一种多取代稠环芳烃衍生物及其制备方法和应用
文献发布时间:2024-04-18 19:58:21
技术领域
本发明涉及有机化合物合成技术领域,具体涉及一种多取代稠环芳烃衍生物及其制备方法和应用。
背景技术
芳烃为苯环化合物的多样化提供了巨大的潜力,苯环化合物存在于药物、农用化学品和液晶中。然而,获取这些高能中间体需要合成前体,这涉及苛刻的条件或多步合成。开发使用更简单的底物和更温和的条件获取芳烃及其相关衍生物是必要的。
多取代芳烃作为芳香族化合物的普遍结构核心,在化学、生物科学和材料科学中具有重要应用。在这些高度官能化芳烃的所有合成方法中,有机催化苯环化是在温和条件下组装结构多样的芳烃结构中最有效和最通用的转化之一,具有出色的化学、区域或立体选择性。
联芳基化合物在药物、天然产物、材料和金属催化剂的配体等方面有着广泛的应用。然而,与自偶联相比,实现两种不同杂芳烃之间的选择性交叉偶联是一个巨大的挑战。稠合多环芳族化合物是用于有机电子应用的有趣材料,为了微调光物理或电化学特性,可以连接各种取代基或将杂原子(如N或S)并入稠合的芳族主链中。
稠环芳烃的应用广泛,多取代稠环芳烃必将应用于更多的领域,因此多取代稠环芳烃具有良好的发展前景。
发明内容
本发明的目的在于提供一种多取代稠环芳烃衍生物及其制备方法,将四炔类化合物与1,2,3,4,5,6-六[2-(三甲基硅烷基)乙炔基]苯反应,得到多取代稠环芳烃衍生物,制备方法简单高效,无需催化剂、控制反应温度就可得到较高产率,制备方法温和、简单、绿色。
本发明还有一个目的在于提供一种多取代稠环芳烃衍生物的应用,用于光学材料。所制备的多取代稠环芳烃衍生物在250nm到400nm区间Abs可达1.5以上,说明该物质的吸光度良好,可较好的用于光学材料的制备。
本发明具体技术方案如下:
本发明提供的一种多取代稠环芳烃衍生物的制备方法,具体为:
将四炔类化合物与1,2,3,4,5,6-六[2-(三甲基硅烷基)乙炔基]苯混合在溶剂中,加热反应,反应结束后分离、纯化,即可得到多取代稠环芳烃衍生物。
所述四炔类化合物、1,2,3,4,5,6-六[2-(三甲基硅烷基)乙炔基]苯的物质的量之比为1:1.0-1.2;
所述1,2,3,4,5,6-六[2-(三甲基硅烷基)乙炔基]苯在溶剂的浓度为0.2~0.3mol/L;
所述溶剂选自甲苯;
所述加热反应是指在100-110℃的条件下反应12-14h;
所述1,2,3,4,5,6-六[2-(三甲基硅烷基)乙炔基]苯的结构式为
多取代稠环芳烃衍生物的制备方法中所述分离、纯化的方法为:将粗产物用饱和氯化钠水溶液和乙酸乙酯萃取,收集有机相,减压旋干,湿法装柱,用体积比为1:60-80的乙酸乙酯:石油醚为洗脱剂进行柱层析分离纯化柱层析分离,得到白色固体产物,即多取代稠环芳烃衍生物。
所述四炔类化合物结构式
所述四炔类化合物的制备方法为:
1)将丙二酸酯与炔丙基溴加入到无水乙腈中冰水浴,以氢化钠为碱,反应,然后分离纯化,即化合物1;
2)将步骤1)中制备的化合物1与苯乙炔基溴混合在CuCl的无水无氧催化体系中,加入正丁胺水溶液和盐酸羟胺,冰浴下搅拌反应,产物分离纯化,即得四炔类化合物。
步骤1)中氢化钠、丙二酸酯、炔丙基溴与无水乙腈的摩尔比为4-5:1:2.1-2.4:20-23。
优选的,步骤1)中所述丙二酸酯选自丙二酸二甲酯。
步骤1)中冰水浴条件下反应温度在0-5℃;反应时间在8小时以上;优选的反应时间8.5h;
步骤1)中所述分离纯化具体为:产物加饱和氯化钠水溶液洗涤,用乙酸乙酯萃取,减压旋干,采用体积比乙酸乙酯:石油醚=1:80的混合溶剂进行柱层析,得到产物,即化合物1。
步骤1)中所述化合物1结构式为:
优选的,所述化合物1结构式为:
步骤2)中步骤1)中所述化合物1:苯乙炔基溴:CuCl:盐酸羟胺的摩尔比=1:2-2.5:0.15-0.16:0.07-0.08。
步骤2)中所述正丁胺水溶液作溶剂,其质量浓度为30%;所述盐酸羟胺为碱,拔二炔上的氢。
步骤2)中,化合物1与正丁胺水溶液的用量比为0.4-0.5mol/L;
步骤2)中,搅拌反应时间至少12小时;
步骤2)中,所述苯乙炔溴的制备方法:取苯乙炔与N-溴代琥珀酰亚胺在丙酮溶剂中,AgNO
步骤2)中,所述分离纯化是指:产物用饱和氯化钠溶液洗涤,用二氯甲烷萃取,减压旋干,用体积比乙酸乙酯:石油醚=1:60-80柱层析分离,得到固体产物,即四炔类化合物;
优选的,四炔类化合物结构式为:
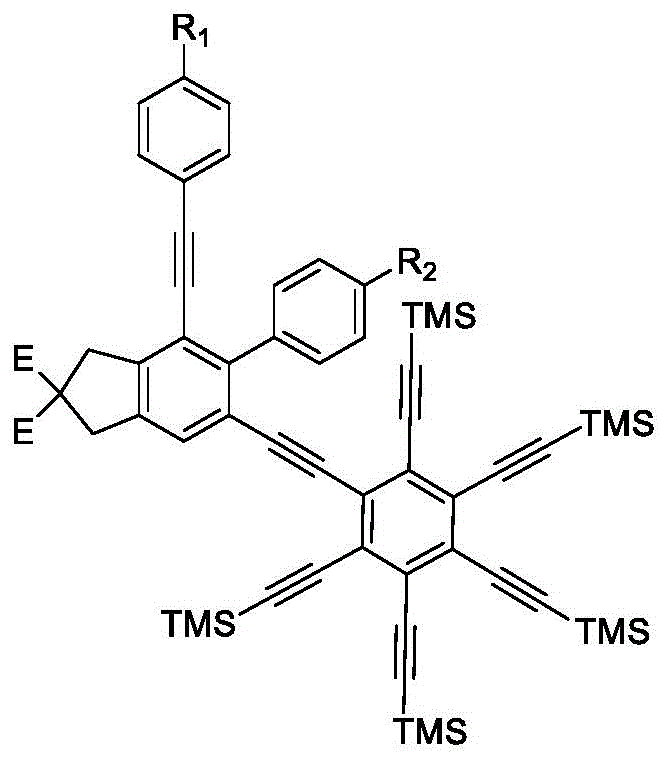
本发明提供的一种多取代稠环芳烃衍生物,采用上述制备方法制备得到;所述多取代稠环芳烃衍生物的结构式为:
优选的,所述多取代稠环芳烃衍生物的结构式为:
本发明提供的一种多取代稠环芳烃衍生物的应用,用于光学材料。
本发明以四炔类化合物和1,2,3,4,5,6-六[2-(三甲基硅烷基)乙炔基]苯为原料,在甲苯溶剂中,100~110℃反应12~14h,在加热的条件下,四炔自身发生HDDA反应形成苯炔中间体,然后苯炔中间体与1,2,3,4,5,6-六[2-(三甲基硅烷基)乙炔基]苯发生环加成反应形成多取代稠环芳烃衍生物,其机理图如图7所示,图7中四炔类化合物a高温下经历[3+2]的HDDA反应生成苯炔化合物b,苯炔化合物b与1,2,3,4,5,6-六[2-(三甲基硅烷基)乙炔基]苯c在甲苯溶剂中,110℃条件下反应12小时,经历[2+2]环加成反应,苯炔化合物b与1,2,3,4,5,6-六[2-(三甲基硅烷基)乙炔基]苯c的共轭体系的端基碳原子彼此头尾相连,形成四元环中间体d。苯炔化合物b的炔基位置更容易受到亲核试剂的攻击,但苯炔化合物b中的侧翼芳基取代基实质上抑制了亲核试剂接近近端炔碳,导致1,2,3,4,5,6-六[2-(三甲基硅烷基)乙炔基]苯的加成位置是d中的状态。由于四元环不稳定,发生开环反应,生成双自由基中间体f。苯炔碳侵蚀三甲基硅基碳上的氢原子,形成新的C-H键,生成新的双自由基中间体g。最后,C-Si键断裂,经过脱二甲基硅烯,g转化为最终产物多取代稠环芳烃衍生物h。
与现有技术相比,本发明提供了一种全新的制备多取代稠环芳烃的方法,生成新的多取代稠环芳烃衍生物。合成的多取代稠环芳烃衍生物具有较高原子经济性,产品在250nm到400nm区间Abs可达1.5以上,说明该物质的吸光度良好,可较好的用于光学材料的制备。
附图说明
图1为多取代稠环芳烃衍生物的结构通式;
图2为实施例1制备的多取代稠环芳烃衍生物的结构式;
图3为实施例1制备的多取代稠环芳烃衍生物的核磁共振氢谱;
图4为实施例1制备的多取代稠环芳烃衍生物的核磁共振碳谱;
图5为实施例1制备过程方程式;
图6为制备四炔类化合物的机理图;
图7为多取代稠环芳烃衍生物的反应机理图;
图8为实施例1制备的多取代稠环芳烃衍生物的紫外光谱图。
具体实施方式
下面结合实施例及说明书附图对本发明进行详细说明。
实施例1
一种多取代稠环芳烃衍生物,其结构式为:
所述多取代稠环芳烃衍生物的制备方法包括以下步骤:
1)以830mmol氢化钠为催化剂,将200mmol丙二酸二甲酯与440mmol炔丙基溴加入到210mL无水乙腈中,冰水浴0-5℃条件下,机械搅拌反应8小时,产物加饱和氯化钠水溶液洗涤,用乙酸乙酯萃取,减压旋干,采用体积比乙酸乙酯:石油醚=1:80的混合溶剂进行柱层析,得到产物,即化合物1,结构式
2)取100mmol苯乙炔(10.2g)与120mmolN-溴代酰琥珀亚胺(21.36g)于二颈烧瓶中,加入适量丙酮溶剂(溶剂淹没固体反应物0.5cm即可),加入5mmol(0.85g)AgNO
3)在500ml三颈烧瓶中,将40mmol化合物1,0.2g盐酸羟胺为碱,混合在0.6gCuCl的无水无氧催化体系中(采取的抽真空放氩气的真空装置,先抽后放,如此重复3次),再以100ml质量分数为30%正丁胺水溶液(63g水+27g正丁胺)作为溶剂,先倒入一半溶剂,再将步骤3)中制得的90mmol苯乙炔溴倒入,最后倒入剩余溶剂,冰浴下,磁力搅拌反应12小时,产物用饱和氯化钠溶液洗涤,用二氯甲烷萃取,收集有机相,减压旋干,柱层析(体积比为乙酸乙酯:石油醚=1:80)得到白色固体产物,即四炔类化合物,结构式见图5。
4)在110℃的条件下,将步骤3)制备的1mmol四炔类化合物在4mL甲苯中与1mmol的1,2,3,4,5,6-六[2-(三甲基硅烷基)乙炔基]苯反应12小时,反应液用饱和氯化钠水溶液和乙酸乙酯萃取,收集有机相,减压旋干,湿法装柱,用体积比为乙酸乙酯:石油醚=1:80柱层析分离,得到白色固体产物,即多取代稠环芳烃衍生物衍生物,柱层析产率约为78%。
所得产物白色粉末产物结构通过
1
13
上述制备的多取代稠环芳烃衍生物衍生物的光学性质,在波长极限为235nm的乙酸乙酯溶剂中测定浓度为7.10mg/ml的多取代稠环芳烃衍生物衍生化合物紫外光谱数据,见图8,250nm到400nm区间Abs可达1.5以上,说明该物质的吸光度良好,可较好的用于光学材料的制备。
上述参照实施例对一种多取代稠环芳烃衍生物衍生物及其制备方法进行的详细描述,是说明性的而不是限定性的,可按照所限定范围列举出若干个实施例,因此在不脱离本发明总体构思下的变化和修改,应属本发明的保护范围之内。
- 一种稠环芳烃加氢生产单环芳烃的多孔Pd复合膜的制备方法
- 一种多取代环庚三烯衍生物的制备方法
- 2,3-稠环吲哚啉衍生物及其制备方法和应用
- 稠杂环取代的苯并茚并芴衍生物及其应用
- 一种1,8-二取代萘系稠环芳烃单硝化衍生物的制备方法
- 一种吩嗪基团取代的稠环芳烃衍生物及其应用