的聚合物作为基础聚合物。
评价实施例1:使用全氟丁烷磺酸的稳定性测试
如下实施根据实施例5获得的基于缩醛的聚合物的稳定性测试。在将0.1g作为无规聚合物的所述基于缩醛的聚合物完全溶解在0.5mL无水四氢呋喃中之后,通过添加8mg全氟丁烷磺酸而实施酸处理。随后,在室温(25℃)下搅拌23小时之后,用乙酸乙酯和碳酸氢钠的水溶液除去所述酸。然后萃取和分离有机层,并且使用旋转蒸发器除去有机溶剂。
图6说明以上描述的酸处理过程。通过所述基于缩醛的聚合物的酸处理,缩醛单元分解。
通过凝胶渗透色谱法分析得自所述酸处理的产物以分析其组成、重均分子量(Mw)、数均分子量(Mn)、和多分散指数(PDI)。分析结果示于图7中。PDI通过方程1评价。
方程1
PDI=Mw/Mn
参考图7,发现基于缩醛的聚合物的Mn、Mw、和PDI与基于缩醛的预聚物的那些相比更大。还发现,在通过酸处理的分解之后,聚合物链的Mw、Mn、和PDI减小。
图7中的阶段A对应于在反应方案7中的阶段A处的基于缩醛的预聚物。
反应方案7
评价实施例2:光刻胶图案形成过程
将12英寸圆形硅晶片基板在UV臭氧清洁系统中预处理10分钟。随后,将在实施例8和对比例1中制备的光刻胶组合物分别旋涂在经预处理的基板上,并且使其在热板上在110℃下经历煅烧30秒(施加后烘烤(PAB))以形成具有约100nm的厚度的光刻胶膜。将掩模和EUV光(ASML NXE-3350)投射到经光刻胶膜涂布的晶片上。随后,使经光刻胶膜涂布的晶片在热板上在90℃下经历曝光30秒(曝光后烘烤,PEB)。将经煅烧的膜在2.38% TMAH显影剂溶液中浸泡30秒。形成各自具有50nm的关键尺寸(临界尺寸,CD)以及线(25nm)和间隔(25nm)的图案。
随后,实施用去离子(DI)水的洗涤10秒以除去未暴露于EUV光的涂布部分,随后干燥,由此形成光刻胶图案。
测量各光刻胶图案的线边缘粗糙度(LER)。测量结果示于表1中。
表1
参考表1,发现与使用对比例1的光刻胶组合物形成的光刻胶图案的那些相比,使用实施例8的光刻胶组合物形成的光刻胶图案具有改善的粗糙度特性和良好的LER特性。
如上所述,根据一种或多种实施方式的基于缩醛的聚合物包括缩醛部分作为对酸不稳定的基团,且因此,当用酸处理时,分子量可有效地减小。当使用含有这样的基于缩醛的聚合物的光刻胶组合物时,经曝光的部分可具有减小的分子量,由此增加对显影溶液的溶解度,同时减小线边缘粗糙度(LER),且因此,可实现高的图案精确度。
应理解,本文中描述的实施方式应仅在描述的意义上考虑且不用于限制的目的。在各实施方式中的特征或方面的描述应典型地被认为可用于其它实施方式中的其它类似特征或方面。尽管已经参照附图描述了一种或多种实施方式,但是本领域普通技术人员将理解,在不背离如由所附权利要求所限定的精神和范围的情况下,可在其中进行形式和细节方面的多种变化。
- 二缩醛组合物、含有该二缩醛组合物的聚烯烃成核剂、含有该二缩醛组合物的聚烯烃树脂组合物、生产该树脂组合物的方法及模制品
- 二缩醛组合物、含有该二缩醛组合物的聚烯烃成核剂、含有该二缩醛组合物的聚烯烃树脂组合物、生产该树脂组合物的方法及模制品