二环吡啶衍生物
文献发布时间:2024-04-18 19:58:26
技术领域
本公开涉及对痕量胺相关受体TAAR1受体具有激动剂活性的二环吡啶衍生物或其药学上允许的盐、以及以该衍生物作为有效成分的精神神经疾病的治疗剂。
背景技术
作为生物体内胺的一种的痕量胺类(TA)包括p-酪胺、β-苯基乙胺、色胺、章鱼胺。与以5-羟色胺、多巴胺、去甲肾上腺素为首的传统生物体内胺类相比,其结构、代谢类似,但在生物体的生理条件下仅存在极微量(非专利文献1)。已知TA在中枢、外周神经系统中对神经传导调节起重要作用。表明TA的调节异常与包括精神分裂症、心境障碍、注意力缺陷多动性障碍、帕金森病、偏头痛、摄食障碍的各种中枢神经系统疾病相关,期待通过改善其调节异常,建立新的疾病治疗方法(非专利文献2)。
TA的受体在人类中报告了9个基因。TAAR1受体为G蛋白共轭型,以TA为配体通过Gαs活化。另外,也有其他胺类、儿茶酚胺代谢体、多巴胺激动剂等作为配体发挥作用的报告(非专利文献1、3)。在下游信号中,已知通过cAMP-PKA/PKC磷酸化多巴胺转运体,抑制多巴胺摄取,PKC依赖性地促进多巴胺释放(非专利文献4、5)。在中枢神经系统中,在包含腹侧被盖区、中缝核的单胺起始核、边缘系统中发现TAAR1受体的表达,还调节谷氨酸神经传递、5-羟色胺神经传递,因此提示TAAR1受体能够作用于各种单胺功能(非专利文献1)。
由于编码TAAR1的基因座6q23是与多种精神疾病相关的区域,并且TAAR1调节的单胺神经传递与精神神经疾病表现出强烈的关联性,因此期待TAAR1受体选择性配体对这些疾病治疗有效。另外,迄今为止报道的显示TAAR1受体激动性的低分子化合物在使用啮齿类的多个疾病模型中显示抗精神病作用、抗抑郁作用,在使用非人类灵长类的评价中也显示认知功能改善作用,因此可期待对人也显示抗精神病作用、抗抑郁作用、认知功能改善作用(非临床文献6)。
现有技术文献
非专利文献
非专利文献1:Beth Borowsky著Proc Natl Acad Sci USA.98,8966-8971,2001
非专利文献2:Branchek,T.A.and Blackburn,T.P.著Curr.Opin.Pharmacol.3,90-97,2003
非专利文献3:James R.Bunzow著Mol Pharmacol.60,1181-1188,2001
非专利文献4:Zhihua Xie and Gregory M.Miller著Journal ofPharmacologyand Experimental Therapeutics 321,128-136,2007
非专利文献5:Zhihua Xie and Gregory M.Miller著Journal ofPharmacologyand Experimental Therapeutics,330,316-325,2009
非专利文献6:FG Revel著Molecular Psychiatry,18,543-556,2013
发明内容
用于解决课题的手段
本公开提供对痕量胺相关受体TAAR1受体具有激动剂活性、作为精神神经疾病治疗剂有用的新型化合物。
本发明人发现下式I所示的化合物或其药学上允许的盐(以下有时称为“本公开的化合物”)对痕量胺相关受体TAAR1受体具有激动剂活性,并提供本公开。
即,本公开例如如下。
[项1]
式I所示的化合物或其药学上允许的盐,
[化1]
[式中,
X为氧原子、硫原子、NR或CR’R”,
n为0或1,
R
R
R
R
R
在此,
该任选被取代的C
该任选被取代的C
其中,不包括以下的(1)~(19):
(1)n为0,X为氧原子,R
(2)n为1,X为氧原子,R
(3)n为1,X为CH
(4)n为1,X为氧原子,R
(5)n为1,X为氧原子,R
(6)n为1,X为氧原子,R
(7)n为1,X为氧原子,R
(8)n为0,X为CH
(9)n为1,X为CH
(10)n为1,X为CH
(11)n为1,X为氧原子,R
(12)n为1,X为氧原子,R
(13)n为0,X为CH
(14)n为1,X为CH
(15)n为0,X为CH
(16)n为1,X为CH
(17)n为1,X为CH
(18)n为0或1,X为CH
(19)n为0,X为CH
[项2]
项1所述的化合物或其药学上允许的盐,其用下式表示
[化2]
[项3]
项1所述的化合物或其药学上允许的盐,其用下式表示,
[化3]
[项4]
项1~3中任一项所述的化合物或其药学上允许的盐,其中,R
[项5]
项1~3中任一项所述的化合物或其药学上允许的盐,其中,R
[项6]
项5所述的化合物或其药学上允许的盐,其中,R
[项7]
项1~6中任一项所述的化合物或其药学上允许的盐,其中,R
[项8]
项1~6中任一项所述的化合物或其药学上允许的盐,其中,R
[项9]
项1~6中任一项所述的化合物或其药学上允许的盐,其中,R
[项10]
项1~6中任一项所述的化合物或其药学上允许的盐,其中,R
[项11]
项1~6中任一项所述的化合物或其药学上允许的盐,其中,R
[项12]
项1~6中任一项所述的化合物或其药学上允许的盐,其中,R
[项13]
项1~6中任一项所述的化合物或其药学上允许的盐,其中,R
[项13-1]
项1~13中任一项所述的化合物或其药学上允许的盐,其中,X为CR’R”,R’和R”为氢原子。
[项13-2]
项1~13中任一项所述的化合物或其药学上允许的盐,其中,X为CR’R”,R’为氢原子,R”为任选被取代的C
[项13-3]
项1~13中任一项所述的化合物或其药学上允许的盐,其中,X为CR’R”,R’和R”为任选被取代的C
[项14]
项1、3~13-3中任一项所述的化合物或其药学上允许的盐,其中,n为1,R
[项15]
项1、3~13-3中任一项所述的化合物或其药学上允许的盐,其中,n为1,R
[项16]
项1、3~13-3中任一项所述的化合物或其药学上允许的盐,其中,n为1,R
[项17]
项1、3~13-3中任一项所述的化合物或其药学上允许的盐,其中,n为1,R
[项18]
项1、3~13-3中任一项所述的化合物或其药学上允许的盐,其中,n为1,R
[项19]
项1、3~13-3中任一项所述的化合物或其药学上允许的盐,其中,n为1,R
[项20]
项1、3~13-3中任一项所述的化合物或其药学上允许的盐,其中,R
[项21]
项1~20中任一项所述的化合物或其药学上允许的盐,其中,R
[项22]
项1~21中任一项所述的化合物或其药学上允许的盐,其中,R
[项22-1]
项1~21中任一项所述的化合物或其药学上允许的盐,其中,R
[项23]
项1~22-1中任一项所述的化合物或其药学上允许的盐,其中,R
[项24]
项1~22-1中任一项所述的化合物或其药学上允许的盐,其中,R
[项25]
项1~22-1中任一项所述的化合物或其药学上允许的盐,其中,R
[项26]
项1~25中任一项所述的化合物或其药学上允许的盐,其中,X为氧原子。
[项27]
项1~25中任一项所述的化合物或其药学上允许的盐,其中,X为硫原子。
[项28]
项1~25中任一项所述的化合物或其药学上允许的盐,其中,X为NR。
[项29]
项1~25中任一项所述的化合物或其药学上允许的盐,其中,X为CR’R”。
[项29-1]
项1~3、5、7~21、22-1和23~29中任一项所述的化合物或其药学上允许的盐,其中,R
R
[项29-2]
项29-1所述的化合物或其药学上允许的盐,其中,R
R
[项30]
项1所述的化合物或其药学上允许的盐,其中,
X为氧原子,
n为0,
R
R
R
R
[项31]
项1-30中任一项所述的化合物或其药学上允许的盐,其中,前述化合物的氢原子为重氢。
[项32]
选自以下的化合物组中的项1所述的化合物或其药学上允许的盐:
1-[(2R,3S)-6-氟-2-甲基-2,3-二氢呋喃并[3,2-b]吡啶-3-基]甲胺、
1-[(2R,3S)-2-甲基-2,3-二氢呋喃并[3,2-b]吡啶-3-基]甲胺、
N-甲基-1-[(2R,3S)-2-甲基-2,3-二氢呋喃并[3,2-b]吡啶-3-基]甲胺、
1-[(2R,3S)-6-氟-2-甲基-2,3-二氢呋喃并[3,2-b]吡啶-3-基]-N-甲基甲胺、
1-[(2R,3S)-6-氟-2-甲基-2,3-二氢呋喃并[3,2-b]吡啶-3-基]-N-(
rel-1-[(3’S)-5’-甲基-3’H-螺[环丙烷-1,2’-呋喃并[3,2-b]吡啶]-3’-基]甲胺、
rel-1-[(3’S)-5’-氟-3’H-螺[环丙烷-1,2’-呋喃并[3,2-b]吡啶]-3’-基]甲胺、
rel-1-[(3S)-2,2,5-三甲基-2,3-二氢呋喃并[3,2-b]吡啶-3-基]甲胺、
rel-1-[(3’S)-3’H-螺[环丙烷-1,2’-呋喃并[3,2-b]吡啶]-3’-基]甲胺、和rac-1-(5-氟-2,2-二甲基-2,3-二氢呋喃并[3,2-b]吡啶-3-基)甲胺。
[项33]
药物组合物,其含有项1~32中任一项所述的化合物或其药学上允许的盐。
[项34]
项33所述的药物组合物,其中,前述药物组合物用于治疗或预防与TAAR1相关的疾病或障碍。
[项35]
项33或34所述的药物组合物,其中,前述药物组合物用于治疗或预防神经或精神障碍。
[项36]
项35所述的药物组合物,其中,前述神经或精神障碍为抑郁症、双相情感障碍、疼痛、精神分裂症、强迫障碍、中毒、社交障碍、注意缺陷/多动障碍、焦虑障碍、运动障碍、癫痫、自闭症、认知功能障碍、阿尔茨海默氏病/帕金森病的精神病、帕金森病的焦躁/攻击性或贪食症。
[项37]
治疗或预防对象的与TAAR1相关的疾病或障碍的方法,其特征在于,将有效量的项1~32中任一项所述的化合物或其药学上允许的盐施与对象。
[项38]
治疗或预防对象的神经或精神障碍的方法,其特征在于,将有效量的项1~32中任一项所述的化合物或其药学上允许的盐施与对象。
[项39]
项38所述的方法,其中,前述神经或精神障碍为抑郁症、双相情感障碍、疼痛、精神分裂症、强迫障碍、中毒、社交障碍、注意缺陷/多动障碍、焦虑障碍、运动障碍、癫痫、自闭症、认知功能障碍、阿尔茨海默氏病/帕金森病的精神病、帕金森病的焦躁/攻击性或贪食症。
[项40]
项1~32中任一项所述的化合物或其药学上允许的盐,其用作药物。
[项41]
项40所述的化合物或其药学上允许的盐,其中,前述药物用于治疗或预防与TAAR1相关的疾病或障碍。
[项42]
项40或41所述的化合物或其药学上允许的盐,其中,前述化合物或其药学上允许的盐用于治疗或预防神经或精神障碍。
[项43]
项42所述的化合物或其药学上允许的盐,其中,前述神经或精神障碍为抑郁症、双相情感障碍、疼痛、精神分裂症、强迫障碍、中毒、社交障碍、注意缺陷/多动障碍、焦虑障碍、运动障碍、癫痫、自闭症、认知功能障碍、阿尔茨海默氏病/帕金森病的精神病、帕金森病的焦躁/攻击性或贪食症。
[项44]
TAAR1激动剂,其为项1~32中任一项所述的化合物或其药学上允许的盐。
[项45]
项1~32中任一项所述的化合物或其药学上允许的盐在TAAR1受体调节中的用途。
[项46]
精神疾病或中枢神经系统疾病的治疗剂,其含有项1~32中任一项所述的化合物或其药学上允许的盐作为有效成分。
[项47]
项46所述的治疗剂,其中,精神疾病或中枢神经系统疾病为包括症状性的器质性精神障碍;使用精神作用物质所导致的精神和行为障碍;精神分裂症,精神分裂症型障碍和妄想性障碍;心境[情感]障碍;神经症性障碍,应激相关障碍和躯体形式障碍;非器质性睡眠障碍;性功能不全,非由器质性障碍或疾病引起;广泛性发育障碍;通常发病于童年与少年期的行为与情绪障碍;锥体外系障碍和异常运动;神经系统的其他变性疾病;或睡眠障碍。
[项48]
项46所述的治疗剂,其中,精神疾病或中枢神经系统疾病为精神分裂症、精神分裂症的阳性症状、精神分裂症的阴性症状、伴随精神病性特征的双相情感障碍、伴随精神病性特征的抑郁障碍、伴随痴呆症的精神病症状、伴随阿尔茨海默氏病的精神病症状、伴随路易体痴呆的精神病症状、伴随帕金森病痴呆症的精神病症状、伴随帕金森病的精神病症状或伴随阿尔茨海默氏病的焦躁、兴奋或攻击性。
[项49]
项46所述的治疗剂,其中,精神疾病或中枢神经系统疾病为精神分裂症、伴随痴呆症的精神病症状、伴随阿尔茨海默氏病的精神病症状、伴随路易体痴呆的精神病症状或伴随阿尔茨海默氏病的焦躁、兴奋或攻击性。
[项50]
治疗精神疾病或中枢神经系统疾病的方法,其包括向需要治疗的患者施与治疗上有效量的项1~32中任一项所述的化合物或其药学上允许的盐。
[项51]
项1~32中任一项所述的化合物或其药学上允许的盐在制造精神疾病或中枢神经系统疾病的治疗剂中的用途。
[项52]
项1~32中任一项所述的化合物或其药学上允许的盐,其用于治疗精神疾病或中枢神经系统疾病。
[项53]
精神疾病或中枢神经系统疾病的治疗剂,其将项1~32中任一项所述的化合物或其药学上允许的盐与选自抗抑郁药、抗焦虑药、精神分裂症治疗药、多巴胺补充剂、多巴胺受体激动剂、帕金森病治疗药、抗癫痫药、镇痛药、激素制剂、偏头痛治疗药、肾上腺素β受体拮抗剂、痴呆症治疗药、心境障碍治疗药、止吐剂、睡眠导入剂和抗痉挛药中的至少一种药物组合而成。
[项54]
含有项1~32中任一项所述的化合物或其药学上允许的盐作为有效成分的治疗剂,其与选自抗抑郁药、抗焦虑药、精神分裂症治疗药、多巴胺补充剂、多巴胺受体激动剂、帕金森病治疗药、抗癫痫药、镇痛药、激素制剂、偏头痛治疗药、肾上腺素β受体拮抗剂、痴呆症治疗药、心境障碍治疗药、止吐剂、睡眠导入剂和抗痉挛药中的至少一种药物组合使用,用于治疗精神疾病或中枢神经系统疾病。
发明效果
本公开的化合物对TAAR1受体具有强的激动剂活性。另外,在优选实施方式中,对作为其他GPCR的多巴胺D2受体、肾上腺素α1受体、肾上腺素α2受体、进一步地hERG通道的抑制作用的选择性高。因此,本公开化合物中优选的化合物可用作安全性高的精神神经疾病治疗剂。
附图说明
[图1]显示实施例27的化合物的苯环利定诱发运动亢进抑制试验(试验例2-1)结果的图。
[图2]显示实施例8的化合物的苯环利定诱发运动亢进抑制试验(试验例2-2)结果的图。
[图3]显示实施例49的化合物的苯环利定诱发运动亢进抑制试验(试验例2-2)结果的图。
具体实施方式
以下,说明本公开的最佳方式。应理解,在本说明书全篇中,除非另外指出,单数形式的表述还包括其复数形式的概念。因此,应理解,除非另外指出,单数冠词(例如,在英语中为“a”、“an”、“the”等)也包括其复数形式的概念。此外,应理解,除非另外指出,本说明书中使用的术语通常以该领域中通常采用的含义使用。因此,除非另有定义,本说明书中使用的所有专业术语和科技术语具有与本公开所属领域的技术人员通常理解的相同的含义。在矛盾的情况下,本说明书(包括定义)优先。
以下适当说明本说明书中特别使用的术语的定义和/或基本技术内容。
本说明书中“或”在采用文章中列举的事项的“至少1个以上”时使用。“或者”也同样。本说明书中,明确记载2个值的“范围内”的情况下,该范围也包括这两个值本身。
“任选被取代的”或“取代的”所定义的基团中的取代基的数量只要可取代即没有限制。另外,除特别指出的情况外,各基团的说明也适用于该基团为其他基团的一部分或取代基的情况。
在本说明书中,也有将“取代基”的定义中的碳的数量标记为例如“C
作为“卤素”,可列举氟、氯、溴和碘。
“C
“C
“C
“3~6元饱和杂环”是指除碳原子以外还含有独立地选自氮原子、氧原子和硫原子中的1~2个原子的由3~6个原子构成的饱和环,包括部分具有不饱和键的杂环和交联结构的杂环。作为“3~6元饱和杂环”,优选列举“4~6元单环饱和杂环”,更优选列举“5或6元单环饱和杂环”。作为“5元或6元单环饱和杂环”的具体例,可列举例如四氢呋喃基、吡咯烷基、咪唑烷基、哌啶基、吗啉基、硫代吗琳基、二氧代硫代吗啉基、六亚甲基亚胺基(hexamethyleneiminyl)、噁唑烷基、噻唑烷基、氧代咪唑烷基、二氧代咪唑烷基、氧代噁唑烷基、二氧代噁唑烷基、二氧代噻唑烷基、四氢呋喃基四氢吡喃基等。作为“4~6元单环饱和杂环”,例如除了作为前述“5元或6元单环饱和杂环”的具体例列举的之外,还可列举氧杂环丁烷基、氮杂环丁烷基等。
“3~6元饱和碳环”是指碳原子数3~6的环状饱和烃,包括部分具有不饱和键的碳环和交联结构的碳环。作为“3~6元饱和碳环”,优选列举“5或6元单环饱和碳环”。作为“5或6元单环饱和碳环”的具体例,可列举例如环戊烷、环己烷等。作为“3~6元饱和碳环”的具体例,例如除了作为前述“5或6元单环饱和碳环”的具体例列举的之外,还可列举环丙烷、环丁烷等。
“C
作为任选被取代的C
作为任选被取代的C
式I表示的本公开的化合物中,X、n、R
作为优选的实施方式,可列举n为0的实施方式。
作为优选的实施方式,可列举n为1的实施方式。
作为优选的实施方式,可列举R
作为优选的实施方式,可列举R
作为优选的实施方式,可列举R
作为优选的实施方式,可列举R
作为优选的实施方式,可列举R
作为优选的实施方式,可列举R
作为优选的实施方式,可列举R
作为优选的实施方式,可列举R
作为优选的实施方式,可列举R
作为优选的实施方式,可列举R
作为优选的实施方式,可列举n为1,R
作为优选的实施方式,可列举n为1,R
作为优选的实施方式,可列举n为1,R
作为优选的实施方式,可列举n为1,R
作为优选的实施方式,可列举n为1,R
作为优选的实施方式,可列举n为1,R
作为优选的实施方式,可列举R
作为优选的实施方式,可列举R
作为优选的实施方式,可列举R
作为优选的实施方式,可列举R
作为优选的实施方式,可列举R
作为优选的实施方式,可列举R
作为优选的实施方式,可列举R
作为优选的实施方式,可列举R
作为优选的实施方式,可列举R
作为优选的实施方式,可列举X为氧原子的实施方式。
作为优选的实施方式,可列举X为硫原子的实施方式。
作为优选的实施方式,可列举X为NR的实施方式。
作为优选的实施方式,可列举X为CR’R”的实施方式。
作为优选的实施方式,可列举R
作为优选的实施方式,可列举R
作为优选的实施方式,可列举X为氧原子,n为0,R
式I表示的化合物也可作为互变异构体存在。因此,本公开的化合物也包括式I表示的化合物的互变异构体。
式I表示的化合物有时具有至少一个不对称碳原子。因此,本公开的化合物不仅包括式I表示的化合物的外消旋体,还包括这些化合物的光学活性体。当式I表示的化合物具有2个以上不对称碳原子时,有时产生立体异构。因此,本公开的化合物还包括这些化合物的立体异构体及其混合物。结构式中,“rac.”的记载表示外消旋体,“chiral”的记载表示光学活性体,“abs.”的记载表示绝对构型。
另外,将式I表示的化合物中的任意1个或2个以上的原子变换为同位素的衍生物也包含在式I表示的化合物中。
例如,将
式I表示的化合物和其药学上允许的盐有时以水合物和/或溶剂化物的形式存在,因此这些水合物或乙醇溶剂化物等溶剂化物也包含在本公开的化合物中。此外,本公开的化合物还包括所有形态的晶型的化合物。
作为药学上允许的盐,式I表示的化合物具有酸性基团时,例如可以举出钠盐、钾盐等碱金属盐;钙盐、镁盐等碱土金属盐;锌盐等无机金属盐;三乙胺、三乙醇胺、三羟基甲基氨基甲烷、氨基酸等有机碱盐等。
式I表示的化合物具有碱性基团时,例如可以举出盐酸盐、氢溴酸盐、硫酸盐、磷酸盐、硝酸盐等无机酸盐;和乙酸盐、丙酸盐、琥珀酸盐、乳酸盐、苹果酸盐、酒石酸盐、柠檬酸盐、马来酸盐、富马酸盐、甲磺酸盐、对甲苯磺酸盐、苯磺酸盐、抗坏血酸盐等有机酸盐等。
以下,举例说明本公开的化合物的制造方法,但本公开当然不限于此。
制造方法
本公开的化合物通过组合下述制造方法和公知的合成方法的方法合成。
也包含反应式中的化合物分别形成盐的情况,作为该盐,例如可以举出与式I表示的化合物的盐相同的盐。另外,这些反应只是单纯的例示,根据熟悉有机合成者的知识,也可以适当地用其他方法制造本公开的化合物。
在下述说明的各制造方法中,即使在没有具体明示保护基的使用的情况下,在存在需要保护的官能团的情况下,也可以根据需要保护该官能团,在反应结束后或进行一系列反应后进行脱保护,从而得到目标物。
作为保护基,可使用文献(T.W.Greene and P.G.M.Wuts,“Protective Groups inOrganic Synthesis”,3rd Ed.,John Wiley and Sons,inc.,New York(1999))等中记载的常规的保护基。更具体地,作为氨基的保护基,例如可以举出苄氧基羰基、叔丁氧基羰基、乙酰基、苄基等。另外,作为羟基的保护,例如可以举出三烷基甲硅烷基、乙酰基、苄基等。
保护基的导入和脱离可以通过有机合成化学中常用的方法(例如T.W.Greene andP.G.M.Wuts,“Protective Groups in Organic Synthesis”,3rd Ed.,John Wiley andSons,inc.,New York(1999)中记载的方法等)或以此为基准的方法进行。
制造方法1
在式I表示的化合物中,式(1a)表示的化合物例如通过下述方法制造。
[化4]
〔式中,n、R
在PG
作为非活性溶剂的具体例,例如可以举出氯仿、二氯甲烷等的卤代烃;苯、甲苯等芳香族烃;乙醚、四氢呋喃、1,4-二噁烷、1,2-二甲氧基乙烷等醚系溶剂;甲醇、乙醇、2-丙醇等低级醇;乙腈、二甲基甲酰胺、N-甲基-2-吡咯烷酮、二甲基亚砜等非质子性极性溶剂;和它们的混合溶剂等。
作为酸的具体例,例如可以举出盐酸、硫酸等无机酸、三氟乙酸等有机酸等。
在PG
作为非活性溶剂的具体例,例如可以举出乙酸乙酯等酯系溶剂;苯、甲苯等芳香族烃;乙醚、四氢呋喃(THF)、1,4-二噁烷、1,2-二甲氧基乙烷等醚系溶剂;甲醇、乙醇、2-丙醇等醇系溶剂;二甲基甲酰胺、N-甲基-2-吡咯烷酮、二甲基亚砜等非质子性极性溶剂;和它们的混合溶剂等。
化合物(5)使化合物(4)在适当非活性溶剂中、在适当的碱存在下与叠氮磷酸二苯酯在室温下反应1小时左右,在50℃~100℃的适当温度下加热搅拌1小时左右后,通过与苯甲醇、叔丁醇等醇在50℃~100℃的适当温度下反应而制造。
另外,化合物(5)使化合物(4)在适当非活性溶剂中、在适当的碱存在下与叠氮磷酸二苯酯在室温下反应1小时左右,在50℃~100℃的适当温度下加热搅拌1小时左右后,与氢氧化钠、氢氧化钾等碱在水存在下反应后,根据需要在适当碱的存在下与二碳酸二叔丁基酯反应也可制造。
反应时间根据反应温度、使用的碱、醇、原料和溶剂等条件而不同,通常为10分钟~48小时。
作为碱的具体例,可列举例如三乙胺、二异丙基乙胺、吡啶等有机碱;碳酸钾、碳酸钠、碳酸铯、碳酸氢钾、碳酸氢钠、磷酸二氢钾、磷酸氢二钾、磷酸钾、磷酸二氢钠、磷酸氢二钠、磷酸钠、氢氧化钾、氢氧化钠、氢化钠等无机碱;甲醇钠、叔丁醇钾等金属醇盐等。
作为非活性溶剂的具体例,例如可以举出氯仿、二氯甲烷等的卤代烃;苯、甲苯等芳香族烃;乙醚、四氢呋喃(THF)、1,4-二噁烷等醚系溶剂;乙腈、丙酮、甲乙酮、二甲基甲酰胺、N-甲基-2-吡咯烷酮、二甲基亚砜等非质子性极性溶剂;和它们的混合溶剂等。
化合物(4)是通过在适当的溶剂中用氢氧化钾、氢氧化钠、氢化钠、氢氧化锂等碱水解化合物(3)而制造的。处理温度通常为约-20℃至所用溶剂沸点的范围的温度。反应时间根据反应温度、使用的碱、原料和溶剂等条件而不同,通常为10分钟~48小时。
作为溶剂的具体例,例如可以举出甲醇、乙醇、2-丙醇等低级醇、水;和它们的混合溶剂等。
化合物(3)通过在适当的非活性溶剂中使化合物(2)在催化剂量的偶氮二异丁腈存在下与三丁基氢化锡反应而制造。反应温度通常为50℃至溶剂沸点范围的温度。反应时间根据反应温度、使用的原料和溶剂等条件而不同,通常为10分钟~48小时。
作为非活性溶剂的具体例,可以举出例如苯、甲苯等芳香族烃;乙醚、四氢呋喃(THF)、1,4-二噁烷等醚系溶剂;和它们的混合溶剂等。
制造方法2
式I所示的化合物中,式(1b)表示的化合物例如通过下述所示的方法制造。
[化5]
〔式中,n、R
在PG
作为非活性溶剂的具体例,例如可以举出氯仿、二氯甲烷等的卤代烃;苯、甲苯等芳香族烃;乙醚、四氢呋喃、1,4-二噁烷、1,2-二甲氧基乙烷等醚系溶剂;甲醇、乙醇、2-丙醇等低级醇;乙腈、二甲基甲酰胺、N-甲基-2-吡咯烷酮、二甲基亚砜等非质子性极性溶剂;和它们的混合溶剂等。
作为酸的具体例,例如可以举出盐酸、硫酸等无机酸、三氟乙酸等有机酸等。
在PG
作为非活性溶剂的具体例,例如可以举出乙酸乙酯等酯系溶剂;苯、甲苯等芳香族烃;乙醚、四氢呋喃(THF)、1,4-二噁烷、1,2-二甲氧基乙烷等醚系溶剂;甲醇、乙醇、2-丙醇等醇系溶剂;二甲基甲酰胺、N-甲基-2-吡咯烷酮、二甲基亚砜等非质子性极性溶剂;和它们的混合溶剂等。
化合物(6)通过使化合物(5)在适当的碱的存在下,在适当的非活性溶剂中与碘甲烷等烷基化剂反应而制造。该反应可以根据需要在相转移催化剂的存在下进行。反应温度通常为约-20℃至所用溶剂沸点的范围的温度。反应时间根据反应温度、使用的碱、原料和溶剂等条件而不同,通常为10分钟~48小时。
碱的具体例可列举例如三乙胺、二异丙基乙胺、吡啶等有机碱;碳酸钾、碳酸钠、碳酸铯、碳酸氢钾、碳酸氢钠、磷酸二氢钾、磷酸氢二钾、磷酸钾、磷酸二氢钠、磷酸氢二钠、磷酸钠、氢氧化钾、氢氧化钠、氢化钠等无机碱;甲醇钠、叔丁醇钾等金属醇盐等。
作为相转移催化剂的具体例,例如可以举出四丁基硫酸氢铵等。
作为非活性溶剂的具体例,例如可以举出氯仿、二氯甲烷等的卤代烃;苯、甲苯等芳香族烃;乙醚、四氢呋喃(THF)、1,4-二噁烷等醚系溶剂;甲醇、乙醇、2-丙醇等低级醇;乙腈、丙酮、甲乙酮、二甲基甲酰胺、N-甲基-2-吡咯烷酮、二甲基亚砜等非质子性极性溶剂;和它们的混合溶剂等。
制造方法3
式(2)所示的化合物例如通过下述方法制造。
[化6]
〔式中,n、R
化合物(2)通过使化合物(8)在适当的非活性溶剂中与乙基(三苯基膦)乙酸酯等(三苯基膦)乙酸酯反应而制造。另外,化合物(2)也可以在适当的碱的存在下通过与乙氧甲酰基甲基三苯基溴化膦等鏻盐、二乙基膦酰基乙酸乙酯等二烷基膦酰基乙酸酯反应来制造。反应温度通常为约-20℃至所用溶剂沸点的范围的温度。反应时间根据反应温度、使用的碱、原料和溶剂等条件而不同,通常为10分钟~48小时。
作为碱的具体例,例如可列举三乙胺、二异丙基乙胺、吡啶等的有机碱;碳酸钾、碳酸钠、碳酸铯、碳酸氢钾、碳酸氢钠、磷酸二氢钾、磷酸氢二钾、磷酸钾、磷酸二氢钠、磷酸氢二钠、磷酸钠、氢氧化钾、氢氧化钠、氢化钠等的无机碱;甲醇钠、叔丁醇钾等金属醇盐等。
作为非活性溶剂的具体例,例如可以举出氯仿、二氯甲烷等的卤代烃;苯、甲苯等芳香族烃;乙醚、四氢呋喃(THF)、1,4-二噁烷等醚系溶剂;乙腈、丙酮、甲乙酮、二甲基甲酰胺、N-甲基-2-吡咯烷酮、二甲基亚砜等非质子性极性溶剂;和它们的混合溶剂等。
化合物(8)通过使化合物(7)在适当的非活性溶剂中与戴斯-马丁氧化剂(Dess-Martin Periodinane)等氧化剂反应而制造。此外,化合物(8)也可以通过在二甲基亚砜中使化合物(7)在三乙胺等三级烷基胺的存在下与三氧化硫吡啶络合物反应来制造。反应温度通常为约-20℃至所用溶剂沸点的范围的温度。反应时间根据反应温度、使用的原料和溶剂等条件而不同,通常为10分钟~48小时。
作为非活性溶剂的具体例,例如可以举出氯仿、二氯甲烷等的卤代烃;苯、甲苯等芳香族烃;乙醚、四氢呋喃(THF)、1,4-二噁烷等醚系溶剂;乙腈、丙酮、甲乙酮、二甲基甲酰胺、N-甲基-2-吡咯烷酮、二甲基亚砜等非质子性极性溶剂;和它们的混合溶剂等。
制造方法4
在式(2)所示的化合物中,式(2a)表示的化合物例如通过下述方法制造。
[化7]
〔式中,n、R
化合物(2a)通过在适当的非活性溶剂中使化合物(9)和式(10)表示的醇在三苯基膦和偶氮二甲酸二异丙酯等偶氮二甲酸酯的存在下反应而制造。反应温度通常为约-20℃至所用溶剂沸点的范围的温度。反应时间根据反应温度、使用的原料和溶剂等条件而不同,通常为10分钟~48小时。
作为非活性溶剂的具体例,例如可以举出氯仿、二氯甲烷等的卤代烃;苯、甲苯等芳香族烃;乙醚、四氢呋喃(THF)、1,4-二噁烷等醚系溶剂;乙腈、丙酮、甲乙酮、二甲基甲酰胺、N-甲基-2-吡咯烷酮、二甲基亚砜等非质子性极性溶剂;和它们的混合溶剂等。
制造方法5
式(7)表示的化合物中,式(7a)表示的化合物例如通过下述方法制造。
[化8]
〔式中,n、R
化合物(7a)通过在适当的非活性溶剂中使化合物(9)和式(11)表示的醇在三苯基膦和偶氮二甲酸二异丙酯等偶氮二甲酸酯的存在下反应,然后用适当的酸处理而制造。反应温度通常为约-20℃至所用溶剂沸点的范围的温度。反应时间根据反应温度、使用的原料和溶剂等条件而不同,通常为10分钟~48小时。
作为非活性溶剂的具体例,例如可以举出氯仿、二氯甲烷等的卤代烃;苯、甲苯等芳香族烃;乙醚、四氢呋喃(THF)、1,4-二噁烷等醚系溶剂;乙腈、丙酮、甲乙基酮、二甲基甲酰胺、N-甲基-2-吡咯烷酮、二甲基亚砜等非质子性极性溶剂;和它们的混合溶剂等。
作为酸的具体例,例如可以举出盐酸、硫酸等无机酸、三氟乙酸等有机酸等。
化合物(7a)通过使化合物(13)在适当的非活性溶剂中与适当的还原剂反应而制造。反应温度通常为约-20℃至所用溶剂沸点的范围的温度。反应时间根据反应温度、使用的原料和溶剂等条件而不同,通常为10分钟~48小时。
作为还原剂的具体例,例如可以举出氢化锂铝、硼氢化钠、氰基硼氢化钠、二异丁基氢化铝等复合氢化合物;硼烷络合物(硼烷-二甲基硫醚络合物或硼烷-四氢呋喃络合物等)等。
作为非活性溶剂的具体例,例如可以举出氯仿、二氯甲烷等的卤代烃;苯、甲苯等芳香族烃;乙醚、四氢呋喃(THF)、1,4-二噁烷等醚系溶剂;和它们的混合溶剂等。
化合物(13)通过在适当的非活性溶剂中使化合物(9)和式(12)表示的烷基化剂在适当的碱存在下反应而制造。该反应可以根据需要在相转移催化剂的存在下进行。反应温度通常为约-20℃至所用溶剂沸点的范围的温度。反应时间根据反应温度、使用的碱、原料和溶剂等条件而不同,通常为10分钟~48小时。
作为碱的具体例,例如可举出三乙胺、二异丙基乙胺、吡啶等的有机碱;碳酸钾、碳酸钠、碳酸铯、碳酸氢钾、碳酸氢钠、磷酸二氢钾、磷酸氢二钾、磷酸钾、磷酸二氢钠、磷酸氢二钠、磷酸钠、氢氧化钾、氢氧化钠、氢化钠等的无机碱;甲醇钠、叔丁醇钾等金属醇盐等。
作为相转移催化剂的具体例,例如可以举出四丁基硫酸氢铵等。
作为非活性溶剂的具体例,例如可以举出氯仿、二氯甲烷等的卤代烃;苯、甲苯等芳香族烃;乙醚、四氢呋喃(THF)、1,4-二噁烷等醚系溶剂;甲醇、乙醇、2-丙醇等低级醇;乙腈、丙酮、甲乙酮、二甲基甲酰胺、N-甲基-2-吡咯烷酮、二甲基亚砜等非质子性极性溶剂;和它们的混合溶剂等。
制造方法6
式(7)所示的化合物中,式(7b)表示的化合物例如通过下述方法制造。
[化9]
〔式中,n、R
化合物(7b)通过使化合物(14)和式(15)表示的硫醇在适当的碱存在下在适当的非活性溶剂中反应而制造。该反应可以根据需要在相转移催化剂的存在下进行。反应温度通常为约-20℃至所用溶剂沸点的范围的温度。反应时间根据反应温度、使用的碱、原料和溶剂等条件而不同,通常为10分钟~48小时。
作为碱的具体例,可列举例如三乙胺、二异丙基乙胺、吡啶等的有机碱;碳酸钾、碳酸钠、碳酸铯、碳酸氢钾、碳酸氢钠、磷酸二氢钾、磷酸氢二钾、磷酸钾、磷酸二氢钠、磷酸氢二钠、磷酸钠、氢氧化钾、氢氧化钠、氢化钠等的无机碱;甲醇钠、叔丁醇钾等金属醇盐等。
作为相转移催化剂的具体例,例如可以举出四丁基硫酸氢铵等。
作为非活性溶剂的具体例,例如可以举出氯仿、二氯甲烷等的卤代烃;苯、甲苯等芳香族烃;乙醚、四氢呋喃(THF)、1,4-二噁烷等醚系溶剂;甲醇、乙醇、2-丙醇等低级醇;乙腈、丙酮、甲乙基酮、二甲基甲酰胺、N-甲基-2-吡咯烷酮、二甲基亚砜非质子性极性溶剂;和它们的混合溶剂等。
制造方法7
式I所示的化合物中,式(1c)所示的化合物例如通过下述方法制造。
[化10]
〔式中,n、R
化合物(1c)通过使化合物(16)在适当的非活性溶剂中与适当的还原剂反应而制造。反应温度通常为约-20℃至所用溶剂沸点的范围的温度。反应时间根据反应温度、使用的原料和溶剂等条件而不同,通常为10分钟~48小时。
作为还原剂的具体例,例如可以举出氢化锂铝、硼氢化钠、氰基硼氢化钠、二异丁基氢化铝等复合氢化合物;硼烷络合物(硼烷-二甲基硫醚络合物或硼烷-四氢呋喃络合物等)等。
作为非活性溶剂的具体例,例如可以举出氯仿、二氯甲烷等的卤代烃;苯、甲苯等芳香族烃;乙醚、四氢呋喃(THF)、1,4-二噁烷等醚系溶剂;和它们的混合溶剂等。
通过适当组合上述制造方法,可以得到在所希望的位置具有所希望的取代基的本公开化合物。上述制造方法中的中间体和产物的分离、纯化可以适当组合通常的有机合成中使用的方法,例如过滤、萃取、洗涤、干燥、浓缩、结晶、各种色谱等来进行。另外,中间体也可以不特别纯化而供于下一步反应。
上述制造方法中的原料化合物或中间体根据反应条件等例如也有能够以盐酸盐等盐的形态存在的物质,可直接或以游离的形式使用。原料化合物或中间体以盐的形态得到,在想以游离的形式使用或取得原料化合物或中间体的情况下,可以将它们溶解或悬浮在适当的溶剂中,例如通过用碳酸氢钠水溶液等碱等中和,转换为游离的形式。
在式I所示的化合物或其药学上允许的盐中,有时存在如酮烯醇体的互变异构体、位置异构体、几何异构体或光学异构体等异构体,包含它们在内的所有可能的异构体和该异构体的任意比例的混合物也包含在本公开中。
另外,光学异构体可以在上述制造方法的适当工序中,通过实施使用光学活性柱的方法、分级结晶法等公知的分离工序来分离。另外,作为起始原料也可以使用光学活性体。
想要取得式I表示的化合物的盐时,在得到式I表示的化合物的盐的情况下直接纯化即可,另外,在式I表示的化合物以游离的形式得到的情况下,将式I表示的化合物溶解或悬浮在适当的溶剂中,加入酸或碱形成盐即可。
本公开的化合物对痕量胺相关受体TAAR1受体具有激动剂活性,具有与现有的精神疾病治疗药物不同的作用机理,因此可以对各种精神疾病提供药物治疗的新选择。即,本公开的化合物对精神疾病的治疗有效。另外,本公开的化合物对中枢神经系统疾病也有效。
作为被期待有效性的精神疾病或中枢神经系统疾病,可列举例如国际疾病分类第10版(ICD-10)中的F00-F09:包括症状性的器质性精神障碍,F10-F19:使用精神作用物质所导致的精神和行为障碍,F20-F29:精神分裂症,精神分裂症型障碍和妄想性障碍,F30-F39:心境[情感]障碍,F40-F48:神经症性障碍,应激相关障碍和躯体形式障碍,F51:非器质性睡眠障碍,F52:性功能不全,非由器质性障碍或疾病引起,F84:广泛性发育障碍,F90-F98:通常发病于童年与少年期的行为与情绪障碍,G20-G26:锥体外系障碍和异常运动,G30-G32:神经系统的其他变性疾病,G47:睡眠障碍等。
F00-F09:作为包括症状性的器质性精神障碍的具体例,可列举例如阿尔茨海默氏病的痴呆症血管性痴呆症、路易体痴呆、帕金森病的痴呆症、伴随脑损伤等疾病的精神障碍、脑功能不全和身体疾病导致的其它精神障碍等。
F10-F19:作为使用精神作用物质所导致的精神和行为障碍的具体例,可列举例如使用各种物质导致的震颤性谵妄、精神病性障碍、健忘症候群等。
F20-F29:作为精神分裂症,精神分裂症型障碍和妄想性障碍的具体例,可列举例如妄想型精神分裂症、单纯性精神分裂症、妄想性障碍等。
F30-F39:作为心境[情感]障碍的具体例,可列举例如躁狂发作、双相情感障碍、抑郁发作等。
F40-F48:作为神经症性障碍,应激相关障碍和躯体形式障碍的具体例,可列举例如恐怖性焦虑障碍、强迫障碍、躯体形式障碍等。
F51:作为非器质性睡眠障碍的具体例,可列举例如非器质性失眠症、梦游症、噩梦等。
F52:作为性功能不全,非由器质性障碍或疾病引起的具体例,可列举例如性欲缺乏或性欲丧失、详情不明的性功能障碍等。
F84:作为广泛性发育障碍的具体例,可列举例如自闭症、精神发育迟缓和刻板动作相关的多动障碍等。
F90-F98:作为通常发病于童年与少年期的行为与情绪障碍的具体例,可列举例如多动障碍、行为障碍、行为和情绪的混合性障碍等。
G20-G26:作为锥体外系障碍和异常运动的具体例,可列举例如帕金森病、继发性帕金森氏综合征、运动障碍、脊髓小脑变性症等。
G30-G32:作为神经系统的其他变性疾病的具体例,例如可以举出阿尔茨海默氏病、额颞叶痴呆症、额颞叶变性、路易体痴呆、老人性脑变性、进行性核上性麻痹等。
G47:作为睡眠障碍的具体例,可列举例如入睡和维持睡眠障碍[失眠症]、睡眠觉醒节律障碍、发作性睡病和猝倒等。
本公开的化合物也可用于伴随这些疾病的各种症状(精神病症状、不安、攻击性、易刺激性和易怒性、睡眠障碍、抑郁症状、焦虑症状、认知功能障碍等)的治疗或复发的防止。
作为被期待有效性的精神疾病或中枢神经系统疾病,优选列举精神分裂症、精神分裂症的阳性症状、精神分裂症的阴性症状、伴随精神病性特征的双相情感障碍、伴随精神病性特征的抑郁障碍、伴随痴呆症的精神病症状、伴随阿尔茨海默氏病的精神病症状、伴随路易体痴呆的精神病症状,伴随帕金森病痴呆症的精神病症状,伴随帕金森病的精神病症状,或伴随阿尔茨海默氏病的焦躁、兴奋或攻击性,更优选精神分裂症、伴随痴呆症的精神病症状,伴随阿尔茨海默氏病的精神病症状与伴随路易体痴呆的精神病症状或伴随阿尔茨海默氏病的焦躁、兴奋或攻击性。
本公开的化合物对痕量胺相关受体TAAR1受体显示激动剂活性(试验例1)。在本公开的化合物的优选实施方案中,由于作为QT延长引起的心律失常的表达指标的hERG通道抑制活性弱(试验例3),因此可以期待对心血管系统的影响小。即,药理作用的表达浓度和副作用的表达浓度背离。
本公开的化合物可以经口或非经口施与。经口施与时,可以以通常使用的施与方式施与。非经口性地,可以以局部施与剂、注射剂、经皮剂、经鼻剂等的形式施与。作为经口剂或直肠施与剂,例如可以举出胶囊、片剂、丸剂、散剂、扁囊剂、栓剂、液体制剂等。作为注射剂,例如可以举出无菌的溶液或混悬液等。作为局部施与剂,例如可以举出霜剂、软膏、洗剂、经皮剂(通常的贴剂、基质剂)等。
上述剂型以通常的方法与药学上允许的赋形剂、添加剂一起配制。作为药学上允许的赋形剂、添加剂,可列举载体、粘合剂、香料、缓冲剂、增稠剂、着色剂、稳定剂、乳化剂、分散剂、悬浮化剂、防腐剂等。
作为药学上允许的载体,可举出例如碳酸镁、硬脂酸镁、滑石、砂糖、乳糖、果胶、糊精、淀粉、明胶、黄蓍胶、甲基纤维素、羧甲基纤维素钠、低熔点蜡、可可脂等。胶囊可以通过将本公开的化合物与药学上允许的载体一起加入其中来制剂化。本公开的化合物可与药学上允许的赋形剂一起混合或不含赋形剂地放入胶囊中。扁囊剂也可以用同样的方法制造。
作为注射用液体制剂,可以举出溶液、悬浮液、乳剂等。例如可以举出水溶液、水-丙二醇溶液等。液体制剂也能够以可以含水的聚乙二醇或/和丙二醇的溶液的形式制造。适合经口施与的液体制剂可以将本公开的化合物加入水中,根据需要添加着色剂、香料、稳定剂、甜味剂、增溶剂、增粘剂等来制造。另外,适合经口施与的液体制剂也可以通过将本公开的化合物与分散剂一起加入水中,使其粘稠来制造。增稠剂可列举药学上允许的天然或合成胶、树脂、甲基纤维素、羧甲基纤维素钠或已知的悬浮化剂等。
剂量根据各个化合物以及患者的疾病、年龄、体重、性别、症状、施与途径等而变化,但通常对于成人(体重50kg),将本公开的化合物以0.1~1000mg/天、优选1~300mg/天1天1次或分成2~3次施与。另外,也可以每隔几天~几周施与1次。
本公开的化合物可与其它药物组合使用,以增强其效果和/或减轻副作用。以下,可以与本公开化合物组合使用的药物简称为组合使用药剂。
作为组合使用药剂的具体例,可列举例如抗抑郁药、抗焦虑药、精神分裂症治疗药、多巴胺补充剂、多巴胺受体激动剂、帕金森病治疗药、抗癫痫药、镇痛药、激素制剂、偏头痛治疗药、肾上腺素β受体拮抗剂、痴呆症治疗药、心境障碍治疗药、止吐剂、睡眠导入剂、抗痉挛药等。作为组合使用药剂,优选列举选择性5-羟色胺再摄取抑制剂等抗焦虑药。
本公开的化合物和组合使用药剂的施与期间没有限定,可以将它们对施与对象同时施与,也可以隔时间差施与。另外,也可以制成本公开化合物和组合使用药剂的合剂。组合使用药剂的施与量可以以临床上使用的剂量为基准适当选择。另外,本公开化合物与组合使用药剂的配合比可以根据施与对象、施与途径、对象疾病、症状、组合等适当选择。例如施与对象为人时,相对于1重量份本公开化合物,使用0.01~100重量份组合使用药剂即可。另外,为了抑制其副作用,可以与止吐剂、睡眠导入剂、抗痉挛药等药剂(组合使用药剂)组合使用。
实施例
以下,通过参考例、实施例和试验例更具体地说明本公开,但当然本公开不限于上述说明,也不限于以下的参考例、实施例和试验例等。因此,本公开的范围不限于本说明书中具体记载的实施方式和实施例,仅由权利要求书限定。另外,以下的参考例和实施例中所示的化合物名未必遵循IUPAC命名法。另外,化合物的鉴定使用质子核磁共振吸收光谱(
[表1]
本说明书有时使用以下的缩写。
Me:甲基
Et:乙基
DMF:N,N-二甲基甲酰胺
THF:四氢呋喃
tert-:叔
TBS:ter叔丁基二甲基甲硅烷基
CDCl
DMSO-D
CD
质子核磁共振谱使用JEOL公司生产的FT-NMR测定装置(400MHz)测定。化学位移值记为δ值(ppm)。作为NMR中使用的符号,s表示单峰,d表示双峰,dd表示双双重峰,dt表示双三重峰,t表示三重峰,q表示四重峰,m表示多重峰,br表示宽峰,brs表示宽的单峰,和J表示耦合常数。
实施例1
rac-1-[(3R,4S)-3-甲基-3,4-二氢-2H-吡喃并[3,2-b]吡啶-4-基]甲胺二盐酸盐
[化11]
向参考例1-1的化合物(461mg)、甲醇(4.4mL)和水(2.2mL)的混合物中在室温下加入氢氧化钠(235mg)。在60℃搅拌5小时后,加入3mol/L盐酸直至水层达到pH5。浓缩反应溶液后,将浓缩残渣溶解在甲醇中,过滤不溶物并浓缩。
向得到的浓缩残渣(406mg)的甲苯溶液(28mL)中在室温下加入三乙胺(0.819mL)和叠氮磷酸二苯酯(0.842mL)。在室温下搅拌30分钟后,将反应溶液升温至100℃。在100℃搅拌1小时后,浓缩反应溶液。向浓缩残渣和四氢呋喃(20mL)的混合物中在冰冷却下加入5mol/L氢氧化钠水溶液(6.66mL)。在室温搅拌3小时后,向反应混合物中加入二碳酸二叔丁酯(1.28g)。在室温下搅拌15小时后,向反应混合物中加入水(20mL),用氯仿(20mL×3次)萃取,用无水硫酸镁干燥后,过滤并浓缩。将浓缩残渣用硅胶柱色谱(己烷/乙酸乙酯)分离纯化。
向得到的纯化物(31mg)和乙酸乙酯(1.0mL)的混合物中加入4mol/L氯化氢-乙酸乙酯(1.0mL),在室温下搅拌2小时。然后,滤取析出的固体,用乙醚(1.0mL)洗涤,通过减压干燥得到标题化合物(26mg)。
1
实施例2
rac-1-[(3S,4S)-3-甲基-3,4-二氢-2H-吡喃并[3,2-b]吡啶-4-基]甲胺二盐酸盐
[化12]
通过与实施例1同样的方法,由参考例1-2的化合物得到标题化合物。
1
实施例3
rel-1-[(3R,4R)-3-甲基-3,4-二氢-2H-吡喃并[3,2-b]吡啶-4-基]甲胺二盐酸盐
[化13]
将参考例5的化合物通过手性HPLC进行光学拆分,得到第1峰的化合物。向该化合物(48.0mg)的乙酸乙酯溶液(1.0mL)中在室温下加入4mol/L氯化氢-乙酸乙酯(0.43mL),在室温下搅拌2小时。然后,滤取析出的固体,通过减压干燥得到标题化合物。
[手性HPLC条件]
柱:IC 4.6cm x25cm
溶剂:己烷(90%)-含有二乙胺(0.1%)的IPA(10%),
流速:1.0mL/min
保留时间=3.8min(第1峰)
[α]
1
实施例4
rel-1-[(3S,4S)-3-甲基-3,4-二氢-2H-吡喃并[3,2-b]吡啶-4-基]甲胺二盐酸盐
[化14]
将参考例5的化合物通过手性HPLC进行光学拆分,得到第2峰的化合物。向该化合物(44.8mg)的乙酸乙酯溶液(1.0mL)中在室温下加入4mol/L氯化氢-乙酸乙酯(0.40mL),在室温下搅拌2小时。然后,滤取析出的固体,通过减压干燥得到标题化合物。
[手性HPLC条件]
柱:IC 4.6cm x25cm
溶剂:己烷(90%)-含有二乙胺(0.1%)的IPA(10%),
流速:1.0mL/min
保留时间=4.5min(第2峰)
[α]
1
实施例5
N-甲基-1-[(2R,4S)-2-甲基-3,4-二氢-2H-吡喃并[3,2-b]吡啶-4-基]甲胺二盐酸盐
[化15]
向参考例6-1的化合物(203mg,0.728mmol)和四氢呋喃(10.0mL)的混合物中在冰冷却下加入55%氢化钠(95.0mg)。在冰冷却下搅拌30分钟后,加入碘甲烷(0.453mL,7.28mmol)。在室温下搅拌3小时后,加入饱和氯化铵水溶液(10mL),用乙酸乙酯(10mL×2次)萃取,用无水硫酸镁干燥后,过滤并浓缩。将浓缩残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化并浓缩。
向得到的产物(192mg)和乙酸乙酯(3.0mL)的混合物中在冰冷却下加入4mol/L氯化氢-乙酸乙酯(3.0mL),在室温下搅拌2小时。然后,滤取析出的固体,通过减压干燥,得到标题化合物(142mg)。
1
实施例6~7
按照实施例5中记载的方法,由对应的参考例的化合物得到实施例6~7的化合物。
[表2]
实施例8
1-[(2R,3S)-6-氟-2-甲基-2,3-二氢呋喃并[3,2-b]吡啶-3-基]甲胺二盐酸盐
[化16]
向参考例13的化合物(98mg,3.47mmol)和2-丙醇(3.5mL)的混合物中加入4mol/L氯化氢-环戊基甲基醚(2mL),在室温下搅拌2小时。浓缩反应溶液后,将得到的固体由2-丙醇重结晶,得到标题化合物(39.7mg)。
[α]
1
实施例9~25
按照实施例8中记载的方法,由对应的参考例的化合物得到实施例9~25的化合物。
[表3-1]
[表3-2]
[表3-3]
实施例27
1-[(2R,3S)-2-甲基-2,3-二氢呋喃并[3,2-b]吡啶-3-基]甲胺二盐酸盐
[化17]
在氮气氛中向参考例37的化合物(23.6g,79.0mmol)和甲醇(158mL)的混合物中加入10%钯-碳(8.00g)。在室温、氢气氛中搅拌5小时后,进行硅藻土过滤并浓缩。
向浓缩残渣和2-丙醇(158mL)的混合物中加入4mol/L氯化氢-环戊基甲基醚(43.5mL,174mmol),在室温下搅拌15分钟。浓缩反应溶液后,将得到的粗结晶由2-丙醇和甲醇重结晶,得到标题化合物(9.57g)。
[α]
1
实施例28~32
按照实施例27中记载的方法,由对应的参考例的化合物得到实施例28~32的化合物。
[表4]
实施例33
rac-1-[(5aS,9aR,10S)-6,7,8,9,9a,10-六氢-5aH-[1]苯并吡喃并[3,2-b]吡啶-10-基]甲胺二盐酸盐
[化18]
向参考例50的化合物(123mg,0.497mmol)的甲苯溶液(7.1mL)中加入三乙胺(0.208mL,1.49mmol)和叠氮磷酸二苯酯(0.214mL,0.995mmol),在室温下搅拌30分钟。在90℃搅拌1小时后,向反应混合物中在冰冷却下滴加5mol/L氢氧化钠水溶液(1.69mL),在室温下搅拌2小时。向反应混合物中加入6mol/L盐酸至pH2并浓缩。向浓缩残渣中加入甲醇,通过过滤除去不溶物后,进行浓缩。向残渣的氯仿溶液(10mL)中加入三乙胺(0.208mL,1.49mmol)和二碳酸二叔丁酯(0.346mL,1.49mmol)。在室温搅拌1小时后,向反应混合物中加入水(30mL),用氯仿(30mL×2次)萃取,用无水硫酸钠干燥后,过滤并浓缩。将浓缩残渣用硅胶柱色谱(己烷/乙酸乙酯)分离纯化。向得到的产物的乙酸乙酯溶液(1.0mL)中加入4mol/L氯化氢-乙酸乙酯(1.0mL),在室温下搅拌2小时。浓缩反应混合物后,用氨基硅胶柱色谱(氯仿/甲醇)分离纯化,得到标题化合物(13mg)。
1
实施例34
1-[(2R,3S)-2,7-二甲基-2,3-二氢呋喃并[3,2-b]吡啶-3-基]甲胺二盐酸盐
[化19]
通过与实施例33同样的方法,由参考例51的化合物得到标题化合物。
1
实施例35
1-[(2R,3S)-2,5-二甲基-2,3-二氢呋喃并[3,2-b]吡啶-3-基]甲胺
[化20]
向参考例52的化合物(24.4mg,0.0878mmol)的乙酸乙酯溶液(1.0mL)中加入4mol/L氯化氢-乙酸乙酯(1.0mL)。在室温下搅拌2小时后,滤取析出的固体并干燥。向得到的固体(20mg)的氯仿混悬液(1.0mL)中加入三乙胺(0.039mL,0.279mmol)和三氟乙酸酐(0.013mL,0.096mmol),在室温下搅拌2小时。向反应混合物中加入水(30mL),用氯仿(30mL×2次)萃取,用无水硫酸钠干燥后,过滤并浓缩。将残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化。向得到的产物、甲醇(0.9mL)和水(0.1mL)的混合物中加入碳酸钾(55.0mg,0.398mmol),在50℃搅拌2小时。向反应混合物中加入水(30mL),用氯仿(30mL×2次)萃取,用无水硫酸钠干燥后,过滤并浓缩。将残渣用氨基硅胶柱色谱(氯仿/甲醇)分离纯化,得到标题化合物(8.2mg)。
1
实施例36
rac-1-[(2R,3R)-2-甲基-2,3-二氢呋喃并[3,2-b]吡啶-3-基]甲胺二盐酸盐
[化21]
向参考例38的10∶1的非对映异构体混合物(1.66g,8.59mmol)的甲苯溶液(57.3mL)中在室温下加入三乙胺(3.59mL,25.8mmol)和叠氮磷酸二苯酯(3.69mL,17.2mmol),在室温下搅拌30分钟。在90℃搅拌1小时后,向反应混合物中在冰冷却下滴加5mol/L氢氧化钠水溶液(29.2mL)。将反应混合物用2小时升温至室温后,加入6mol/L盐酸直至水层达到pH5。将反应混合物浓缩后,将浓缩残渣溶解在甲醇中,过滤不溶物并浓缩。接着,向浓缩残渣和氯仿(100mL)的混合物中加入三乙胺(3.59mL,25.8mmol)和二碳酸二叔丁酯(5.63g,25.8mmol)。在室温下搅拌1小时后,向反应混合物中加入水(100mL),用氯仿(50mL×2次)萃取,用无水硫酸镁干燥后,过滤并浓缩。将浓缩残渣用硅胶柱色谱(己烷/乙酸乙酯)分离纯化。将得到的产物的一部分(100mg)进一步用反相硅胶柱色谱(水/乙腈/三氟乙酸)分离纯化。
向得到的产物(10mg)和乙酸乙酯(1.0mL)的混合物中加入4mol/L氯化氢-乙酸乙酯(0.1mL),在室温下搅拌1小时。然后,滤取析出的固体,通过减压干燥得到标题化合物(7.0mg)。
1
实施例37
1-[(2R,3S)-6-氯-2-甲基-2,3-二氢呋喃并[3,2-b]吡啶-3-基]甲胺二盐酸盐
[化22]
向参考例53的化合物(342mg,1.50mmol)的甲苯溶液(2.2mL)中在室温下加入三乙胺(0.314mL,2.25mmol)和叠氮磷酸二苯酯(0.388mL,1.80mmol),在室温下搅拌30分钟。在90℃搅拌1小时后,向反应混合物中在冰冷却下滴加5mol/L氢氧化钠水溶液(5.11mL)。将反应混合物用2小时升温至室温后,加入6mol/L盐酸直至水层达到pH5。将反应混合物浓缩后,将浓缩残渣溶解在甲醇中,过滤不溶物并浓缩。接着,向浓缩残渣和氯仿(2.2mL)的混合物中加入三乙胺(0.628mL,4.51mmol)和二碳酸二叔丁酯(984mg,4.51mmol)。在室温下搅拌1小时后,向反应混合物中加入水(4.0mL),用氯仿(4mL×2次)萃取,用无水硫酸镁干燥后,过滤并浓缩。将浓缩残渣用硅胶柱色谱(己烷/乙酸乙酯)分离纯化。
向得到的化合物(219mg)和2-丙醇(1.5mL)的混合物中加入4mol/L氯化氢-环戊基甲基醚(1.83mL),在室温下搅拌1小时。浓缩反应溶液后,将得到的粗结晶由2-丙醇和甲醇重结晶,得到标题化合物(66mg)。
1
实施例38
1-[(2R,3S)-5-氯-2-甲基-2,3-二氢呋喃并[3,2-b]吡啶-3-基]甲胺二盐酸盐
[化23]
通过与实施例37同样的方法,由参考例54的化合物得到标题化合物。
1
实施例39
rel-1-[(4R)-3,4-二氢-2H-噻喃并[3,2-b]吡啶-4-基]甲胺二盐酸盐
[化24]
将参考例15的化合物通过手性HPLC进行光学拆分,得到第1峰的化合物。向该化合物(20.0mg,0.0713mmol)的乙酸乙酯溶液(1.0mL)中加入4mol/L氯化氢-乙酸乙酯(0.178mL),在室温下搅拌2小时。然后,滤取析出的固体,通过减压干燥得到标题化合物(12mg)。
[手性HPLC条件]
柱:IC
溶剂:己烷(90%)-含有二乙胺(0.1%)的IPA(10%)
流速:1.0mL/min
保留时间=7.7min(第1峰)
[α]
1
实施例40
rel-1-[(4S)-3,4-二氢-2H-噻喃并[3,2-b]吡啶-4-基]甲胺二盐酸盐
[化25]
将参考例15的化合物通过手性HPLC进行光学拆分,得到第2峰的化合物。然后,通过与实施例39同样的方法,得到标题化合物。
[手性HPLC条件]
柱:IC
溶剂:己烷(90%)-含有二乙胺(0.1%)的IPA(10%)
流速:1.0mL/min
保留时间=9.2min(第2峰)
[α]
1
实施例41
rel-1-[(3R)-2,3-二氢噻吩并[3,2-b]吡啶-3-基]甲胺二盐酸盐
[化26]
将参考例18的化合物通过手性HPLC进行光学拆分,得到第1峰的化合物。然后,通过与实施例39同样的方法,得到标题化合物。
[手性HPLC条件]
柱:IC
溶剂:己烷(90%)-IPA(10%)
流速:1.0mL/min
保留时间=7.67min(第1峰)
1
实施例42
rel-1-[(3S)-2,3-二氢噻吩并[3,2-b]吡啶-3-基]甲胺二盐酸盐
[化27]
将参考例18的化合物通过手性HPLC进行光学拆分,得到第2峰的化合物。然后,通过与实施例39同样的方法,得到标题化合物。
[手性HPLC条件]
柱:IC
溶剂:己烷(90%)-IPA(10%)
流速:1.0mL/min
保留时间=9.19min(第2峰)
1
实施例43
rel-1-[(2R,3S)-2-乙基-2,3-二氢呋喃并[3,2-b]吡啶-3-基]甲胺二盐酸盐
[化28]
将参考例33的化合物通过手性HPLC进行光学拆分,得到第1峰的化合物。然后,通过与实施例39同样的方法,得到标题化合物。
[手性HPLC条件]
柱:IC
溶剂:含有二乙胺(0.1%)的己烷(95%)-含有二乙胺(0.1%)的IPA(5%)
流速:1.0mL/min
保留时间=9.72min(第1峰)
1
实施例44
rel-1-[(2S,3R)-2-乙基-2,3-二氢呋喃并[3,2-b]吡啶-3-基]甲胺二盐酸盐
[化29]
/>
将参考例33的化合物通过手性HPLC进行光学拆分,得到第2峰的化合物。然后,通过与实施例39同样的方法,得到标题化合物。
[手性HPLC条件]
柱:IC
溶剂:含有二乙胺(0.1%)的己烷(95%)-含有二乙胺(0.1%)的IPA(5%)
流速:1.0mL/min
保留时间=12.64min(第2峰)
1
实施例45
rac-1-(3,4-二氢-2H-噻喃并[3,2-b]吡啶-4-基)-N-甲基甲胺二盐酸盐
[化30]
向参考例15的化合物(244mg,0.870mmol)和N,N-二甲基甲酰胺(2.2mL)的混合物中在冰冷却下加入55%氢化钠(144mg)。在冰冷却下搅拌10分钟后,加入碘甲烷(0.544mL,8.70mmol)。在室温下搅拌2小时后,加入饱和氯化铵水溶液(5mL),用2∶1-己烷/乙酸乙酯(5mL×3次)萃取,用无水硫酸镁干燥后,过滤并浓缩。将浓缩残渣用硅胶柱色谱(己烷/乙酸乙酯)分离纯化。
向得到的化合物(150mg)和乙酸乙酯(1.1mL)的混合物中加入4mol/L氯化氢-乙酸乙酯(1.1mL),在室温下搅拌2小时。然后,滤取析出的固体,用乙醚(4.0mL)洗涤,通过减压干燥得到标题化合物(101mg)。
1
实施例46
N-甲基-1-[(2R,3S)-2-甲基-2,3-二氢呋喃并[3,2-b]吡啶-3-基]甲胺二盐酸盐
[化31]
向实施例27的化合物(10mg,0.042mmol)和氯仿(0.10mL)的混合物中加入三乙胺(0.024mL,0.169mmol)和二碳酸二叔丁酯(18mg,0.084mmol)。在室温下搅拌1小时后,浓缩反应混合物。将浓缩残渣用硅胶柱色谱(己烷/乙酸乙酯)分离纯化。
向得到的化合物(101mg,0.382mmol)和N,N-二甲基甲酰胺(0.764mL)的混合物中在冰冷却下加入55%氢化钠(50.0mg)。在冰冷却下搅拌10分钟后,加入碘甲烷(0.239mL,3.82mmol)。在室温下搅拌1小时后,加入水(2mL),用氯仿(2mL×3次)萃取,用无水硫酸镁干燥后,过滤并浓缩。将浓缩残渣用硅胶柱色谱(己烷/乙酸乙酯)分离纯化。
向得到的化合物(50mg)和乙酸乙酯(1.0mL)的混合物中加入4mol/L氯化氢-乙酸乙酯(0.955mL),在室温下搅拌2小时。然后,滤取析出的固体,通过减压干燥得到标题化合物(45mg)。
1
实施例47~48
按照实施例46记载的方法,由对应实施例的化合物得到实施例47~48的化合物。
[表5]
实施例49
1-[(2R,3S)-6-氟-2-甲基-2,3-二氢呋喃并[3,2-b]吡啶-3-基]-N-甲基甲胺二氢溴酸盐
[化32]
将参考例55的化合物(39.5g,120mmol)和30%氢溴酸/乙酸(117mL)的混合物在室温下搅拌3小时。然后,浓缩混合物。向浓缩残渣中加入乙酸乙酯,搅拌后滤取固体。将得到的固体和乙醇(211mL)的混合物在75℃加热搅拌,确认固体全部溶解后,冷却至60℃,确认固体析出后,滴加己烷(633mL)。然后,将混合物慢慢冷却至0℃,滤取固体,用在0℃冷却的己烷/乙醇=9/1(60mL)洗涤,通过干燥得到标题化合物(34.2g)。
[α]
1
实施例50~51
按照实施例46记载的方法,由对应实施例的化合物得到实施例50~51的化合物。
[表6]
实施例52~61
按照实施例27记载的方法,由对应的参考例的化合物得到实施例52~61的化合物。
[表7-1]
[表7-2]
实施例62
rac-1-[(2R,3R,4R)-2,3-二甲基-3,4-二氢-2H-吡喃并[3,2-b]吡啶-4-基]甲胺二盐酸盐
[化33]
向参考例88-1的化合物(120mg,0.368mmol)的甲醇溶液(1.2mL)中加入10%钯-碳(60mg)。在室温、氢气气氛中搅拌4小时后,进行硅藻土过滤并浓缩。向浓缩残渣中加入4mol/L盐酸-乙酸乙酯(0.5mL),用乙醚研制,以与杂质的混合物形式得到标题化合物的盐酸盐。向混合物的四氢呋喃混悬液(3.68mL)中加入三乙胺(0.154mL)和二碳酸二叔丁酯(96mg)。在室温下搅拌2小时后,加入水,用乙酸乙酯萃取,用无水硫酸钠干燥后,过滤并浓缩。将浓缩残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化,得到纯化物(38.7mg)。向该纯化物(38.7mg)的乙酸乙酯溶液(0.4mL)中加入4mol/L盐酸-乙酸乙酯(0.331mL)。在室温下搅拌2小时后,滤取析出的固体,通过减压干燥得到白色固体形式的标题化合物(24.2mg)的盐酸盐。
1
实施例63~66
按照实施例62记载的方法,由对应的参考例的化合物得到实施例63~66的化合物。
[表8]
实施例67~79
按照实施例8记载的方法,由对应的参考例的化合物得到实施例67~79的化合物。
[表9-1]
[表9-2]
实施例80~87
按照实施例49记载的方法,由对应的参考例的化合物得到实施例80~87的化合物。
[表10-1]
[表10-2]
实施例88
rac-1-(2-甲氧基-5,6,7,8-四氢喹啉-8-基)甲胺二盐酸盐
[化34]
向参考例91的化合物(9mg,0.028mmol)的甲醇溶液(0.28mL)中在室温下加入肼一水合物(0.007mL,0.140mmol)。在50℃搅拌2小时后,浓缩反应混合物。向浓缩残渣中加入1mol/L盐酸,滤去析出的固体,用水洗涤后,浓缩滤液。用乙醚洗涤浓缩残渣,通过减压干燥得到标题化合物(7mg)。
1
实施例89
rac-1-[4-(4-甲基苯基)-5,6,7,8-四氢喹啉-8-基]甲胺二盐酸盐
[化35]
通过与实施例88同样的方法,由参考例101的化合物得到标题化合物。
1
实施例90
rac-1-(5’-甲基-3’H-螺[环丙烷-1,2’-呋喃并[3,2-b]吡啶]-3’-基)甲胺二盐酸盐
[化36]
向参考例95的化合物(300mg,0.870mmol)、碳酸铯(850mg,2.61mmol)、三甲基环三硼氧烷(0.365mL)、甲苯(2.9mL)、水(1.45mL)的混合物中加入(2-二环己基膦基-2′,4′,6′-三异丙基-1,1′-联苯基)[2-(2‘-氨基-1,1’-联苯基)]钯(II)甲磺酸盐(73.6mg,0.087mmol)。在90℃搅拌5小时后,加入饱和氯化铵水溶液(30mL)。用乙酸乙酯(30mL×2次)萃取,用无水硫酸钠干燥后,过滤并浓缩。将浓缩残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化,得到纯化物(128mg)。向该纯化物(128mg)中在冰冷却下加入5.1mol/L氢溴酸-乙酸(0.387mL)。在室温下搅拌2小时后,浓缩反应混合物。将浓缩残渣通过氨基硅胶柱色谱(氯仿/甲醇)分离纯化。向得到的纯化物的2-丙醇溶液(1.0mL)中加入4mol/L盐酸-乙酸乙酯(3.0mL),滤取析出的固体,得到标题化合物(44.4mg)。
1
实施例91
rac-1-[4-(环丁氧基)-6,7-二氢-5H-环戊烷并[b]吡啶-7-基]甲胺二盐酸盐
[化37]
向参考例100的化合物(50.0mg,0.189mmol)、环丁醇(0.045mL,0.567mmol)的甲苯溶液(1.89mL)中加入氰基亚甲基三丁基膦(0.149mL,0.567mmol)。在100℃搅拌3小时后,向反应混合物中加入水(30mL),用乙酸乙酯(30mL×2次)萃取,用无水硫酸钠干燥后,过滤并浓缩。将浓缩残渣用硅胶柱色谱(己烷/乙酸乙酯)分离纯化。向得到的纯化物中加入4mol/L氢溴酸-乙酸(0.500mL)。在室温下搅拌2小时后,浓缩反应混合物。将浓缩残渣通过反相硅胶柱色谱(水/乙腈)分离纯化。向得到的纯化物的2-丙醇溶液(1.0mL)中加入4mol/L盐酸-乙酸乙酯(1.0mL)并浓缩,得到标题化合物(8.2mg)。
1
实施例92
rac-1-(4-甲基-5,6,7,8-四氢喹啉-8-基)甲胺
[化38]
向参考例104的化合物(40.2mg,0.233mmol)的四氢呋喃溶液(1.2mL)中在冰冷却下加入0.91mol/L硼烷-四氢呋喃络合物的四氢呋喃溶液(0.769mL,0.700mmol)。在室温下搅拌2小时后,向反应混合物中加入甲醇(1.0mL),进一步加入浓盐酸(1.0mL)。在50℃搅拌1小时后,向反应混合物中加入1mol/L氢氧化钠水溶液,用氯仿/甲醇=4/1(30mL×2次)萃取,用无水硫酸钠干燥后,过滤并浓缩。将浓缩残渣通过氨基硅胶柱色谱(氯仿/甲醇)分离纯化,得到标题化合物(33.5mg)。
1
实施例93-1、实施例93-2
实施例93-1
rac-1-[(5R,7S)-5-甲基-6,7-二氢-5H-环戊烷并[b]吡啶-7-基]甲胺
实施例93-2
rac-1-[(5R,7R)-5-甲基-6,7-二氢-5H-环戊烷并[b]吡啶-7-基]甲胺
[化39]
通过与实施例92同样的方法,由参考例108的化合物以实施例93-1与实施例93-2的1∶1非对映异构体混合物形式得到标题化合物。
1
实施例94
rel-1-[(2S,3R)-2-甲基(2-
[化40]
向实施例57的化合物(112mg,0.470mmol)和三乙胺(0.328mL,2.35mmol)的四氢呋喃溶液(2.4mL)中加入二碳酸二叔丁酯(113mg,0.517mmol)。在室温搅拌15小时后,向反应混合物中加入水(30mL),用乙酸乙酯(30mL×2次)萃取,用无水硫酸钠干燥后,过滤并浓缩。将浓缩残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化,得到纯化物(120mg)。将得到的纯化物通过手性HPLC进行光学拆分,得到第1峰的化合物(48.7mg)。然后,通过与实施例39同样的方法,得到标题化合物(42.9mg)。
[手性HPLC条件]
柱:IC
溶剂:含有二乙胺(0.1%)的己烷(90%)-含有二乙胺(0.1%)的IPA(10%)
流速:1.0mL/min
保留时间=6.80min(第1峰)
1
实施例95
rel-1-[(2R,3S)-2-甲基(2-
[化41]
通过与实施例94同样的方法,由实施例57的化合物得到Boc体后,通过手性HPLC进行光学拆分,得到第2峰的化合物。然后,通过与实施例39同样的方法,得到标题化合物。
[手性HPLC条件]
柱:IC
溶剂:含有二乙胺(0.1%)的己烷(90%)-含有二乙胺(0.1%)的IPA(10%)
流速:1.0mL/min
保留时间=8.43min(第2峰)
1
实施例96
rel-1-[(3’R)-3’H-螺[环丙烷-1,2’-呋喃并[3,2-b]吡啶]-3’-基]甲胺二盐酸盐
[化42]
通过与实施例94同样的方法,由实施例58的化合物得到Boc体后,通过手性HPLC进行光学拆分,得到第1峰的化合物。然后,通过与实施例39同样的方法,得到标题化合物。
[手性HPLC条件]
柱:IC
溶剂:己烷(90%)-含有二乙胺(0.1%)的IPA(10%)
流速:1.0mL/min
保留时间=7.41min(第1峰)
1
实施例97
rel-1-[(3’S)-3’H-螺[环丙烷-1,2’-呋喃并[3,2-b]吡啶]-3’-基]甲胺二盐酸盐
[化43]
通过与实施例94同样的方法,由实施例58的化合物得到Boc体后,通过手性HPLC进行光学拆分,得到第2峰的化合物。然后,通过与实施例39同样的方法,得到标题化合物。
[手性HPLC条件]
柱:IC
溶剂:己烷(90%)-含有二乙胺(0.1%)的IPA(10%)
流速:1.0mL/min
保留时间=8.21min(第2峰)
1
实施例98
rel-1-[(3’R)-6’-氯-3’H-螺[环丙烷-1,2’-呋喃并[3,2-b]吡啶]-3’-基]甲胺二盐酸盐
[化44]
将参考例89的化合物通过手性HPLC进行光学拆分,得到第1峰的化合物。然后,通过与实施例39同样的方法,得到标题化合物。
[手性HPLC条件]
柱:AY-H
溶剂:己烷(90%)-IPA(10%)
流速:1.0mL/min
保留时间=5.04min(第1峰)
1
实施例99
rel-1-[(3’S)-6’-氯-3’H-螺[环丙烷-1,2’-呋喃并[3,2-b]吡啶]-3’-基]甲胺二盐酸盐
[化45]
将参考例89的化合物通过手性HPLC进行光学拆分,得到第2峰的化合物。然后,通过与实施例39同样的方法,得到标题化合物。
[手性HPLC条件]
柱:AY-H
溶剂:己烷(90%)-IPA(10%)
流速:1.0mL/min
保留时间=7.34min(第2峰)
1
实施例100
rel-1-[(3’R)-5’-甲基-3’H-螺[环丙烷-1,2’-呋喃并[3,2-b]吡啶]-3’-基]甲胺二盐酸盐
[化46]
通过与实施例94同样的方法,由实施例90的化合物得到Boc体后,通过手性HPLC进行光学拆分,得到第1峰的化合物。然后,通过与实施例39同样的方法,得到标题化合物。
[手性HPLC条件]
柱:AY-H
溶剂:己烷(95%)-IPA(5%)
流速:1.0mL/min
保留时间=5.09min(第1峰)
1
实施例101
rel-1-[(3’S)-5’-甲基-3’H-螺[环丙烷-1,2’-呋喃并[3,2-b]吡啶]-3’-基]甲胺二盐酸盐
[化47]
通过与实施例94同样的方法,由实施例90的化合物得到Boc体后,通过手性HPLC进行光学拆分,得到第2峰的化合物。然后,通过与实施例39同样的方法,得到标题化合物。
[手性HPLC条件]
柱:AY-H
溶剂:己烷(95%)-IPA(5%)
流速:1.0mL/min
保留时间=6.03min(第2峰)
1
实施例102
rel-1-[(3’R)-5’-氟-3’H-螺[环丙烷-1,2’-呋喃并[3,2-b]吡啶]-3’-基]甲胺二氢溴酸盐
[化48]
将参考例96的化合物通过手性HPLC进行光学拆分,得到第1峰的化合物。然后,通过与实施例49同样的方法,得到标题化合物。
[手性HPLC条件]
柱:IG
溶剂:己烷(50%)-EtOH(50%)
流速:1.0mL/min
保留时间=5.36min(第1峰)
1
实施例103
rel-1-[(3’S)-5’-氟-3’H-螺[环丙烷-1,2’-呋喃并[3,2-b]吡啶]-3’-基]甲胺二氢溴酸盐
[化49]
将参考例96的化合物通过手性HPLC进行光学拆分,得到第2峰的化合物。然后,通过与实施例49同样的方法,得到标题化合物。
[手性HPLC条件]
柱:IG
溶剂:己烷(50%)-EtOH(50%)
流速:1.0mL/min
保留时间=7.14min(第1峰)
1
实施例104
rel-1-[(3R)-2,2,5-三甲基-2,3-二氢呋喃并[3,2-b]吡啶-3-基]甲胺二盐酸盐
[化50]
通过与实施例94同样的方法,由实施例83的化合物得到Boc体后,通过手性HPLC进行光学拆分,得到第1峰的化合物。然后,通过与实施例39同样的方法,得到标题化合物。
[手性HPLC条件]
柱:IC
溶剂:己烷(95%)-IPA(5%)
流速:1.0mL/min
保留时间=5.575min(第1峰)
1
实施例105
rel-1-[(3S)-2,2,5-三甲基-2,3-二氢呋喃并[3,2-b]吡啶-3-基]甲胺二盐酸盐
[化51]
通过与实施例94同样的方法,由实施例83的化合物得到Boc体后,通过手性HPLC进行光学拆分,得到第2峰的化合物。然后,通过与实施例39同样的方法,得到标题化合物。
[手性HPLC条件]
柱:IC
溶剂:己烷(95%)-IPA(5%)
流速:1.0mL/min
保留时间=6.539min(第2峰)
1
实施例106
rac-1-(5-氟-2,2-二甲基-2,3-二氢呋喃并[3,2-b]吡啶-3-基)甲胺二氢溴酸盐
[化52]
通过与实施例49同样的方法,由参考例118的化合物得到标题化合物。
1
参考例1-1、参考例1-2
参考例1-1
rac-乙基[(3R,4R)-3-甲基-3,4-二氢-2H-吡喃并[3,2-b]吡啶-4-基]乙酸酯
参考例1-2
rac-乙基[(3S,4R)-3-甲基-3,4-二氢-2H-吡喃并[3,2-b]吡啶-4-基]乙酸酯
[化53]
向参考例2的化合物(1.64g,5.22mmol)的甲苯溶液(26.0mL)中在室温下加入三丁基氢化锡(2.58mL,8.87mmol)和偶氮二异丁腈(0.086g,0.522mmol)。在90℃搅拌1小时后,浓缩反应混合物。将残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化,得到标题化合物的参考例1-1(0.461g)和参考例1-2(0.624g)。
参考例1-1
1
参考例1-2
1
参考例2
rac-乙基(2E)-5-[(2-溴吡啶-3-基)氧]-4-甲基戊-2-烯酸酯
[化54]
向参考例3的化合物(1.72g,7.05mmol)和甲苯(23.5mL)的混合物中在室温下加入乙基(三苯基膦)乙酸酯(2.58g,7.40mmol)。在90℃搅拌2小时后,浓缩反应溶液,将残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化,得到标题化合物(1.64g)。
1
参考例3
rac-3-[(2-溴吡啶-3-基)氧]-2-甲基丙醛
「化55]
向参考例4的化合物(3.65g,14.8mmol)和氯仿(49.4mL)的混合物中在室温下加入戴斯-马丁氧化剂(3.59g,8.45mmol)。在室温搅拌1小时后,向反应混合物中在冰冷却下加入饱和碳酸氢钠水溶液(30mL)和饱和硫代硫酸钠水溶液(30mL),用氯仿(50mL×2次)萃取,用无水硫酸镁干燥后,过滤并浓缩。将浓缩残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化,得到标题化合物(1.72g)。
1
参考例4
rac-3-[(2-溴吡啶-3-基)氧]-2-甲基丙烷-1-醇
[化56]
向2-甲基-1,3-丙二醇(1.13g,12.6mmol)、2-溴-3-羟基吡啶(2.19g,12.6mmol)、三苯基膦(3.30g,12.6mmol)和N,N-二甲基甲酰胺(9.75mL)的混合物中在冰冷却下加入偶氮二甲酸二异丙酯(2.45mL,12.6mmol)。在室温下搅拌5小时后,加入甲醇并浓缩。将残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化,得到标题化合物(3.65g)。
1
参考例5
rac-叔丁基{[(3S,4S)-3-甲基-3,4-二氢-2H-吡喃并[3,2-b]吡啶-4-基]甲基}氨基甲酸酯
[化57]
向实施例2的化合物(22.0mg)的氯仿溶液(1.0mL)中加入三乙胺(0.049mL)和二碳酸二叔丁酯(0.041mL)。在室温搅拌2小时后,通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化,得到标题化合物(20.4mg)。
1
参考例6-1、参考例6-2
参考例6-1
叔丁基{[(2R,4S)-2-甲基-3,4-二氢-2H-吡喃并[3,2-b]吡啶-4-基]甲基}氨基甲酸酯
参考例6-2
叔丁基{[(2R,4R)-2-甲基-3,4-二氢-2H-吡喃并[3,2-b]吡啶-4-基]甲基}氨基甲酸酯
[化58]
向参考例7的化合物(5.37g,24.3mmol)、甲醇(53.9mL)和水(27.0mL)的混合物中在室温下加入氢氧化钠(1.94g,48.5mmol)。在60℃搅拌2小时后,向反应混合物中加入3mol/L盐酸直至水层达到pH5。浓缩反应溶液后,将浓缩残渣溶解在甲醇中,过滤不溶物并浓缩。
向得到的残渣(5.79g)的甲苯溶液(400mL)中在室温下加入三乙胺(11.7mL,84.0mmol)和叠氮磷酸二苯酯(12.0mL,55.9mmol)。在室温下搅拌30分钟后,将反应混合物在90℃搅拌1小时。向反应混合物中在冰冷却下滴加5mol/L氢氧化钠水溶液(84.0mL)。将反应混合物用2小时升温至室温后,加入6mol/L盐酸使其为pH7,进行浓缩。将浓缩残渣溶解在甲醇中,过滤不溶物并浓缩。向浓缩残渣和氯仿(100mL)的混合物中加入三乙胺(11.7mL,84.0mmol)和二碳酸二叔丁酯(18.3g,84.0mmol)。在室温搅拌1小时后,向反应混合物中加入水(100mL),用氯仿(100mL×2次)萃取,用无水硫酸镁干燥后,过滤并浓缩。将浓缩残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化,得到标题化合物的参考例6-1(1.32g)和参考例6-2(3.34g)。
参考例6-1
1
参考例6-2
1
参考例7
甲基[(2R)-2-甲基-3,4-二氢-2H-吡喃并[3,2-b]吡啶-4-基]乙酸酯
[化59]
向参考例8的化合物(7.90g,26.3mmol)的甲苯溶液(132mL)中在室温下加入三丁基氢化锡(11.7mL,44.7mmol)和偶氮二异丁腈(0.432g,2.63mmol)。在90℃搅拌1小时后,浓缩反应混合物。将残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化,以3∶1的非对映异构体混合物形式得到标题化合物(5.37g)。
主非对映异构体
1
参考例8
乙基(2E,5R)-5-[(2-溴吡啶-3-基)氧]己-2-烯酸酯
[化60]
向参考例9的化合物(8.91g,36.2mmol)和氯仿(120mL)的混合物中在冰冷却下加入戴斯-马丁氧化剂(21.5g,50.7mmol)。在室温下搅拌1小时后,向反应混合物中在冰冷却下加入饱和碳酸氢钠水溶液(75mL)和饱和硫代硫酸钠水溶液(75mL),用氯仿(75mL×2次)萃取,用无水硫酸镁干燥后,过滤并浓缩。向浓缩残渣和甲苯(100mL)的混合物中在室温下加入乙基(三苯基膦)乙酸酯(12.7g,38.0mmol)。在80℃搅拌2小时后,浓缩反应混合物。将残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化,得到标题化合物(7.90g)。
1
参考例9
(3R)-3-[(2-溴吡啶-3-基)氧]丁烷-1-醇
[化61]
向参考例10的化合物(13.5g,37.5mmol)和甲醇(135mL)的混合物中在冰冷却下加入6mol/L盐酸(18.7mL)。在室温下搅拌2小时后,向反应混合物中在冰冷却下加入饱和碳酸氢钠水溶液(250mL),用氯仿(150mL×3次)萃取,用无水硫酸镁干燥后,过滤并浓缩。将残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化,得到标题化合物(8.91g)。
1
参考例10
2-溴-3-{[(2R)-4-{[叔丁基(二甲基)甲硅烷基]氧}丁烷-2-基]氧}吡啶
[化62]
向(S)-4-((叔丁基二甲基甲硅烷基)氧)-2-丁醇(8.22g,40.2mmol)、2-溴-3-羟基吡啶(7.00g,40.2mmol)、三苯基膦(11.6g,44.3mmol)和四氢呋喃(134mL)的混合物中在冰冷却下加入偶氮二甲酸二异丙酯(8.60mL,44.3mmol)。在室温下搅拌15小时后,向反应混合物中加入甲醇并浓缩。将浓缩残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化并浓缩,得到标题化合物(13.5g)。
1
参考例11
叔丁基{[(2S,4S)-2-甲基-3,4-二氢-2H-吡喃并[3,2-b]吡啶-4-基]甲基}氨基甲酸酯
[化63]
通过参考例6-2至参考例10同样的方法,由(R)-4-((叔丁基二甲基甲硅烷基)氧)-2-丁醇得到标题化合物。
1
参考例12
rac-叔丁基{[(2R,4S)-2-甲基-3,4-二氢-2H-吡喃并[3,2-b]吡啶-4-基]甲基}氨基甲酸酯
[化64]
通过与参考例6-1至参考例10同样的方法,由4-((叔丁基二甲基甲硅烷基)氧)-2-丁醇得到标题化合物。
1
参考例13
叔丁基{[(2R,3S)-6-氟-2-甲基-2,3-二氢呋喃并[3,2-b]吡啶-3-基]甲基}氨基甲酸酯
[化65]
/>
向参考例14的化合物(282mg,1.33mmol)的甲苯溶液(13mL)中在室温下加入三乙胺(0.56mL,4.00mmol)和叠氮磷酸二苯酯(0.57mL,2.67mmol),在室温下搅拌30分钟。在90℃搅拌1小时后,在冰冷却下向反应混合物中加入5mol/L氢氧化钠水溶液(4.0mL,20.0mmol)。在室温下搅拌2小时后,加入12mol/L盐酸(1.7mL)进行中和,加入2-丙醇(10mL)进行浓缩。将浓缩残渣溶解在甲醇中,过滤不溶物并浓缩。向浓缩残渣和氯仿(5.0mL)的混合物中加入三乙胺(0.56mL,4.0mmol)和二碳酸二叔丁酯(873mg,4.0mmol)。在室温下搅拌4小时后,浓缩反应混合物。将浓缩残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化,得到标题化合物(98mg)。
1
参考例14
[(2R,3S)-6-氟-2-甲基-2,3-二氢呋喃并[3,2-b]吡啶-3-基]乙酸
[化66]
向参考例40的化合物(375mg,2.6mmol)、2-溴-5-氟吡啶-3-醇(500mg,2.60mmol)、三苯基膦(751mg,2.86mmol)和四氢呋喃(8.7mL)的混合物中在冰冷却下加入偶氮二甲酸双(2-甲氧基乙基)酯(671mg,2.86mmol)。在室温下搅拌2小时后,向反应混合物中加入甲醇并浓缩。向浓缩残渣中加入甲苯(500mL),用水(200mL×3次)洗涤,用无水硫酸镁干燥后,过滤并浓缩。向浓缩残渣的甲苯溶液(9.0mL)中在室温下加入三丁基氢化锡(1.16mL,4.42mmol)和偶氮二异丁腈(42.7mg,0.26mmol)。在90℃搅拌1小时后,浓缩反应混合物。向浓缩残渣和四氢呋喃(10mL)的混合物中在室温下加入4mol/L氢氧化钠水溶液(2.08mL,10.4mmol)。在60℃搅拌4小时后,向反应混合物中加入水(10mL),将水层用乙醚(10mL×2次)洗涤。然后,加入4mol/L盐酸(10.4mL)直至水层达到pH5,用氯仿(200mL×5次)进行萃取。将合并的有机层用无水硫酸镁干燥后,过滤并浓缩,达到标题化合物(281mg)。
1
参考例15
rac-叔丁基[(3,4-二氢-2H-噻喃并[3,2-b]吡啶-4-基)甲基]氨基甲酸酯
[化67]
向参考例16的化合物(980mg,4.39mmol)、甲醇(9.8mL)和水(4.9mL)的混合物中在室温下加入氢氧化钠(351mg,8.78mmol)。在60℃搅拌2小时后,在冰冷却下加入3mol/L盐酸直至水层达到pH5。将反应混合物浓缩后,将浓缩残渣溶解在甲醇中,过滤不溶物并浓缩。
向得到的化合物(1.10g)的甲苯溶液(35.0mL)中在室温下加入三乙胺(2.20mL,15.8mmol)和叠氮磷酸二苯酯(2.26mL,10.5mmol),在室温搅拌30分钟。在90℃搅拌1小时后,向反应混合物中在冰冷却下滴加5mol/L氢氧化钠水溶液(17.9mL)。将反应混合物用2小时升温至室温后,加入6mol/L盐酸并中和,浓缩。将浓缩残渣溶解在甲醇中,过滤不溶物并浓缩。向浓缩残渣和氯仿(30mL)的混合物中加入三乙胺(2.20mL,15.8mmol)和二碳酸二叔丁酯(3.66g,15.8mmol)。在室温下搅拌1小时后,向反应混合物中加入水(50mL),用氯仿(50mL×2次)萃取,用无水硫酸镁干燥后,过滤并浓缩。将浓缩残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化,得到标题化合物(560mg)。
1
参考例16
rac-甲基(3,4-二氢-2H-噻喃并[3,2-b]吡啶-4-基)乙酸酯
[化68]
向参考例17的化合物(1.66g,6.69mmol)和氯仿(22.3mL)的混合物中在冰冷却下加入戴斯-马丁氧化剂(2.98g,7.02mmol)。在冰冷却下搅拌1小时后,向反应混合物中加入乙基(三苯基膦)乙酸酯(2.46g,7.36mmol)。在室温下搅拌1小时后,向反应混合物中在冰冷却下加入饱和碳酸氢钠水溶液(15mL)和饱和硫代硫酸钠水溶液(15mL),用氯仿(20mL×2次)萃取,用无水硫酸镁干燥后,过滤并浓缩。将浓缩残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化并浓缩。
向得到的化合物(1.62g,5.36mmol)的甲苯溶液(26.8mL)中在室温下加入三丁基氢化锡(2.39mL,9.11mmol)和偶氮二异丁腈(0.088g,0.536mmol)。在90℃搅拌1小时后,浓缩反应混合物。将残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化,得到标题化合物(0.980g)。
1
参考例17
3-[(2-溴吡啶-3-基)硫基]丙烷-1-醇
[化69]
向2-溴-3-氟吡啶(3.00g,17.1mmol)、3-巯基-1-丙醇(1.43g,15.5mmol)和N,N-二甲基甲酰胺(15.5mL)的混合物中加入碳酸钾(8.00g)。在室温下搅拌24小时后,进行硅藻土过滤,进行浓缩。将浓缩残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化,得到标题化合物(1.66g)。
1
参考例18~22
按照参考例15~17中记载的方法,由对应的化合物得到参考例18~22的化合物。
[表11]
参考例25-1、参考例25-2
参考例25-1
叔丁基{[(2R,4R)-2,6-二甲基-3,4-二氢-2H-吡喃并[3,2-b]吡啶-4-基]甲基}氨基甲酸酯
参考例25-2
叔丁基{[(2R,4S)-2,6-二甲基-3,4-二氢-2H-吡喃并[3,2-b]吡啶-4-基]甲基}氨基甲酸酯
[化70]
通过与参考例6-1、6-2至参考例10同样的方法,由2-溴-3-羟基-6-甲基吡啶得到标题化合物的参考例25-1和参考例25-2。
参考例25-1
LC-MS:R.T.=0.745min 0bsMS=293.0[M+1]
参考例25-2
LC-MS:R.T.=0.650min ObsMS=294.3[M+2]
参考例26-1、参考例26-2
参考例26-1
叔丁基{[(2S,3R)-2-甲基-2,3-二氢呋喃并[3,2-b]吡啶-3-基]甲基}氨基甲酸酯
参考例26-2
叔丁基{[(2S,3S)-2-甲基-2,3-二氢呋喃并[3,2-b]吡啶-3-基]甲基}氨基甲酸酯
[化71]
通过与参考例6-1、参考例6-2至参考例7同样的方法,由参考例27的化合物得到标题化合物的参考例26-1和参考例26-2。
参考例26-1
1
参考例26-2
LC-MS:R.T.=1.42min 0bsMS=265[M+1]
参考例27
甲基(2E,4S)-4-[(2-溴吡啶-3-基)氧]戊-2-烯酸酯
[化72]
向参考例28的化合物(1.56g,6.00mmol)和二氯甲烷(12.0mL)的混合物中在-78℃加入1.03mol/L二异丁基氢化铝的甲苯溶液(17.47mL,17.99mmol)。在-78℃搅拌2小时后,加入饱和氯化铵水(50mL),将不溶物通过硅藻土过滤除去。用乙酸乙酯萃取后,用无水硫酸镁干燥,过滤并浓缩。向浓缩残渣和甲苯的混合物中在室温加入乙基(三苯基膦)乙酸酯(2.005g,6.00mmol)。在室温搅拌1小时后,浓缩反应混合物。将残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化,得到标题化合物(0.54g)。
1
参考例28
(2S)-2-[(2-溴吡啶-3-基)氧]丙酸甲酯
[化73]
向(R)-(+)-乳酸甲基(1.20g,11.5mmol)、2-溴-3-羟基吡啶(2.00g,11.5mmol)、三苯基膦(3.32g,12.6mmol)和四氢呋喃(38mL)的混合物中在冰冷却下加入偶氮二甲酸二异丙酯(1.83mL,12.6mmol)。在室温下搅拌15小时后,向反应混合物中加入甲醇并浓缩。将浓缩残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化并浓缩,得到标题化合物(13.5g)。
1
参考例29
rac-叔丁基{[(2R,3S)-2-(丙烷-2-基)-2,3-二氢呋喃并[3,2-b]吡啶-3-基]甲基}氨基甲酸酯
[化74]
通过与参考例6-1、参考例6-2和参考例7同样的方法,由参考例30的化合物得到标题化合物。
1
参考例30
rac-甲基(2E)-4-[(2-溴吡啶-3-基)氧]-5-甲基己-2-烯酸酯
[化75]
向参考例31的化合物(1.18g,4.54mmol)和二甲基亚砜(15.0mL)的混合物中在室温下加入三乙胺(1.90mL,13.6mmol)和三氧化硫吡啶络合物(2.17g,13.6mmol)。在室温搅拌3小时后,向反应混合物中加入水(200mL),用乙酸乙酯(200mL×2次)萃取。将有机层用无水硫酸镁干燥、过滤并浓缩。向浓缩残渣和甲苯(15mL)的混合物中在室温下加入乙基(三苯基膦)乙酸酯(1.72g,5.16mmol)。在室温下搅拌12小时后,浓缩反应溶液,将残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化,得到标题化合物(0.77g)。
1
参考例31
rac-2-[(2-溴吡啶-3-基)氧]-3-甲基丁烷-1-醇
[化76]
在-78℃向参考例32的化合物(1.34g,4.43mmol)和四氢呋喃(15.0mL)的混合物中加入1.0mol/L二异丁基氢化铝的甲苯溶液(4.88mL,4.88mmol)。在室温搅拌3小时后,在冰冷却下加入1.0mol/L二异丁基氢化铝的甲苯溶液(4.88mL,4.88mmol)。在冰冷却下搅拌2小时后,向反应混合物中加入水(30mL)和10%硫酸氢钾水溶液(30mL),用乙酸乙酯(50mL×2次)萃取。将有机层用无水硫酸镁干燥、过滤并浓缩。将浓缩残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化,得到标题化合物(1.19g)。
1
参考例32
rac-甲基2-[(2-溴吡啶-3-基)氧]-3-甲基丁酸酯
[化77]
向2-溴-3-羟基吡啶(2.00g,11.5mmol)和N,N-二甲基甲酰胺(30mL)的混合物中加入碳酸钾(1.91g,13.8mmol)和2-溴-3-甲基丁酸乙酯(2.25mL,13.8mmol)。在50℃搅拌4小时后,向反应混合物中加入水(200mL),用乙酸乙酯(200mL×2次)萃取后,将有机层用无水硫酸镁干燥,过滤并浓缩。将浓缩残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化并浓缩,得到标题化合物(1.36g)。
1
参考例33~34
按照参考例29~32中记载的方法,由对应化合物得到参考例33~34的化合物。
[表12]
参考例35-1、参考例35-2
参考例35-1
rac-叔丁基{[(2S,4R)-2-苯基-3,4-二氢-2H-吡喃并[3,2-b]吡啶-4-基]甲基}氨基甲酸酯
参考例35-2
rac-叔丁基{[(2S,4S)-2-苯基-3,4-二氢-2H-吡喃并[3,2-b]吡啶-4-基]甲基}氨基甲酸酯
[化78]
通过与参考例6-1、参考例6-2至参考例10同样的方法,由3-((叔丁基二甲基甲硅烷基)氧)-1-苯基-1-丙醇得到标题化合物的参考例35-1和参考例35-2。
参考例35-1
1
参考例35-2
1
参考例36
rac-叔丁基{[(2S,3S)-2-(羟基甲基)-2,3-二氢呋喃并[3,2-b]吡啶-3-基]甲基}氨基甲酸酯
[化79]
向实施例32的化合物(42.0mg,0.157mmol)的二氯甲烷混悬液(2.0mL)中在冰冷却下加入三乙胺(0.044mL,0.314mmol)和1.0mol/L三溴化硼的二氯甲烷溶液(0.330mL,0.330mmol)。在室温搅拌4小时后,向反应混合物中在冰冷却下加入甲醇(2.0mL)。在冰冷却下搅拌10分钟后,浓缩反应混合物。向浓缩残渣、四氢呋喃(1.0mL)和水(1.0mL)的混合物中加入碳酸钾(217mg,1.57mmol)和二碳酸二叔丁酯(51.5mg,0.236mmol)。在室温下搅拌3小时后,向反应混合物中加入水(30mL),用乙酸乙酯(30mL×2次)萃取,用无水硫酸镁干燥后,过滤并浓缩。将残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化,得到标题化合物(22.9mg)。
1
参考例37
苄基{[(2R,3S)-2-甲基-2,3-二氢呋喃并[3,2-b]吡啶-3-基]甲基}氨基甲酸酯
[化80]
向参考例38的化合物(28.1g,145mmol)的甲苯溶液(208mL)中在室温下加入三乙胺(30.4mL,218mmol)和叠氮磷酸二苯酯(37.5mL,175mmol),在室温下搅拌30分钟。在90℃搅拌1小时后,向反应混合物加入苄基醇(22.5mL,218mmol)。在90℃搅拌3小时后,向反应混合物中在冰冷却下加入饱和碳酸氢钠水溶液(250mL),用乙酸乙酯(150mL×2次)萃取,用无水硫酸镁干燥后,过滤并浓缩。将浓缩残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化并浓缩,以10∶1的非对映异构体混合物形式得到标题化合物(23.6g)。
主非对映异构体
1
参考例38
叔丁基{[(2S,4S)-2-苯基-3,4-二氢-2H-吡喃并[3,2-b]吡啶-4-基]甲基}氨基甲酸酯
[化81]
向参考例39的化合物(45.0g,150mmol)的甲苯溶液(499mL)中在室温下加入三丁基氢化锡(66.8mL,255mmol)和偶氮二异丁腈(2.46g,15.0mmol)。在90℃搅拌1小时后,浓缩反应混合物。向浓缩残渣、四氢呋喃(333mL)和水(167mL)的混合物中在室温下加入氢氧化钠(24.0g,599mmol)。在60℃搅拌3小时后,向反应混合物加入水(333mL),将水层用乙醚(167mL×2次)洗涤。然后,加入5mol/L盐酸(120mL)直至水层达到pH5,用氯仿(200mL×5次)进行萃取。将合并的有机层用无水硫酸镁干燥后,过滤并浓缩,以10∶1的非对映异构体混合物形式得到标题化合物(28.1g)。
主非对映异构体
1
参考例39
乙基(2E,4R)-4-[(2-溴吡啶-3-基)氧]戊-2-烯酸酯
[化82]
向参考例40的化合物(26.0g,180mmol)、2-溴-3-羟基吡啶(31.4g,180mmol)、三苯基膦(52.0g,198mmol)和四氢呋喃(515mL)的混合物中在冰冷却下加入偶氮二甲酸双(2-甲氧基乙基)酯(46.5g,198mmol)。在室温下搅拌2小时后,向反应混合物中加入甲醇并浓缩。向浓缩残渣中加入甲苯(500mL),用水(200mL×3次)洗涤,用无水硫酸镁干燥后,过滤并浓缩。向浓缩残渣中加入己烷/乙醚(4/1,250mL),滤去析出的固体,浓缩滤液。将浓缩残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化并浓缩,得到标题化合物(45.0g)。
1
参考例40
乙基(2E,4S)-4-羟基戊-2-烯酸酯
[化83]
在-35℃向参考例41的化合物(23.8g,167mmol)和甲醇(478mL)的混合物中加入硼氢化钠(7.59g,201mmol)。用1小时升温至0℃后,向反应混合物中在冰冷却下加入饱和氯化铵水溶液(500mL),用乙酸乙酯(500mL×3次)萃取,用无水硫酸镁干燥后,过滤并浓缩。将浓缩残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化并浓缩,得到标题化合物(17.0g)。
1
参考例41
乙基(4S)-4-羟基戊-2-烯酸酯
[化84]
向(S)-(-)-3-丁炔-2-醇(14.5g,207mmol)、双(三甲基甲硅烷基)胺(18.4g,114mmol)和四氢呋喃(104mL)的混合物中在室温加入浓硫酸(0.110mL)。在65℃搅拌3小时后,将反应混合物冷却至-78℃。在-78℃滴加2.69mol/L正丁基锂(100mL,269mmol)。在-78℃搅拌30分钟后,滴加氯甲酸乙酯(26.6mL,279mmol)的四氢呋喃溶液(59mL)。在-78℃搅拌1小时后,将反应混合物用1小时升温至室温。向反应混合物中加入6mol/L硫酸(108mL),在室温搅拌15小时。向反应混合物中加入水(300mL),用乙酸乙酯(200mL×3次)萃取,用无水硫酸镁干燥后,过滤并浓缩。将浓缩残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化并浓缩,得到标题化合物(23.4g)。
1
参考例42~45
按照参考例37~39中记载的方法,由对应化合物得到参考例42~45的化合物。
[表13]
参考例46
rac-苄基{[(2S,3S)-2-(甲氧基甲基)-2,3-二氢呋喃并[3,2-b]吡啶-3-基]甲基}氨基甲酸酯
[化85]
通过与参考例37同样的方法,由参考例47得到标题化合物。
1
参考例47
rac-[(2S,3S)-2-(甲氧基甲基)-2,3-二氢呋喃并[3,2-b]吡啶-3-基]乙酸
[化86]
向参考例48的化合物(324mg,1.37mmol)的甲醇溶液(5.0mL)中加入水(5.0mL)和氢氧化钠(546mg,13.7mmol),在室温搅拌30分钟。然后,向反应混合物中加入6mol/L盐酸直至pH4并浓缩。将浓缩残渣通过硅胶柱色谱(氯仿/甲醇)分离纯化,得到标题化合物(258mg)。
1
参考例48
rac-甲基[(2S,3S)-2-(甲氧基甲基)-2,3-二氢呋喃并[3,2-b]吡啶-3-基]乙酸酯
[化87]
通过与参考例1-1同样的方法,由参考例49的化合物得到标题化合物。
1
参考例49
rac-甲基(2E)-4-[(2-溴吡啶-3-基)氧]-5-甲氧基戊-2-烯酸酯
[化88]
通过与参考例30至参考例32同样的方法,由甲基2-溴-3-甲氧基丙酸酯得到标题化合物。
1
参考例50
rac-[(5aS,9aR,10R)-6,7,8,9,9a,10-六氢-5aH-[1]苯并吡喃并[3,2-b]吡啶-10-基]乙酸
[化89]
通过与参考例8至参考例10和参考例14同样的方法,由2-(((叔丁基二甲基甲硅烷基)氧)甲基)环己烷-1-醇得到标题化合物。
LC-MS:R.T.=1.353min 0bsMS=248.4[M+1]
参考例51
[(2R,3S)-2,7-二甲基-2,3-二氢呋喃并[3,2-b]吡啶-3-基]乙酸
[化90]
通过与参考例38至参考例39同样的方法,由2-溴-3-羟基-4-甲基吡啶得到标题化合物。
LC-MS:R.T.=0.449min 0bsMS=208.1[M+1]
参考例52
叔丁基{[(2R,3S)-2,5-二甲基-2,3-二氢呋喃并[3,2-b]吡啶-3-基]甲基}氨基甲酸酯
[化91]
通过与参考例13至参考例14同样的方法,由2-溴-3-羟基-6-甲基吡啶得到标题化合物。
1
参考例53~54
按照参考例38~39中记载的方法,由对应的化合物得到参考例53~54的化合物。
[表14]
参考例55
苄基{[(2R,3S)-6-氟-2-甲基-2,3-二氢呋喃并[3,2-b]吡啶-3-基]甲基}甲基氨基甲酸酯
[化92]
向55%氢化钠(12.0g,276mmol)的N-甲基吡咯烷酮(491mL)的混悬液中在冰冷却下加入参考例56的化合物(58.2g,184mmol)的N-甲基吡咯烷酮溶液(123mL)。搅拌30分钟后,在冰冷却下向反应混合物中加入碘甲烷(23.0mL,368mmol),在室温下搅拌2小时。然后,向反应混合物中在冰冷却下加入水(500mL),用乙酸乙酯(1000mL)萃取,用水(200mL×2次)洗涤,用无水硫酸钠干燥后,过滤并浓缩。将残渣通过硅胶柱色谱(己烷∶乙酸乙酯)分离纯化后,得到标题化合物(39.5g)。
1
参考例56
苄基{[(2R,3S)-6-氟-2-甲基-2,3-二氢呋喃并[3,2-b]吡啶-3-基]甲基}氨基甲酸酯
[化93]
向参考例14的化合物(33.6g,159mmol)的甲苯溶液(227mL)中在室温下加入三乙胺(33.2mL,239mmol)和叠氮磷酸二苯酯(41.0mL,191mmol)。在室温搅拌1小时后,将反应溶液升温至90℃。在90℃搅拌20分钟后,向反应溶液中加入苄基醇(18.0mL,175mmol)。在90℃搅拌3.5小时后,向反应混合物中在冰冷却下加入饱和碳酸氢钠水溶液(400mL),用乙酸乙酯(200mL)萃取。将有机层用水(400mL)洗涤,用无水硫酸钠干燥后,过滤并浓缩,得到标题化合物(52g)。
1
参考例57~59
按照参考例7至参考例10、参考例37和参考例47中记载的方法,由对应的化合物得到参考例57~59的化合物。
[表15]
参考例60
rac-叔丁基{[6-(三氟甲基)-3,4-二氢-2H-吡喃并[3,2-b]吡啶-4-基]甲基}氨基甲酸酯
[化94]
通过与参考例1至参考例4和参考例6同样的方法,由丙烷-1,3-二醇和2-溴-6-(三氟甲基)吡啶-3-醇得到得到标题化合物。
LC-MS:R.T.=1.133min 0bsMS=333.2[M+1]
参考例61
rac-叔丁基[(1,2,3,4-四氢-1,5-萘啶-4-基)甲基]氨基甲酸酯
[化95]
向参考例62的化合物(261mg,1.01mmol)和四氢呋喃(5.0mL)的混合物中加入0.90mol/L四氢呋喃-硼烷.四氢呋喃溶液(2.24mL,2.01mmol)。在60℃搅拌2小时后,向反应混合物中加入甲醇(2.5mL),搅拌15分钟。浓缩反应混合物后,将浓缩残渣溶解于2.0mol/L盐酸(2.52mL,5.03mmol)中,在60℃搅拌2小时。将反应混合物浓缩后,将浓缩残渣溶解于氯仿(5.0mL)中,加入三乙胺(0.701mL,5.03mmol)和二碳酸二叔丁酯(330mg,1.51mmol)。在室温下搅拌1天后,向反应混合物中加入水(5.0mL),用氯仿(3.0mL×2次)萃取,用无水硫酸镁干燥后,过滤并浓缩。将浓缩残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化,得到标题化合物(100mg)。
1
参考例62
rac-叔丁基4-氰基-3,4-二氢-1,5-萘啶-1(2H)-甲酸酯
[化96]
向参考例63的化合物(660mg,2.11mmol)、三甲基氰硅烷(0.523mL,4.21mmol)和乙腈(14mL)的混合物中加入1.0mol/L四丁基氟化铵(4.21mL,4.21mmol),在70℃搅拌3小时。将反应混合物浓缩后,将浓缩残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化,得到白色固体形式的标题化合物(261mg)。
1
参考例63
rac-叔丁基4-溴-3,4-二氢-1,5-萘啶-1(2H)-甲酸酯
[化97]
向1,1-二甲基乙基3,4-二氢-1,5-萘啶-1(2H)-甲酸酯(560mg,2.39mmol)、N-溴代琥珀酰亚胺(510mg,2.87mmol)和四氯化碳(12mL)的混合物中加入偶氮二异丁腈(7.85mg,0.0480mmol),加热回流5小时。将反应混合物冷却至室温后,滤去析出的固体,浓缩滤液。将浓缩残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化,得到标题化合物(510mg)。
1
参考例64~65
按照参考例37至参考例39记载的方法,由对应的化合物得到参考例64~65的化合物。
[表16]
参考例66
2-碘-4,6-二甲基吡啶-3-醇
[化98]
/>
向4,6-二甲基-3-羟基吡啶(99.8mg,0.810mmol)、水(12mL)和四氢呋喃(2.0mL)的混合物中在0℃加入碘(247mg,0.972mmol)。在室温搅拌25小时后,向反应混合物中加入1mol/L盐酸(4mL),用乙酸乙酯(10mL×3次)萃取,用无水硫酸镁干燥后,过滤并浓缩。将有机层用0.1mol/L硫代硫酸钠水溶液(10mL×3次)洗涤,用无水硫酸钠干燥后,过滤并浓缩。将浓缩残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化,得到标题化合物(140mg)。
1
参考例67
苄基{2-[(2R,3S)-2-甲基-2,3-二氢呋喃并[3,2-b]吡啶-3-基]丙烷-2-基}氨基甲酸酯
[化99]
向参考例68的化合物(78.0mg,0.313mmol)和甲醇(0.70mL)、水(0.35mL)的混合物中在室温下加入氢氧化钠(37.5mg,0.939mmol)。在60℃搅拌2小时后,加入3.0mol/L盐酸直至水层达到pH5。浓缩反应溶液后,将浓缩残渣溶解在甲醇中,滤去不溶物,再次浓缩。
向得到的浓缩残渣(69.0mg)的甲苯溶液(0.45mL)中在室温下加入三乙胺(0.130mL,0.936mmol)和叠氮磷酸二苯酯(0.0800mL,0.374mmol)。在室温下搅拌30分钟后,将反应溶液升温至90℃。在90℃搅拌1小时后,向反应溶液中加入苄基醇(0.0482mL,0.468mmol)。在90℃搅拌3小时后,向反应混合物中在冰冷却下加入饱和碳酸氢钠水溶液(1.0mL),用乙酸乙酯(1.0mL×2次)萃取,用无水硫酸镁干燥后,过滤并浓缩。将浓缩残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化,得到标题化合物(53.0mg)。
1
参考例68
乙基2-甲基-2-[(2R,3S)-2-甲基-2,3-二氢呋喃并[3,2-b]吡啶-3-基]丙酸酯
[化100]
向参考例69的化合物(150mg,0.678mmol)的四氢呋喃溶液(1.7mL)中在-78℃下滴加1.0mol/L双(三甲基硅烷基)氨基锂甲苯溶液(1.63mL,1.63mmol)。在-78℃搅拌30分钟后,加入碘甲烷(1.63mL,1.63mmol)。进一步搅拌1小时后,用1小时将反应混合物升温至室温。向反应混合物中在冰冷却下加入饱和氯化铵水溶液(3.4mL),用乙酸乙酯(1.7mL×2次)萃取,用无水硫酸镁干燥后,过滤并浓缩。将浓缩残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化,得到标题化合物(78.0mg)。
1
参考例69
乙基[(2R,3S)-2-甲基-2,3-二氢呋喃并[3,2-b]吡啶-3-基]乙酸酯
[化101]
向参考例39的化合物(6.58g,21.9mmol)的甲苯溶液(73mL)中在室温加入三丁基氢化锡(9.77mL,37.3mmol)和偶氮二异丁腈(0.360g,2.19mmol)。在90℃搅拌1小时后,浓缩反应混合物。将残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化,以5:1的非对映异构体混合物形式得到标题化合物(3.87g)。
主非对映异构体
1
参考例70-1、参考例70-2
参考例70-1
苄基{(1S)-1-[(2R,3S)-2-甲基-2,3-二氢呋喃并[3,2-b]吡啶-3-基]乙基}氨基甲酸酯
参考例70-2
苄基{(1R)-1-[(2R,3S)-2-甲基-2,3-二氢呋喃并[3,2-b]吡啶-3-基]乙基}氨基甲酸酯
[化102]
通过与参考例67同样的方法,由参考例71的化合物得到标题化合物的参考例70-1和参考例70-2。
参考例70-1
1
参考例70-2
1
参考例71-1、参考例71-2
参考例71-1
乙基(2S)-2-[(2R,3S)-2-甲基-2,3-二氢呋喃并[3,2-b]吡啶-3-基]丙酸酯
参考例71-2
乙基(2R)-2-[(2R,3S)-2-甲基-2,3-二氢呋喃并[3,2-b]吡啶-3-基]丙酸酯
[化103]
向参考例69的化合物(800mg,3.62mmol)的四氢呋喃溶液(9.0mL)中在-78℃滴加1.0mol/L双(三甲基硅烷基)氨基锂四氢呋喃溶液(3.80mL,3.80mmol)。在-78℃搅拌30分钟后,加入碘甲烷(0.270mL,4.34mmol)。进一步搅拌1小时后,用1小时将反应混合物升温至室温。向反应混合物中在冰冷却下加入饱和氯化铵水溶液(18mL),用乙酸乙酯(18mL×2次)萃取,用无水硫酸镁干燥后,过滤并浓缩。将浓缩残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化,得到参考例71-1和参考例71-2的1.4∶1的非对映异构体混合物形式的标题化合物(826mg)。
主非对映异构体
1
参考例72~73
按照参考例1至参考例4和参考例6中记载的方法,由对应化合物得到参考例72~73的化合物。
[表17]
参考例74
rac-苄基{[(2R,3S)-2-甲基(2-
[化104]
通过与参考例8至参考例10和参考例37至参考例38同样的方法,由参考例75的化合物得到标题化合物。
1
参考例75
rac-1-{[叔丁基(二甲基)甲硅烷基]氧}(2-
[化105]
向丙烷-2-d-1,2-二醇(1.04g,13.5mmol)的氯仿溶液(45mL)中在室温加入咪唑(0.919g,13.5mmol)和叔丁基二甲基氯硅烷(1.83g,12.2mmol)。在室温搅拌2小时后,向反应混合物中加入水(200mL),用氯仿(200mL×2次)萃取,用无水硫酸钠干燥后,过滤并浓缩。将残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化,得到标题化合物(1.51g)。
1
参考例76-1、参考例76-2
参考例76-1
rac-苄基{[(2R,4R)-2-乙基-3,4-二氢-2H-吡喃并[3,2-b]吡啶-4-基]甲基}氨基甲酸酯
参考例76-2
rac-苄基{[(2R,4S)-2-乙基-3,4-二氢-2H-吡喃并[3,2-b]吡啶-4-基]甲基}氨基甲酸酯
[化106]
通过与参考例8和参考例37至参考例38同样的方法,由参考例77的化合物得到标题化合物的参考例76-1和参考例76-2。
参考例76-1
1
参考例76-2
1
参考例77
rac-3-[(2-溴吡啶-3-基)氧]戊烷-1-醇
[化107]
向参考例78的化合物(1.73g,6.31mmol)的氯仿溶液(13mL)中在冰冷却下加入1mol/L三溴化硼的二氯甲烷溶液(12.6mL,12.6mmol)。在室温搅拌2小时后,向反应混合物中加入饱和碳酸氢钠水溶液(100mL),用氯仿(100mL×2次)萃取,用无水硫酸钠干燥后过滤并浓缩。将残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化,得到标题化合物(0.541g)。
1
参考例78
rac-2-溴-3-[(1-甲氧基戊烷-3-基)氧]吡啶
[化108]
通过与参考例10同样的方法,由1-甲氧基戊烷-3-醇得到标题化合物。
1
参考例79
rac-苄基[(3’H-螺[环丙烷-1,2’-呋喃并[3,2-b]吡啶]-3’-基)甲基]氨基甲酸酯
[化109]
通过与参考例30至参考例31和参考例37至参考例38同样的方法,由参考例80的化合物得到标题化合物。
1
参考例80
甲基1-[(2-溴吡啶-3-基)氧]环丙烷-1-甲酸酯
[化110]
向参考例81的化合物(2.65g,7.51mmol)的四氢呋喃溶液(75mL)中在0℃加入叔丁醇钾(0.842g,7.51mmol)。在室温搅拌90分钟后,进一步加入叔丁醇钾(0.168g,1.50mmol)。在室温搅拌20分钟后,加水,用乙酸乙酯萃取,用无水硫酸钠干燥后,过滤并浓缩。将浓缩残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化,得到标题化合物(1.69g)。
1
参考例81
rac-甲基4-溴-2-[(2-溴吡啶-3-基)氧]丁酸酯
[化111]
向2-溴-3-羟基吡啶(1.67g,9.62mmol)的N,N-二甲基甲酰胺溶液(48.1mL)中在室温加入碳酸钾(2.66g,19.24mmol)和2,4-二溴丁酸甲酯(3.0g,11.54mmol)。在室温搅拌3小时后,向反应混合物中加入水,用乙酸乙酯萃取。将有机层用水洗涤3次,将有机层用无水硫酸钠干燥后,过滤并浓缩。将浓缩残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化,得到标题化合物(2.75g)。
1
参考例82
叔丁基{[(2R,3S)-2-甲基-2,3-二氢呋喃并[3,2-b]吡啶-3-基]甲基}丙-2-烯-1-基氨基甲酸酯
[化112]
向参考例83的化合物(50mg,0.189mmol)的四氢呋喃溶液(48.1mL)中在0度加入55%氢化钠(24.76mg,0.567mmol)。在室温搅拌90分钟后,加入烯丙基溴(0.048mL,0.567mmol)。在室温搅拌90分钟后,加入水,用乙酸乙酯萃取,用无水硫酸钠干燥后,过滤并浓缩。将浓缩残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化,得到标题化合物(34.8mg)。
1
参考例83
叔丁基{[(2R,3S)-2-甲基-2,3-二氢呋喃并[3,2-b]吡啶-3-基]甲基}氨基甲酸酯
[化113]
通过与实施例5同样的方法,由实施例27的化合物得到标题化合物。
1
参考例84
叔丁基{[(2R,3S)-2-甲基-2,3-二氢呋喃并[3,2-b]吡啶-3-基]甲基}丙基氨基甲酸酯
[化114]
向参考例82的化合物(34.8mg,0.114mmol)的甲醇溶液(1.0mL)中加入10%钯/碳(25mg)。在氢气氛中在室温搅拌2小时后,进行硅藻土过滤并浓缩。将浓缩残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化,得到标题化合物(32.0mg)。
1
参考例85~87
按照参考例82中记载的方法,由对应化合物得到参考例85~87的化合物。
[表18]
参考例88-1、参考例88-2、参考例88-3、参考例88-4
参考例88-1
rac-苄基{[(2R,3R,4R)-2,3-二甲基-3,4-二氢-2H-吡喃并[3,2-b]吡啶-4-基]甲基}氨基甲酸酯
参考例88-2
rac-苄基{[(2R,3S,4R)-2,3-二甲基-3,4-二氢-2H-吡喃并[3,2-b]吡啶-4-基]甲基}氨基甲酸酯
参考例88-3
rac-苄基{[(2S,3R,4R)-2,3-二甲基-3,4-二氢-2H-吡喃并[3,2-b]吡啶-4-基]甲基}氨基甲酸酯
参考例88-4
rac-苄基{[(2S,3S,4R)-2,3-二甲基-3,4-二氢-2H-吡喃并[3,2-b]吡啶-4-基]甲基}氨基甲酸酯
[化115]
通过与参考例8至参考例10、参考例37至参考例38和参考例75同样的方法,由2-甲基丁烷-1,3-二醇得到标题化合物的参考例88-1、参考例88-2、参考例88-3和参考例88-4。
参考例88-1
1
参考例88-2
1
参考例88-3
1
参考例88-4
1
参考例89
rac-叔丁基[(6’-氯-3’H-螺[环丙烷-1,2’-呋喃并[3,2-b]吡啶]-3’-基)甲基]氨基甲酸酯
[化116]
通过与参考例6至参考例7和参考例30至参考例31同样的方法,由参考例90的化合物得到标题化合物。
1
参考例90
甲基1-[(2-溴-5-氯吡啶-3-基)氧]环丙烷-1-甲酸酯
[化117]
通过与参考例80至参考例81同样的方法,由2-溴-5-氯-3-羟基吡啶得到标题化合物。
1
参考例91
rac-2-[(2-甲氧基-5,6,7,8-四氢喹啉-8-基)甲基]-1H-异吲哚-1,3(2H)-二酮
[化118]
向参考例92的化合物(110mg,0.337mmol)、二甲基亚砜(1.68mL)、甲醇(1.68mL)的混合物中在室温下加入甲醇钠(364mg,6.73mmol)。在100℃搅拌后,向反应混合物中加水,用乙酸乙酯萃取,用硫酸钠干燥后,过滤并浓缩。将浓缩残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化,得到标题化合物(9mg)。
1
参考例92
rac-2-[(2-氯-5,6,7,8-四氢喹啉-8-基)甲基]-1H-异吲哚-1,3(2H)-二酮
[化119]
向参考例93的化合物(440mg,1.427mmol)中在室温下加入磷酰氯(2.0mL,21.46mmol)。在90℃搅拌4小时后,向反应混合物中加入冰水。然后加入4mol/L氢氧化钠,用乙酸乙酯萃取,用硫酸钠干燥后,过滤并浓缩。将浓缩残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化,得到标题化合物(110mg)。
1
参考例93
rac-2-[(1-氧代-5,6,7,8-四氢-1λ
[化120]
向参考例94的化合物(608mg,2.080mmol)的氯仿溶液(10.4mL)中在0℃加入70%3-氯过氧苯甲酸(564mg,2.29mmol)。在0℃搅拌4小时后,向反应混合物中加入饱和碳酸氢钠水,用氯仿萃取,用硫酸钠干燥后,过滤并浓缩,得到标题化合物(646mg)。
1
参考例94
rac-2-[(5,6,7,8-四氢喹啉-8-基)甲基]-1H-异吲哚-1,3(2H)-二酮
[化121]
向5,6,7,8-四氢喹啉-8-基甲胺(400mg,2.47mmol)的氯仿溶液(25mL)中加入邻苯二甲酸酐(548mg,3.70mmol)。在70℃搅拌5小时后,浓缩反应混合物。将浓缩残渣通过硅胶色谱(己烷/乙酸乙酯)分离纯化,得到标题化合物(608mg)。
1
参考例95~96
按照参考例79至参考例81中记载的方法,由对应的化合物得到参考例95~96的化合物。
[表19]
参考例97
rac-叔丁基[(4-氯-6,7-二氢-5H-环戊烷并[b]吡啶-7-基)甲基]氨基甲酸酯
[化122]
向参考例98的化合物(49.1mg,0.275mmol)的甲苯溶液(1.374mL)中在0℃加入1mol/L二异丁基氢化铝的甲苯溶液(0.825mL,0.825mmol)。在室温搅拌3小时后,向反应混合物中加入30%酒石酸钾钠水溶液、饱和碳酸氢钠水、乙酸乙酯。向该混合溶液中在室温加入二碳酸二叔丁酯(90mg,0.412mmol)。在室温搅拌1小时后,向反应混合物中加入水,用乙酸乙酯萃取。将有机层用无水硫酸钠干燥后,过滤并浓缩。将浓缩残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化,得到淡黄色油状物形式的标题化合物(30.9mg)。
1
参考例98
rac-4-氯-6,7-二氢-5H-环戊烷并[b]吡啶-7-甲腈
[化123]
向4-氯-6,7-二氢-5H-环戊烷并[B]吡啶-7-醇(100mg,0.590mmol)和丙酮氰醇(0.081mL,0.884mmol)的甲苯溶液(1.965mL)中加入氰基亚甲基三丁基膦(0.464mL,1.769mmol)。在60℃搅拌1小时后,向反应混合物中加入水,用乙酸乙酯萃取,用无水硫酸钠干燥后,过滤并浓缩。将浓缩残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化,得到标题化合物(49.1mg)。
1
参考例99
rac-叔丁基[(4-乙氧基-6,7-二氢-5H-环戊烷并[b]吡啶-7-基)甲基]氨基甲酸酯
[化124]
向参考例100的化合物(50mg,0.189mmol)、乙醇(0.055mL,0.946mmol)、三苯基膦(74.4mg,0284mmol)的甲苯溶液(1.89mL)中在0℃加入偶氮二甲酸双(2-甲氧基乙基)酯。在室温搅拌1小时后,向反应混合物中加入甲醇,浓缩。将浓缩残渣通过硅胶柱色谱(己烷/乙酸乙酯)进行分离纯化,得到标题化合物(35mg)。
1
参考例100
rac-叔丁基[(4-羟基-6,7-二氢-5H-环戊烷并[b]吡啶-7-基)甲基]氨基甲酸酯
[化125]
向参考例97的化合物(248mg,0.877mmol)和碳酸钾(606mg,4.39mmol)的N-甲基-2-吡咯烷酮混悬液(1.46mL)中加入乙酰氧肟酸(198mg,2.63mmol)。在100℃搅拌6小时后,将反应混合物进行硅藻土过滤并浓缩。将浓缩残渣通过硅胶柱色谱(氯仿/甲醇)分离纯化,得到标题化合物(128mg)。
1
参考例101
rac-2-{[4-(4-甲基苯基)-5,6,7,8-四氢喹啉-8-基]甲基}-1H-异吲哚-1,3(2H)-二酮
[化126]
向参考例102的化合物(100mg,0.306mmol)、4,5-双(二苯基膦基)-9,9-二甲基呫吨(53.1mg,0.092mmol)、4-甲基苯基硼酸(125mg,0.918mmol)和碳酸铯(199mg,0.612mmol)的甲苯混悬液(2.3mL)中加入三(二亚苄基丙酮)二钯(0)(56mg,0.061mmol)。在130℃搅拌后,向反应混合物中加入水,用乙酸乙酯萃取,用无水硫酸钠干燥后,过滤并浓缩。将浓缩残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化,得到标题化合物(26.5mg)。
1
参考例102
rac-2-[(4-氯-5,6,7,8-四氢喹啉-8-基)甲基]-1H-异吲哚-1,3(2H)-二酮
[化127]
向参考例93的化合物(500mg,1.62mmol)中在室温下加入磷酰氯(1.5mL,16.22mmol)。在90℃搅拌4小时后,向反应混合物中加入冰水。然后,加入4mol/L氢氧化钠,用乙酸乙酯萃取,用硫酸钠干燥后,过滤并浓缩。将浓缩残渣通过硅胶柱色谱(己烷/乙酸乙酯)分离纯化,得到标题化合物(256mg)。
1
参考例103-1、参考例103-2
参考例103-1
rac-苄基{[(3R,4S)-3-乙基-3,4-二氢-2H-吡喃并[3,2-b]吡啶-4-基]甲基}氨基甲酸酯
参考例103-2
rac-苄基{[(3S,4S)-3-乙基-3,4-二氢-2H-吡喃并[3,2-b]吡啶-4-基]甲基}氨基甲酸酯
[化128]
通过与参考例2至参考例4和参考例37至参考例38同样的方法,由2-乙基丙烷-1,3-二醇得到标题化合物的参考例103-1和参考例103-2。
参考例103-1
1
参考例103-2
1
参考例104
rac-4-甲基-5,6,7,8-四氢喹啉-8-甲腈
[化129]
通过与参考例62同样的方法,由参考例105的化合物得到标题化合物。
1
参考例105
rac-8-溴-4-甲基-5,6,7,8-四氢喹啉
[化130]
向参考例106的化合物(87.0mg,0.530mmol)的氯仿溶液(5.3mL)中加入三溴化磷(0.0747mL,0.795mmol)。在70℃2小时后,向反应混合物中加入饱和碳酸氢钠水溶液(30mL),用氯仿(30mL×2次)萃取,用无水硫酸钠干燥后,通过过滤并浓缩得到标题化合物(118mg)。
1
参考例106
rac-4-甲基-5,6,7,8-四氢喹啉-8-醇
[化131]
向参考例107的化合物(85.5mg,0.530mmol)的甲醇溶液(5.3mL)中在冰冷却下加入硼氢化钠(30.1mg,0.796mmol)。在室温搅拌3小时后,向反应混合物中加入饱和氯化铵水溶液(30mL),用氯仿(30mL×2次)萃取,用无水硫酸钠干燥后,过滤并浓缩,由此得到标题化合物(93.7mg)。
1
参考例107
4-甲基-6,7-二氢喹啉-8(5H)-酮
[化132]
将4-氯-6,7-二氢喹啉-8(5H)-酮(302mg,1.67mmol)、三甲基环三硼氧烷(0.756mL,5.45mmol)、1,1’-双(二苯基膦基)二茂铁氯化钯(II)(130mg,0.159mmol)、碳酸钾(388mg,2.81mmol)和二氯乙烷(2.0mL)的混合物在微波照射下在120℃搅拌2小时。然后,将反应混合物通过硅胶柱色谱(氯仿/甲醇)分离纯化,得到标题化合物(85.5mg)。
1
参考例108-1、参考例108-2
参考例108-1
rac-(5R,7R)-5-甲基-6,7-二氢-5H-环戊烷并[b]吡啶-7-甲腈
参考例108-2
rac-(5R,7S)-5-甲基-6,7-二氢-5H-环戊烷并[b]吡啶-7-甲腈
[化133]
通过与参考例104至参考例105同样的方法,由参考例109的化合物以参考例108-1和参考例108-2的1∶1非对映异构体混合物形式得到标题化合物。
1
参考例109-1、参考例109-2
参考例109-1
rac-(5R,7R)-5-甲基-6,7-二氢-5H-环戊烷并[b]吡啶-7-醇
参考例109-2
rac-(5R,7S)-5-甲基-6,7-二氢-5H-环戊烷并[b]吡啶-7-醇
[化134]
在冰冷却下向参考例110的化合物(232mg,1.55mmol)的氯仿溶液(2.0mL)中加入三氟乙酸酐(3.0mL,21.4mmol)。在室温搅拌20小时后,向反应混合物中加入1mol/L氢氧化钠水溶液(30mL),用氯仿(30mL×2次)萃取,用无水硫酸钠干燥后,过滤并浓缩,由此以参考例109-1和参考例109-2的1∶1非对映异构体混合物形式得到标题化合物(125mg)。
1
参考例110
rac-5-甲基-1-氧代-6,7-二氢-5H-1λ
[化135]
通过与参考例93同样的方法,由5-甲基-6,7-二氢-5H-环戊烷并[b]吡啶得到标题化合物。
1
参考例111-1、参考例111-2
参考例111-1
rac-叔丁基{[(5R,8S)-5-甲基-5,6,7,8-四氢喹啉-8-基]甲基}氨基甲酸酯
参考例111-2
rac-叔丁基{[(5R,8R)-5-甲基-5,6,7,8-四氢喹啉-8-基]甲基}氨基甲酸酯
[化136]
通过与参考例61同样的方法,由参考例112的化合物以参考例111-1和参考例111-2的非对映异构体混合物形式得到标题化合物。
1
参考例112-1、参考例112-2
参考例112-1
rac-(5R,8R)-5-甲基-5,6,7,8-四氢喹啉-8-甲腈
参考例112-2
rac-(5R,8S)-5-甲基-5,6,7,8-四氢喹啉-8-甲腈
[化137]
通过与参考例104至参考例105、和参考例109至参考例110同样的方法,由5-甲基-5,6,7,8-四氢喹啉以参考例112-1和参考例112-2的非对映异构体混合物形式得到标题化合物。
1
参考例113~114
按照参考例5和参考例55中记载的方法,由对应实施例的化合物得到参考例113~114的化合物。
[表20]
参考例115
rac-苄基[(2,2,5-三甲基-2,3-二氢呋喃并[3,2-b]吡啶-3-基)甲基]氨基甲酸酯
[化138]
通过与参考例30至参考例32、和参考例37至参考例38同样的方法,由2-溴-6-甲基吡啶-3-醇和乙基2-溴-2-甲基丙酸酯得到标题化合物。
1
参考例116
rac-苄基{[(6S,7S)-3-氯-6-甲基-6,7-二氢-5H-环戊烷并[b]吡啶-7-基]甲基}氨基甲酸酯
[化139]
通过与参考例30和参考例37至参考例38同样的方法,由参考例117的化合物得到标题化合物。
1
参考例117
rac-3-(2-溴-5-氯吡啶-3-基)-2-甲基丙烷-1-醇
[化140]
向2-溴-5-氯烟醛(999mg,4.30mmol)和甲苯(11mL)的混合物中在冰冷却下加入2-(三苯基膦基)丙酸乙酯(1.64g,4.52mmol)。在90℃搅拌3小时后,浓缩反应混合物。将浓缩残渣用硅胶柱色谱(己烷/乙酸乙酯)分离纯化。
向得到的纯化物(1.29g)和四氢呋喃(42.5mL)的混合物中在冰冷却下用5分钟滴加4mol/L硼氢化锂四氢呋喃溶液(1.28mL,5.10mmol)。在室温搅拌15分钟后,在60℃搅拌2小时,在冰冷却下加入甲醇(5mL)和1mol/L盐酸(10mL)。用乙酸乙酯(20mL×3次)萃取,用无水硫酸钠干燥后,过滤并浓缩。将浓缩残渣通过氨基硅胶柱色谱(己烷/乙酸乙酯)分离纯化,得到标题化合物(469mg)。
1
参考例118
rac-苄基[(5-氟-2,2-二甲基-2,3-二氢呋喃并[3,2-b]吡啶-3-基)甲基]氨基甲酸酯
[化141]
通过与参考例30至参考例32和参考例37至参考例38同样的方法,由2-溴-6-氟吡啶-3-醇和乙基2-溴-2-甲基丙酸酯得到标题化合物。
1
试验例1:对人型TAAR1受体的激动剂活性评价
TAAR1为结合于G蛋白质(Gαs)的G蛋白质共轭型受体,激动剂对TAAR1受体的活化诱导细胞内cAMP水平的增加。因此,使用cAMP测定法评价受试化合物对人TAAR1受体的激动剂活性。
购买了表达人型TAAR1受体的细胞cAMP Hunter
受试化合物%=100×{(Csamp)-(Cblank)}/{(Ctyramine)-(Cblank)}
Csamp:受试化合物计数、Ctyramine:100μM酪胺计数、Cblank:空白计数
[表21-1]
[表21-2]
[表21-3]
[表21-4]
/>
[表21-5]
试验例2-1:苯环利定诱发运动亢进抑制试验
使用8周龄的C57BL/6J雄性小鼠。在受试化合物的施与液制备中,使用0.5%甲基纤维素溶液作为溶剂混悬使用,在苯环利定的施与液制备中,使用生理食盐水作为溶剂溶解使用。
苯环利定诱发运动亢进抑制试验利用室町机械公司制造的Supermex、数据集录程序CompACT AMS和透明塑料制笼如下实施。
将动物放入上述笼中,开始测定运动量。30分钟后,安静地取出小鼠,经口施与化合物施与液(溶剂或受试化合物混悬液),将小鼠放回笼中。施与30分钟后皮下施与苯环利定施与液或生理食盐水。施与两种后,迅速将小鼠返回各通道的运动量测定笼,继续进行运动量测定。在运动量测定中,测量间隔5分钟,测量开始后进行120分钟。使用运动量测定开始30分钟后到120分钟后的90分钟的数据作为试验结果,分别对个体的90分钟的运动量进行总计。
在试验化合物施与组和溶剂施与组中,进行了parametric Dunnett型多重比较(显著水平:两侧5%)。试验化合物施与组与溶剂施与组相比,显示出显著的运动量的抑制时,判断为具有抗精神病作用。
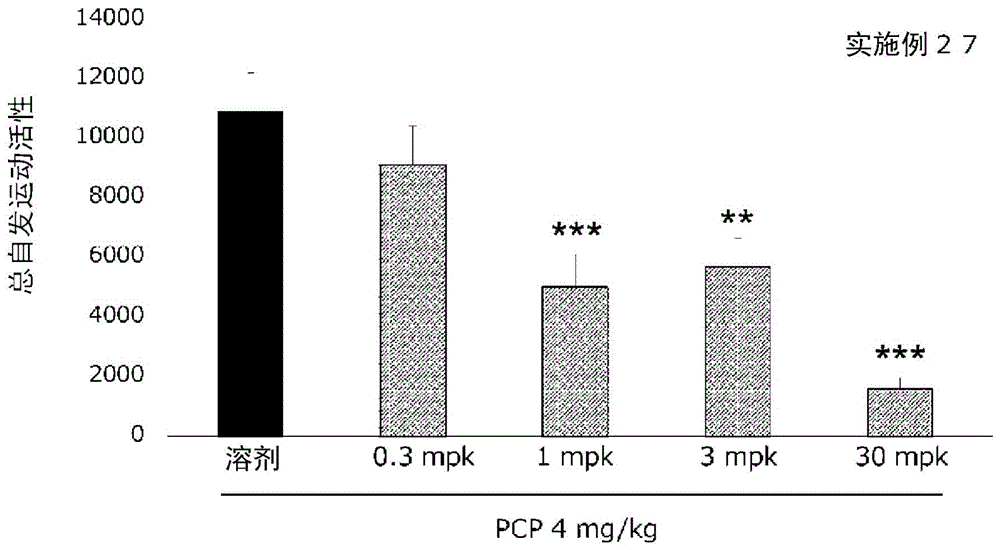
使用实施例27的化合物的上述试验的结果如图1所示。
试验例2-2:苯环利定诱发运动亢进抑制试验
使用8周龄的C57BL/6J雄性小鼠。在受试化合物的施与液制备中,使用生理食盐水作为溶剂混悬使用,在苯环利定的施与液制备中,使用生理食盐水作为溶剂溶解使用。
使用具有光电组件和电动计数器的活动计,如下进行苯环利定诱发运动亢进抑制试验。
将皮下施与了化合物施与液(溶剂或受试化合物混悬液)的小鼠放入上述装置中,开始运动量测定。测定开始30分钟后,安静地取出小鼠,皮下施与苯环利定施与液或生理食盐水。施与后迅速将小鼠返回装置内,持续60分钟测定运动量。运动量测定将测量间隔设为5分钟。使用开始运动量测定30分钟后到90分钟后的60分钟的数据作为试验结果,将各个体60分钟的运动量总计。
在试验化合物施与组和溶剂施与组中,进行了parametric Dunnett型多重比较(显著水平:两侧5%)。试验化合物施与组与溶剂施与组相比,显示出显著的运动量的抑制时,判断为具有抗精神病作用。
使用实施例8和实施例49的化合物的上述试验的结果如图2和图3所示。
试验例3:hERG通道抑制活性的评价
本公开的化合物的hERG通道抑制作用是使用CHO细胞通过采用自动膜片钳系统的全细胞膜片钳法测量,所述CHO细胞强制表达了与人快速活性型延迟整流钾电流(IKr)有关的hERG通道。
(细胞混悬液的制备)
将从ChanTest公司购买的hERG-CHO细胞在CO
(溶液制备)
如下制备用于测定的细胞外液、细胞内液。
细胞外液:2mmol/L CaCl
HEPES、4mmol/L KCl、145mmol/L NaCl、10mmol/L葡萄糖
细胞内液:10mmol/L HEPES、10mmol/L EGTA、20mmol/L KCl、130mmol/L KF
受试物质溶液:将受试物质溶解于DMSO中,使其为2mmol/L或20mmol/L,制备受试物质溶液。此外,用细胞外液将受试物质溶液稀释200倍,用细胞外液将其阶段稀释,由此制备计算hERG抑制IC50值所需的各浓度的受试物质溶液并应用。
(电流值测定和数据分析)
将细胞混悬液、细胞外液、细胞内液和测定板设置在自动膜片钳系统中,采用全细胞膜片钳法实施hERG电流测定。电压协议将保持电位设为-80mV,将去极化脉冲从-50mV到+20mV施加5秒钟后,将复极化脉冲在-50mV施加5秒钟,返回保持电位。各脉冲间隔为15秒。数据分析使用Qube用分析软件(Sophion Sophion公司制造)。对各受试物质递增地适用4个浓度,将在各适用浓度的最终3次刺激中得到的最大外向电流(Peaktail current)的平均值作为评价数据。
另外,根据各受验物质的各浓度下的电流对适用前值的抑制率,使用该软件,通过Hill方程(Hill equation)算出IC
结果如下表所示。
[表22-1]
[表22-2]
[表22-3]
试验例4:对副作用相关受体的结合活性评价
本公开化合物对副作用相关受体(例如,多巴胺D2受体、肾上腺素α1A受体)的结合亲和性可以通过以下方法测定。
使用表达人型目标受体的CHO细胞膜组分,如下实施结合评价试验。将溶解于二甲基亚砜(DMSO)中的受试化合物、用缓冲液稀释的各种受体膜标本、和对各目标受体具有强结合活性的[3H]标记配体混合,分别在室温下孵育后,迅速添加到玻璃纤维过滤板(Multiscreen FB,Millipore公司制)上进行减压过滤。使用液体闪烁计数器(PerkinElmer公司制造)测定残留在过滤器上的放射活性。结合抑制率根据下式计算。在对受体膜标本的非特异性结合量的计算中,使用对目标受体具有强结合活性的对照化合物代替受试物质。
对目标受体的结合抑制率(%)=100-100×{(受试物质存在下的[
(注释)
如上所述,使用本公开的优选实施方式对本公开进行了例示,但是应理解本公开仅应通过权利要求书来解释其范围。本申请对作为日本申请的特愿2021-66825(2021年4月10日申请)和特愿2021-150394(2021年9月15日申请)要求优先权,其内容整体在本说明书中作为参考被援用。可以理解,在本说明书中引用的专利、专利申请、科学文献以及其他文献,其内容本身与在本说明书中具体记载的相同,其内容应作为对本说明书的参考而被援用。
产业实用性
本公开的化合物对痕量胺相关受体TAAR1受体具有激动剂活性,因此对精神疾病的治疗有效。另外,本公开的化合物对中枢神经系统疾病也有效。
- 作为PI3Kβ抑制剂的二环吡啶、二环吡嗪、和二环嘧啶衍生物
- 8-氮杂二环3.2.1辛烷-,8-氮杂二环3.2.1辛-6-烯,9-氮杂二环3.3.1壬烷-,9-氮杂-3-氧杂二环3.3.1壬烷-,9-氮杂-3-硫杂二环3,3,1壬烷衍生物,其制备方法及其作为杀虫剂的用途