合成吸附剂、抗体的纯化方法和抗体的制造方法
文献发布时间:2024-04-18 19:58:53
技术领域
本发明涉及一种合成吸附剂、抗体的纯化方法和抗体的制造方法。
背景技术
近年来,生物医药品由于基因重组技术的发达而迅速发展。其中,抗体医药品是生产量逐年增加的医药品。抗体医药品与其他低分子医药品相比是非常昂贵的医药品。作为抗体医药品价格昂贵的原因之一,可举出抗体的纯化成本巨大。
抗体这样的蛋白质一般而言是通过培养插入有载体(vector)的重组细胞而产生的,该载体包含对作为目标的蛋白质进行编码的基因。在培养液中除了包含作为目标的蛋白质以外,还包含来自多种多样的培养基的成分、来自宿主细胞的成分、来自蛋白质的副产物等杂质。因此,需要将杂质分离除去至作为医药品所要求的纯度为止,并对目标蛋白质进行纯化。
如果利用蛋白质分解酶木瓜蛋白酶将抗体分解,则抗体的特定部位被切断,被分为称为Fab区域和Fc区域的部分。作为可与Fc区域特异性结合的蛋白质,可举出蛋白质(protein)A。利用该蛋白质的特异性而仅吸附抗体并将抗体与杂质分离的方法是使用蛋白质A的亲和色谱。该分离方法能够从包含多种多样的杂质的培养液中选择性地仅捕捉作为目标的抗体。因此,可用于大部分的抗体医药品的纯化。
专利文献1中公开了一种使用亲和色谱的抗体的纯化方法。另一方面,在专利文献2中公开了一种不使用亲和色谱而利用活性碳的纯化方法。
专利文献1:日本特表平5-504579号公报
专利文献2:国际公开第2014/024514号
与利用了一般的离子交换作用或疏水性相互作用的分离剂相比,专利文献1中公开的使用了蛋白质A等的亲和色谱的亲和分离剂非常昂贵。因此,大量使用亲和分离剂而生产的抗体医药品也变得价格昂贵。
如专利文献2公开所示,通过采用不使用亲和色谱而利用活性碳的纯化方法,也能够降低抗体的制造成本。但是,该方法中,与亲和色谱相比,作为目标的抗体的回收率大幅低。
如上所述,以往的抗体的纯化方法难以实现低成本且高生产。
发明内容
本发明的目的在于提供一种合成吸附剂,能够降低抗体的制造成本,且在工业规模上也有效地生产抗体。另外,本发明的目的在于提供一种抗体的纯化方法和抗体的制造方法,能够降低抗体的制造成本,且在工业规模上也有效地生产抗体。
本发明人等发现,通过将微分细孔容积(mL/g)的最大值超过规定值的合成吸附剂用于抗体的纯化,能够实现低成本且高生产的抗体的纯化方法,从而完成了本发明。
即,本发明的主旨如下所述。
[1]一种合成吸附剂,利用以下方法测定的、0.5psia~30.0psia的压力条件下的微分细孔容积(mL/g)的最大值超过0.05mL/g。
<微分细孔容积(mL/g)的测定方法>
1.将放入有干燥后的该合成吸附剂的试样容器减压至10Pa以下,且将JIS K8572中规定的汞减压至10Pa以下并进行脱泡后,以0.5psia的压力填充至该试样容器中。
2.对于填充有该汞的试样容器,使压力从0.5psia阶段性地上升至30.0psia,测定此时的汞压入量。
3.求出微分细孔容积(mL/g),上述微分细孔容积是用基于上述2中测定的汞压入量而算出的使压力上升了一阶段时的汞压入量的增加量除以测定合成吸附剂量而得的。
[2]根据[1]所述的合成吸附剂,是抗体纯化用的合成吸附剂。
[3]根据[1]或[2]所述的合成吸附剂,其中,具有细孔。
[4]根据[3]所述的合成吸附剂,其中,上述细孔的半径为3nm~15nm。
[5]根据[1]~[4]中任一项所述的合成吸附剂,其为疏水性。
[6]根据[1]~[5]中任一项所述的合成吸附剂,其中,包含选自苯乙烯系树脂和丙烯酸系树脂中的至少一种。
[7]根据[1]~[6]中任一项所述的合成吸附剂,其中,体积平均粒径为1μm~500μm。
[8]根据[1]~[7]中任一项所述的合成吸附剂,其中,利用以下方法求出的校正正圆度的值超过0.10。
<校正正圆度的值的测定和算出方法>
1.使用光学显微镜拍摄合成吸附剂。
2.使用图像解析软件WinROOF2018制作所拍摄的合成吸附剂的近似圆。
3.按照JIS B0621,利用与该近似圆同心的两个同心的几何圆即同心二圆夹持。
4.求出上述3的同心二圆的间隔为最小时的上述同心二圆中的外周侧的圆的半径与内周侧的圆的半径之差作为正圆度。
5.将求出的正圆度除以上述近似圆的半径。
6.对100个点以上进行上述1~5的测定和算出,将其平均值作为校正正圆度。
[9]根据[1]~[8]中任一项所述的合成吸附剂,其中,压溃强度为100gf/粒~2000gf/粒。
[10]根据[2]~[9]中任一项所述的合成吸附剂,其中,上述抗体为单克隆抗体。
[11]根据[10]所述的合成吸附剂,其中,上述单克隆抗体的分子量为100000以上。
[12]根据[10]或[11]所述的合成吸附剂,其中,上述单克隆抗体为免疫球蛋白G。
[13]一种抗体的纯化方法,在将包含抗体和杂质的混合物与[1]~[12]中任一项所述的合成吸附剂混合后进行过滤。
[14]一种抗体的纯化方法,包括如下工序:使包含抗体和杂质的混合物通过[1]~[12]中任一项所述的合成吸附剂,回收未被吸附剂吸附的包含抗体的非吸附级分。
[15]一种抗体的纯化方法,具有下述工序(i)和工序(ii):
工序(i):接触工序,使包含抗体和杂质的混合溶液与[1]~[12]中任一项所述的合成吸附剂接触,
工序(ii):分离工序,在上述工序(i)之后,将上述混合溶液与上述合成吸附剂分离。
[16]根据[15]所述的抗体的纯化方法,其中,在上述工序(i)的前段具有下述工序(A):
工序(A):离子交换树脂接触工序,使包含抗体和杂质的混合溶液与阴离子交换树脂和/或阳离子交换树脂接触。
[17]根据[13]~[16]中任一项所述的抗体的纯化方法,其中,上述杂质包含分子量50000以下的物质。
[18]根据[13]~[17]中任一项所述的抗体的纯化方法,其中,上述杂质包含选自来自宿主细胞的蛋白质、来自抗体的聚合物、来自抗体的分解物和核酸中的至少一种。
[19]根据[13]~[18]中任一项所述的抗体的纯化方法,其中,在pH为3~8的液体中进行上述混合物与合成吸附剂的混合、上述混合物在上述合成吸附剂中的通过、或者上述混合溶液与合成吸附剂的接触。
[20]一种抗体的制造方法,包括[13]~[19]中任一项所述的抗体的纯化方法。
另外,本发明的第二主旨如下所述。
<1>一种合成吸附剂,是抗体纯化用的破碎的合成吸附剂。
<2>根据<1>所述的合成吸附剂,其中,具有细孔。
<3>根据<2>所述的合成吸附剂,其中,上述细孔的半径为3nm~15nm。
<4>根据<1>~<3>中任一项所述的合成吸附剂,其为疏水性。
<5>根据<1>~<4>中任一项所述的合成吸附剂,其中,包含选自苯乙烯系树脂和丙烯酸系树脂中的至少一种。
<6>根据<1>~<5>中任一项所述的合成吸附剂,其中,体积平均粒径为1μm~500μm。
<7>根据<1>~<6>中任一项所述的合成吸附剂,其中,上述抗体为单克隆抗体。
<8>根据<7>所述的合成吸附剂,其中,上述单克隆抗体的分子量为100000以上。
<9>根据<7>或<8>所述的合成吸附剂,其中,上述单克隆抗体为免疫球蛋白G。
<10>一种抗体的纯化方法,其中,在将包含抗体和杂质的混合物与<1>~<9>中任一项所述的合成吸附剂混合后进行过滤。
<11>一种抗体的纯化方法,包括如下工序:使包含抗体和杂质的混合物通过<1>~<9>中任一项所述的合成吸附剂,回收未被合成吸附剂吸附的包含抗体的非吸附级分。
<12>根据<10>或<11>所述的抗体的纯化方法,其中,上述杂质包含分子量50000以下的物质。
<13>根据<10>~<12>中任一项所述的抗体的纯化方法,其中,上述杂质包含选自来自宿主细胞的蛋白质、来自抗体的聚合物、来自抗体的分解物和核酸中的至少一种。
<14>根据<10>~<13>中任一项所述的抗体的纯化方法,其中,在pH为3~8的液体中进行上述混合物与合成吸附剂的混合。
<15>一种抗体的制造方法,包括<10>~<14>中任一项所述的抗体的纯化方法。
本发明的合成吸附剂能够降低抗体的纯化、制造成本,且在工业规模上也有效地生产抗体。
本发明的抗体的纯化方法和抗体的制造方法能够降低抗体的纯化、制造成本,且在工业规模上也有效地生产抗体。
附图说明
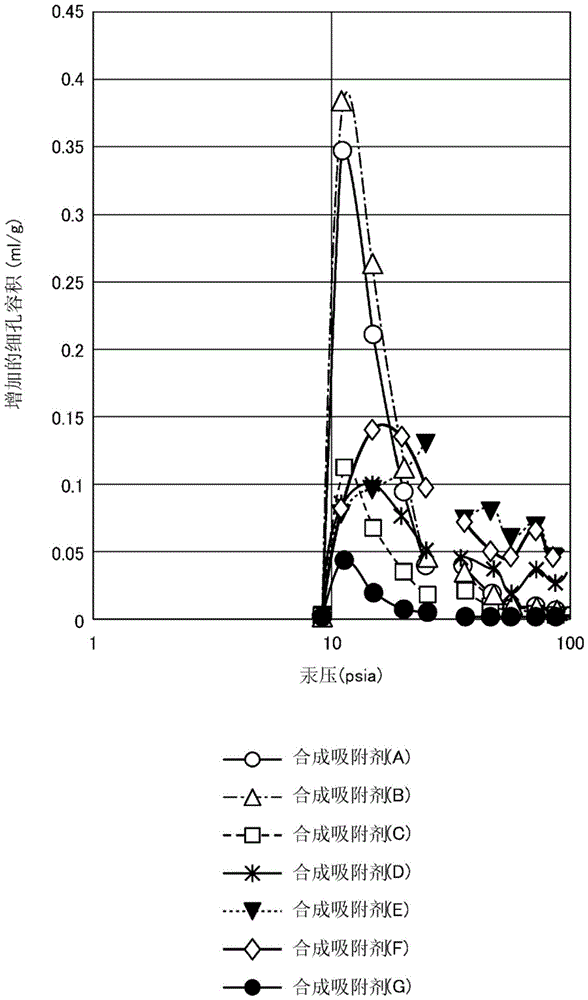
图1是表示实施例和比较例中的合成吸附剂(A)~(G)的微分细孔容积(mL/g)的测定值的图表。
具体实施方式
以下对本发明进行详述。本发明并不限定于以下的实施方式,可在其主旨的范围内进行各种变更而实施。
在本说明书中使用“~”的表述的情况下,作为包含其前后的数值或物性值的表述而使用。
在本说明书中,“(甲基)丙烯酸”是指“丙烯酸”、“甲基丙烯酸”或该两者。“(甲基)丙烯酸酯”是指“丙烯酸酯”、“甲基丙烯酸酯”或该两者。对于“(甲基)丙烯酰基”也同样。
(合成吸附剂)
本发明的合成吸附剂的特征在于,利用以下方法测定的0.5psia~30.0psia的压力条件下的微分细孔容积(mL/g)的最大值超过0.05mL/g。
<微分细孔容积(mL/g)的测定方法>
1.将放入有干燥后的该合成吸附剂的试样容器减压至10Pa以下,且将JIS K8572中规定的汞减压至10Pa以下并进行脱泡后,以0.5psia的压力填充至该试样容器中。
2.对于填充有该汞的试样容器,使压力从0.5psia阶段性地上升至30.0psia,测定此时的汞压入量。
3.求出微分细孔容积(mL/g),该微分细孔容积(mL/g)是用基于上述2中测定的汞压入量而算出的使压力上升了一阶段时的汞压入量的增加量除以测定合成吸附剂量而得的。
汞压入法是一种空隙或细孔容积的测定方法,其利用由于汞的表面张力大,因此,仅在大气压力下无法进入空隙或细孔的情况,通过利用压力将汞压入来进行测定。测定中,使压力阶段性地上升,利用静电电容式等的检测器检测所注入的汞量,使用增加的汞的注入量来算出细孔容积。
使压力阶段性地上升并对汞压入量进行测定,将用基于所测定的汞压入量而算出的使压力上升了一阶段时的汞压入量的增加量除以测定合成吸附剂量而得的值称为微分细孔容积(mL/g)。
在微分细孔容积(mL/g)的测定中,使压力阶段性地上升时的每次测定的压力的上升幅度虽然也取决于所使用的细孔容积测定装置的规格,但从能够准确地掌握本发明的微分细孔容积(mL/g)的方面出发,优选为2psia~10psia,特别优选为2psia~5psia的范围。
在本发明中,关于微分细孔容积(mL/g),具体而言,如后述公开的实施例的项中记载所示,使用细孔分布测定装置(机种名“AutoPore IV 9520”,MicromeriticsInstruments Corporation公司制造),通过汞压入法,并根据使测定压力阶段性地上升时的汞压入量的增加量求出微分细孔容积(mL/g)。作为一个例子的汞压入量的测定压力值优选为以下的压力。
第一测定压力值:9.0±0.5psia
第二测定压力值:11.0±0.5psia
第三测定压力值:15.0±0.5psia
第四测定压力值:20.0±0.5psia
第五测定压力值:25.0±0.5psia
本发明的合成吸附剂的特征在于,在0.5psia~30.0psia的范围的测定压力下的微分细孔容积(mL/g)的最大值超过0.05mL/g。微分细孔容积(mL/g)的最大值超过0.05mL/g的合成吸附剂在合成吸附剂粒子表面具有杂质除去性优异的细孔,杂质的除去性、抗体的纯化效率优异。由于杂质的除去性优异,因此本发明的合成吸附剂的微分细孔容积(mL/g)的最大值超过0.05mL/g,优选为0.1mL/g以上,更优选为0.2mL/g以上,进一步优选为0.3mL/g以上。另一方面,关于微分细孔容积(mL/g)的最大值的上限,没有特别限制,但从测定的准确性或合成吸附剂的物理强度的观点考虑,通常为1.0mL/g以下。
微分细孔容积(mL/g)的最大值超过0.05mL/g的合成吸附剂例如可通过如下方式得到:如后所述将市售的合成吸附剂(合成树脂)粒子或所制造的合成吸附剂(合成树脂)粒子破碎以便得到满足这样的微分细孔容积(mL/g)的最大值的合成吸附剂。
合成吸附剂是指优化了母体结构、细孔结构、表面积、表面极性等的、通过物理相互作用或化学相互作用而从溶液中吸附特定的有机化合物的合成物质。
在本发明中,合成吸附剂是指有机系合成吸附剂。
作为合成吸附剂的材料,例如可举出苯乙烯系树脂、丙烯酸系树脂、酚系树脂、酰胺系树脂等。这些合成吸附剂的材料可以单独使用一种,也可以并用两种以上。在这些合成吸附剂的材料中,从成本低且疏水性优异的方面出发,优选为苯乙烯系树脂、丙烯酸系树脂,更优选为苯乙烯系树脂。
疏水性是对水分子亲和性小的非极性基团在水溶液中相互聚集的性质。将利用该引力的相互作用称为疏水性相互作用。
作为疏水性的官能团,例如可举出烷基、苯基、苄基等烃的官能团等。
在本说明书中,苯乙烯系树脂在构成苯乙烯系树脂的全部单体单元100质量%中,来自芳香族乙烯基单体的构成单元为50质量%以上。从杂质向合成吸附剂的吸附量、合成吸附剂的物理强度、细孔的形成性优异的方面出发,该比例优选为80质量%以上。苯乙烯系树脂也可以包含来自芳香族乙烯基单体的构成单元以外的构成单元。
作为芳香族乙烯基单体,例如可举出苯乙烯、甲基苯乙烯、乙基苯乙烯、α-甲基苯乙烯、氯苯乙烯、氯甲基苯乙烯、溴丁基苯乙烯等芳香族单乙烯基单体;二乙烯基苯、双(乙烯基苯基)乙烷、二乙烯基萘、2,4,6-三乙烯基乙基苯等交联性芳香族乙烯基单体等。这些芳香族乙烯基单体可以单独使用一种,也可以并用两种以上。
在这些芳香族乙烯基单体中,从杂质向合成吸附剂的吸附量、合成吸附剂的物理强度、细孔的形成性优异的方面出发,优选包含交联性芳香族乙烯基单体,更优选并用芳香族单乙烯基单体和交联性芳香族乙烯基单体,进一步优选并用苯乙烯和二乙烯基苯。
在构成苯乙烯系树脂的全部单体单元100质量%中,苯乙烯系树脂中的来自芳香族单乙烯基单体的构成单元的含有率优选为0质量%~99质量%,更优选为20质量%~80质量%。如果来自芳香族单乙烯基单体的构成单元的含有率为上述下限值以上,则合成吸附剂的柔软性优异。如果来自芳香族单乙烯基单体的构成单元的含有率为上述上限值以下,则能够抑制合成吸附剂在溶剂中的溶解或溶胀、收缩。
在构成苯乙烯系树脂的全部单体单元100质量%中,苯乙烯系树脂中的来自交联性芳香族乙烯基单体的构成单元的含有率优选为1质量%~100质量%,更优选为20质量%~80质量%。如果来自交联性芳香族乙烯基单体的构成单元的含有率为上述下限值以上,则能够抑制合成吸附剂在溶剂中的溶解或溶胀、收缩。如果来自交联性芳香族乙烯基单体的构成单元的含有率为上述上限值以下,则合成吸附剂的柔软性优异。
从合成吸附剂的疏水性优异的方面出发,在构成苯乙烯系树脂的全部单体单元100质量%中,苯乙烯系树脂中的除芳香族单乙烯基单体单元和交联性芳香族乙烯基单体单元以外的来自其他单体的构成单元的含有率优选为10质量%以下,更优选为1质量%以下,进一步优选为0质量%以下。
在本说明书中,丙烯酸系树脂在构成丙烯酸系树脂的全部单体单元100质量%中来自(甲基)丙烯酸酯的构成单元为50质量%以上。从抗体的回收率优异的方面出发,该比例优选为80质量%以上。丙烯酸系树脂也可以包含来自(甲基)丙烯酸酯的构成单元以外的构成单元。
作为(甲基)丙烯酸酯,例如可举出(甲基)丙烯酸甲酯、(甲基)丙烯酸乙酯、(甲基)丙烯酸丁酯、(甲基)丙烯酸硬脂基酯、(甲基)丙烯酸2-乙基己酯、(甲基)丙烯酸环己酯等(甲基)丙烯酸烷基酯;(甲基)丙烯酸羟基乙酯、(甲基)丙烯酸羟基丙酯、甘油单(甲基)丙烯酸酯等含羟基的(甲基)丙烯酸酯;(甲基)丙烯酸缩水甘油酯、(甲基)丙烯酸4,5-环氧基丁酯、(甲基)丙烯酸9,10-环氧基硬脂基酯等含环氧基的(甲基)丙烯酸酯;(甲基)丙烯酰胺、二甲基(甲基)丙烯酰胺、羟基乙基(甲基)丙烯酰胺等(甲基)丙烯酰胺类;(甲基)丙烯腈等含氰基的(甲基)丙烯酸酯;乙二醇二(甲基)丙烯酸酯等亚烷基二(甲基)丙烯酸酯;聚乙二醇二(甲基)丙烯酸酯等聚亚烷基二醇二(甲基)丙烯酸酯;N,N’-亚烷基双(甲基)丙烯酰胺、二(甲基)丙烯酸甘油酯、三羟甲基丙烷三(甲基)丙烯酸酯等交联性(甲基)丙烯酸酯等。这些(甲基)丙烯酸酯可以单独使用一种,也可以并用两种以上。
在这些(甲基)丙烯酸酯中,从合成吸附剂的官能团化容易、合成吸附剂的物理强度优异的方面出发,优选包含交联性(甲基)丙烯酸酯,更优选包含乙二醇二(甲基)丙烯酸酯。
在构成丙烯酸系树脂的全部单体单元100质量%中,丙烯酸系树脂中的交联性(甲基)丙烯酸酯单元的含有率优选为1质量%~100质量%,更优选为20质量%~80质量%。如果交联性(甲基)丙烯酸酯单元的含有率为上述下限以上,则能够抑制合成吸附剂在溶剂中的溶解或溶胀、收缩。如果交联性(甲基)丙烯酸酯单元的含有率为上述上限以下,则合成吸附剂的柔软性优异。
本发明的合成吸附剂通过将利用悬浮聚合或乳液聚合等公知的聚合方法而制造的合成树脂粒子或市售品破碎而得到。
破碎是指通过物理力使合成吸附剂变细。
作为用于破碎的器具,只要能够通过物理力使合成吸附剂变细就没有特别限定,例如可举出研钵、混合机、球磨机、锤磨机、针磨机等。在这些用于破碎的器具中,从能够将合成吸附剂粉碎得细、容易控制粒径分布的方面出发,优选为球磨机。
从高效地增加比表面积、杂质的除去性优异的方面出发,本发明的合成吸附剂优选具有细孔。
关于合成吸附剂的细孔的形成,只要在得到合成树脂时的聚合时使用多孔化剂即可。
合成吸附剂的细孔的半径优选为3nm~15nm,更优选为5nm~10nm。如果合成吸附剂的细孔的半径为上述下限值以上,则杂质向合成吸附剂内部的扩散性优异,杂质的除去性优异。如果合成吸附剂的细孔的半径为上述上限值以下,则杂质向合成吸附剂的吸附量优异,能够抑制由抗体向合成吸附剂内部的扩散引起的产率的下降。即,抗体纯化时的抗体的回收率、抗体的生产性优异。
合成吸附剂的细孔的半径为利用氮气且使用细孔分布测定装置而测定的细孔的众数半径。
合成吸附剂的细孔的半径可通过控制多孔化剂的种类或添加量、得到合成树脂时的聚合的速度而进行调整。
合成吸附剂的体积平均粒径优选为1μm~500μm,更优选为10μm~200μm。如果合成吸附剂的体积平均粒径为上述下限值以上,则抗体纯化时的过滤性优异,抗体的回收率、抗体的生产性优异。如果合成吸附剂的体积平均粒径为上述上限值以下,则能够增加合成吸附剂的表面积,杂质的除去性能优异。
合成吸附剂的体积平均粒径为使用激光衍射-散射式粒度分析仪测定而得的值。
作为测定样品,使用使合成吸附剂浸渍于20体积%乙醇水溶液中并通过过滤来除去附着水分而得的样品。
本发明的合成吸附剂的校正正圆度的值优选大于0.10,更优选为0.40以上。如果校正正圆度的值大于0.10,则成吸附剂向细孔内部的扩散性高,杂质的除去性能优异。校正正圆度的上限没有特别限制,但通常为1.00以下。
这里,“正圆度”在JIS B0621中进行了定义,是指“圆形形体从几何学上的正圆的偏差的大小”。正圆度是“在利用两个同心几何圆夹持对象物(圆形形体)时,同心二圆的间隔为最小时的二圆的半径之差”。
本发明的“校正正圆度”是将该正圆度除以求出正圆度时所拍摄的合成吸附剂的近似圆的半径而得的,相当于校正了由粒径引起的正圆度值的变化后的值。表示校正正圆度越接近零越接近几何圆,校正正圆度越大越偏离几何圆。
本发明的校正正圆度利用以下方法求出。应予说明,以下的正圆度与近似圆的半径的值的长度单位相同。
<校正正圆度的值的测定和算出方法>
1.使用光学显微镜拍摄合成吸附剂。
2.使用图像解析软件WinROOF2018制作所拍摄的合成吸附剂的近似圆。
3.按照JIS B0621,利用与该近似圆同心的两个同心的几何圆即同心二圆夹持。
4.求出上述3的同心二圆的间隔为最小时的上述同心二圆中的外周侧的圆的半径与内周侧的圆的半径之差作为正圆度。
5.将求出的正圆度除以上述近似圆的半径。
6.对100个点以上进行上述1~5的测定和算出,将其平均值作为校正正圆度。
合成吸附剂的压溃强度(物理强度)优选为100gf/粒~2000gf/粒,更优选为300gf/粒~1000gf/粒。一般而言,压溃强度越高越难以在使用过程中破碎,因此优选。但是,压溃强度过高的合成吸附剂变得无法耐受由进行了通液的溶剂组成的变化引起的溶胀、收缩,发生破碎。从由合成吸附剂粒子的层叠引起的载荷和由搅拌引起的碰撞等物理冲击强、可耐受由溶剂的组成变化引起的溶胀、收缩的平衡出发言,优选为上述压溃强度的范围。
这里,压溃强度是在某物体受到压溃(压缩)的力时变得无法保持原形(被破坏)的力的平均值。具体而言,压溃强度利用后述公开的实施例的项中记载的方法进行测定。
在后述公开的实施例中,关于压溃强度,对破碎前的合成吸附剂进行了测定。其原因在于:破碎后的合成吸附剂中粒子小,无法得到测定结果。压溃强度是作为材料的强度,因此一般而言如果为相同的组合物,则越小越高。因此,认为在破碎后数值不会变小,破碎前的合成吸附剂的压溃强度的测定值为破碎后的合成吸附剂的测定值以上。
(抗体)
本发明的合成吸附剂优选用于抗体的纯化。
抗体是作为抗原刺激的结果而通过免疫反应在生物体内产生的蛋白质,是指具有与抗原特异性结合的活性。
作为抗体,例如可举出小鼠抗体、美洲鸵抗体、嵌合抗体、人源化抗体、人类抗体、改变了这些的Fc区域等的抗体、对这些抗体进行了多价化的抗体、改变了这些抗体的一部分的抗体等。作为抗体的分子型,例如可举出免疫球蛋白G(IgG)、免疫球蛋白M(IgM)、免疫球蛋白A(IgA)、免疫球蛋白D(IgD)、免疫球蛋白E(IgE)、Fab、Fc、Fc-融合蛋白、VH、VL、VHH、Fab’2、scFv、scFab、scDb、scDbFc等。在这些抗体中,从单一性优异的方面出发,优选来自同一细胞的抗体、同一类抗体,更优选为单克隆抗体。
单克隆抗体是单克隆的抗体生产细胞所生产的抗体,为一级结构、即氨基酸序列均匀。因此,类、子类、同种异型、L链型等也均匀。
作为单克隆抗体,例如可举出免疫球蛋白G(IgG)、免疫球蛋白M(IgM)、免疫球蛋白A(IgA)、免疫球蛋白D(IgD)、免疫球蛋白E(IgE)等。在这些单克隆抗体中,从不易吸附于合成吸附剂的方面出发,优选为IgG、IgD、IgE,更优选为IgG。
单克隆抗体的分子量优选为100000~9000000,更优选为300000~2000000。如果单克隆抗体的分子量为上述下限值以上,则能够抑制由抗体向合成吸附剂内部的扩散引起的产率下降。另外,如果单克隆抗体的分子量为上述上限值以下,则单克隆抗体在水中不易沈淀而容易溶解。
单克隆抗体的分子量为利用分子筛效应的尺寸排阻色谱测定而得的值。
(抗体的纯化方法)
使用本发明的合成吸附剂纯化抗体的方法没有特别限制,例如可举出以下的(1)~(3)的方法。
(1)一种抗体的纯化方法,在将包含抗体和杂质的混合物与本发明的合成吸附剂混合后进行过滤。即,通过过滤而将吸附有杂质的合成吸附剂与溶解于液体的抗体分离。
(2)一种所谓流过(flowthrough)模式的纯化方法,包括如下工序:使包含抗体和杂质的混合物通过本发明的合成吸附剂,回收未被合成吸附剂吸附的包含抗体的非吸附级分。
(3)一种抗体的纯化方法,在下述工序(i)和工序(ii)的前段包括下述工序(A)。
工序(i):接触工序,使包含抗体和杂质的混合溶液与本发明的合成吸附剂接触
工序(ii):分离工序,在上述工序(i)之后,将混合溶液与合成吸附剂分离
工序(A):离子交换树脂接触工序,使包含抗体和杂质的混合溶液与阴离子交换树脂和/或阳离子交换树脂接触
通过利用上述工序(A)进行的脱盐处理,能够提高由本发明的合成吸附剂带来的杂质除去效率、抗体纯化效率、抗体回收效率。应予说明,也可以在上述工序(i)中使用的合成吸附剂中混合上述工序(A)中使用的阴离子交换树脂和/或阳离子交换树脂的混合物。上述工序(i)可以为分批方式,也可以为流过方式,在流过方式的情况下,也可以省略上述工序(ii)。
杂质是为了将抗体用作医药品而需要除去的,例如可举出为了培养重组细胞而需要的培养基成分、除去来自细胞的抗体的蛋白质和代谢物、核酸等。
具体而言,杂质包含选自来自宿主细胞的蛋白质、来自抗体的聚合物、来自抗体的分解物和核酸中的至少一种。
来自宿主细胞的蛋白质是对生产抗体的细胞进行培养时在生长过程中产生的蛋白质、死细胞的细胞片等,例如可举出来自接受了糖链成分的除去、氧化、脱酰胺等的蛋白质的修饰体、从宿主细胞溶出的酶类等。
作为从宿主细胞溶出的酶类,例如可举出去糖酶、蛋白质水解酶、氧化还原酶、氨基酸异构酶等。
作为去糖酶,例如可举出神经氨酸酶(唾液酸酶)、半乳糖苷酶、聚糖酶等。
作为蛋白质分解酶,例如可举出丝氨酸蛋白酶、酯酶、半胱氨酸蛋白酶、胰蛋白酶样蛋白酶、氨肽酶、天冬氨酸蛋白酶、组织蛋白酶等。
作为氧化还原酶,例如可举出硫氧还蛋白还原酶等硫氧还蛋白相关酶。
来自抗体的聚合物是抗体通过热、催化剂、pH条件等条件聚合而得的,例如可举出人丙种球蛋白的热变性聚合物、通过氧化而生成的聚合物等。
来自抗体的分解物是抗体通过蛋白质分解酶、热、pH条件等而使抗体的肽键或二硫键断裂而分解的碎片。
核酸是以由嘌呤、嘧啶碱基、戊糖和磷酸构成的核苷酸为基本单元且磷酸在各核苷酸之间通过二酯键在糖的3’位与5’位的碳之间聚合而成的长链多核苷酸,例如可举出DNA、RNA、它们的分解物等。
杂质的分子量优选为1000~50000,更优选为5000~25000。如果杂质的分子量为上述下限值以上,则杂质具有疏水部,容易吸附于合成吸附剂。另外,如果杂质的分子量为上述上限值以下,则杂质向合成吸附剂内部的扩散性优异。
杂质的分子量为通过利用分子筛效应的尺寸排阻色谱测定而得的值。
上述(1)的方法中的过滤通过使用过滤材料将固体与液体分离来进行。
作为过滤材料,例如可举出亲水性砜系高分子膜、亲水性芳香族醚系高分子膜、亲水性氟系高分子膜、亲水性烯烃系高分子膜、纤维素系膜、(甲基)丙烯酸系高分子膜、(甲基)丙烯腈系高分子膜、乙烯醇系高分子膜等。在这些过滤材料中,从亲水性优异的方面出发,优选为亲水性氟系高分子膜、亲水性砜系高分子膜、纤维素系膜,更优选为亲水性氟系高分子膜。
过滤材料的孔径优选为0.1μm~30μm,更优选为0.2μm~10μm。如果过滤材料的孔径为上述下限值以上,则过滤的通液性优异。另外,如果过滤材料的孔径为上述上限值以下,则固体与液体的分离性优异。
过滤材料的孔径使用扫描式电子显微镜(SEM),通过所得到的图像进行测量。
在上述(3)的方法中,作为上述工序(i)中使包含抗体和杂质的混合溶液与本发明的合成吸附剂接触的方法,可举出将包含抗体和杂质的混合溶液与本发明的合成吸附剂混合的方法。另外,也可以如上述(2)的方法那样采用在填充了本发明的合成吸附剂的柱中通入混合溶液的流过模式。
在上述(3)的方法中,作为上述工序(A)中使用的阴离子交换树脂,可举出在由单乙烯基芳香族单体和交联性芳香族单体构成的交联聚合物中具有季铵(三甲基铵或二甲基乙醇胺)基等交换基团且与氢氧化钠、氢氧化钾等强碱同样地解离而显示出碱性的碱性阴离子交换树脂。特别是优选在全部pH范围(0~14)内显示离子交换性的碱性阴离子交换树脂,优选为强碱性阴离子交换树脂。作为市售的碱性阴离子交换树脂,例如可举出三菱化学株式会社制造的Diaion(注册商标)的SA系列、PA系列、UBA系列。作为碱性阴离子交换树脂的具体的树脂,具有季铵(三甲基铵或二甲基乙醇胺)基作为交换基团,盐形式可举出OH型、Cl型等,这些之中,OH型的脱盐效率优异而优选。
另一方面,作为阳离子交换树脂,是在由单乙烯基芳香族单体和交联性芳香族单体构成的交联聚合物中具有磺酸基(-SO
另外,如果离子交换树脂按照其结构性质大致进行分类,则可分为“凝胶型”、“多孔(porous)(多孔性)型”,但本发明中使用的酸性阳离子交换树脂和碱性阴离子交换树脂优选均为凝胶型。
即,凝胶型的离子交换树脂与多孔型的离子交换树脂相比,单位体积的离子交换容量大,物理强度(压溃强度)高,因此可长期使用。
上述工序(A)的脱盐处理中,可以仅使用阴离子交换树脂和阳离子交换树脂中的一者,但从能够除去阳离子性杂质和阴离子性杂质这两者的方面出发,优选将阴离子交换树脂与阳离子交换树脂混合或组合使用。
上述工序(A)的脱盐处理可通过如下方式进行:通过将包含抗体和杂质的混合溶液与阴离子交换树脂和/或阳离子交换树脂混合,或者通过将包含抗体和杂质的混合溶液通入阴离子交换树脂床和/或阳离子交换树脂床、优选为阴离子交换树脂与阳离子交换树脂的混床。
在任一情况下,从提高由脱盐处理带来的杂质除去效率、抗体纯化效率、抗体回收效率的观点考虑,优选利用上述工序(A)的脱盐处理得到的处理液由于离子性杂质的除去而显示出导电率为10mS/cm以下、特别是5mS/cm以下的低导电率。
在上述(1)~(3)的方法中,混合物与合成吸附剂的混合、混合物在合成吸附剂中通过、或者混合溶液与合成吸附剂的接触优选在pH为3~8的液体中进行,更优选在pH为4~6的液体中进行。如果pH为上述下限值以上,则能够保持抗体的稳定性。另外,如果pH为上述上限值以下,则能够抑制抗体向合成吸附剂的吸附。
因此,在包含抗体和杂质的混合物或混合溶液中可以根据需要添加酸、碱等来调整pH。
(抗体的制造方法)
本发明的抗体的制造方法包括上述的抗体的纯化方法。
本发明的抗体的制造方法除了包括上述的抗体的纯化工序以外,还可以包括细胞的培养工序、利用离子交换树脂进行的纯化工序、利用尺寸排阻色谱进行的纯化工序、利用膜分离进行的纯化工序。
(用途)
本发明的合成吸附剂能够减少降低的制造成本,且在工业规模上也有效地生产抗体。另外,本发明的抗体的纯化方法和抗体的制造方法能够降低抗体的制造成本,且在工业规模上也有效地生产抗体。
实施例
以下,使用实施例对本发明进行更具体的说明,但本发明只要不脱离其主旨,则并不限定于以下的实施例的记载。
(体积平均粒径、粒度分布的测定)
对于合成吸附剂(A)~(G),使用激光衍射-散射式粒度分析仪(机种名“MLS-2000e”,株式会社Seishin企业制造)测定体积平均粒径、粒度分布。
(微分细孔容积的测定)
对于合成吸附剂(A)~(G),通过汞压入法,使用细孔分布测定装置(机种名“AutoPore IV 9520”、Micromeritics Instruments Corporation公司制造),对使压力从0.5psia阶段性地上升至30.0psia时的微分细孔容积进行测定,求出其最大值。
作为一个例子,将合成吸附剂(A)~(G)的实际测定压力和微分细孔容积的测定值示于下述表1。
[表1]
(细孔的半径的测定)
对于合成吸附剂(D)~(G),利用氮气,使用细孔分布测定装置(机种名“ASAP2024”,Micromeritics Instruments Corporation公司制造),对细孔的众数半径进行测定。应予说明,在粉碎前后细孔的半径没有大幅变化。
(校正正圆度的测定和算出)
对于合成吸附剂(A)~(G),使用Nicon SMZ18(NIKON SOLUTIONS株式会社制造)拍摄了显微照片。同心二圆的各半径和近似圆的半径的测定使用图像解析软件WinROOF2018(三谷商事株式会社制造)进行,利用上述的方法算出正圆度以及用近似圆的半径除正圆度而得的校正正圆度。
(压溃强度的测定)
将合成吸附剂(A)~(F)的制造中使用的破碎前的合成吸附剂(合成吸附剂(A)~(C)、(E)使用相同的物质)的粒子浸渍于脱盐水中,在即将测定之前保管于脱盐水中。接着,随机选择最少30粒破碎前的合成吸附剂粒子,利用Chatillon forcemeasurement CS225(AMETEK TEST&CALIBRATION INSTRUMENTS)进行强度测定,算出所有粒子的强度的平均值。应予说明,合成吸附剂(G)的体积粒径(μm)小,难以测定压溃强度。
(杂质的浓度和抗体的回收率的测定)
作为单克隆抗体(免疫球蛋白G,分子量:150000)的培养液,使用使来自中国仓鼠卵巢的细胞(CHO细胞)生产抗体后除去CHO细胞并进行澄清化后的培养液上清液。使用5mol/L的乙酸水溶液,将pH7.4的培养液调整为pH4.5。
接着,将合成吸附剂(A)~(G)分别以各0.2g量取至15mL的容器中,将所制备的培养液分别添加各4mL。在添加后,振荡8小时,进行混合。然后,使用膜滤器(Merck公司制造,孔径0.22μm,PVDF膜)进行过滤来除去合成吸附剂,得到合成吸附剂处理液。
对于来自宿主细胞的蛋白质浓度(HCP浓度)[ng/mL],使用试剂盒(试剂盒名“ELISA试剂盒F550(CHO宿主细胞蛋白质(CHO Host Cell Protein),第三代(3RdGeneration))”,通过ELISA法进行测定。
对于HCP浓度的对数减少值(LRV),根据处理前后的HCP浓度算出。
对于抗体的回收率[%],使用亲和色谱柱(商品名“POROS A20μmColumn”,ThermoFisher Scientific公司制造),通过高效液相色谱HPLC进行测定。溶剂使用磷酸缓冲生理盐水(pH7.4),洗脱液使用20mmol/L的磷酸缓冲液(pH2.8,食盐浓度150mmol/L)。
(导电率的测定)
培养液上清液或培养液上清液的脱盐处理液的导电率通过HORIBA公司的台式pH计F-74导电率(电导率)电极:3552-10D进行测定。
[实施例1]
利用研钵将市售的合成吸附剂(商品名“Sepabeads 825L”,三菱化学株式会社制造,进行了多孔化处理的苯乙烯-二乙烯基苯共聚物)粉碎,使用过滤膜(Merck公司制造,孔径5μm),利用20体积%乙醇水溶液进行清洗,并通过过滤来除去附着水分,得到合成吸附剂(A)。
将所得到的合成吸附剂(A)的评价结果示于表2。
[实施例2]
利用研钵将市售的合成吸附剂(商品名“Sepabeads 825L”,三菱化学株式会社制造,进行了多孔化处理的苯乙烯-二乙烯基苯共聚物)粉碎,利用20μm的筛子进行整粒,使用过滤膜(Merck公司制造,孔径5μm),利用20体积%乙醇水溶液进行清洗,并通过过滤来除去附着水分,得到合成吸附剂(B)。
将所得到的合成吸附剂(B)的评价结果示于表2。
[实施例3]
利用研钵将市售的合成吸附剂(商品名“Sepabeads 825L”,三菱化学株式会社制造,进行了多孔化处理的苯乙烯-二乙烯基苯共聚物)粉碎,利用63μm的筛子进行整粒,使用过滤膜(Merck公司制造,孔径5μm),利用20体积%乙醇水溶液进行清洗,并通过过滤来除去附着水分,得到合成吸附剂(C)。
将所得到的合成吸附剂(C)的评价结果示于表2。
[比较例1]
不将进行了多孔化处理的苯乙烯-二乙烯基苯共聚物粉碎,使用过滤膜(Merck公司制造,孔径5μm),利用20体积%乙醇水溶液进行清洗,并通过过滤来除去附着水分,而得到未粉碎的合成吸附剂(G)。
将所得到的合成吸附剂(G)的评价结果示于表2和表3中。
[表2]
由表2可知,与比较例1中制造的微分细孔容积为0.05mL/g以下的合成吸附剂(G)相比,实施例1~3中制造的微分细孔容积超过0.05mL/g的合成吸附剂(A)~(C)的HCP浓度、LRV、抗体回收率优异。特别是体积平均粒径小的实施例1的合成吸附剂(A)的HCP浓度低,杂质的除去性能优异。另外,体积平均粒径大的实施例3的合成吸附剂(C)的抗体的回收率高,能够有效地生产抗体。
应予说明,将合成吸附剂(A)、合成吸附剂(B)、合成吸附剂(C)和合成吸附剂(G)的微分细孔容积的测定值的测定压力与微分细孔容积(增加的细孔容积)的关系示于图1中。由图1可知,合成吸附剂(A)、合成吸附剂(B)和合成吸附剂(C)与合成吸附剂(G)相比,微分细孔容积的最大值明显大。
[实施例4]
利用研钵将市售的合成吸附剂(商品名“Sepabeads 850”,三菱化学株式会社制造,进行了多孔化处理的苯乙烯-二乙烯基苯共聚物,细孔的半径5nm)粉碎,使用过滤膜(Merck公司制造,孔径5μm),利用20体积%乙醇水溶液进行清洗,并通过过滤来除去附着水分,得到合成吸附剂(D)。
将所得到的合成吸附剂(D)的评价结果示于表3。
[实施例5]
利用研钵将市售的合成吸附剂(商品名“Sepabeads 825L”,三菱化学株式会社制造,进行了多孔化处理的苯乙烯-二乙烯基苯共聚物,细孔的半径7nm)粉碎,使用过滤膜(Merck公司制造,孔径5μm),利用20体积%乙醇水溶液进行清洗,并通过过滤来除去附着水分,得到合成吸附剂(E)。
将所得到的合成吸附剂(E)的评价结果示于表3。
[实施例6]
利用研钵将市售的合成吸附剂(商品名“Sepabeads HP21”,三菱化学株式会社制造,进行了多孔化处理的苯乙烯-二乙烯基苯共聚物,细孔的半径11nm)粉碎,使用过滤膜(Merck公司制造,孔径5μm),利用20体积%乙醇水溶液进行清洗,并通过过滤来除去附着水分,得到合成吸附剂(F)。
将所得到的合成吸附剂(F)的评价结果示于表3。
[表3]
由表3可知,与比较例1中制造的未粉碎的合成吸附剂(G)相比,实施例4~6中制造的合成吸附剂(D)~(F)的HCP浓度、LRV、抗体回收率优异。特别是细孔的半径大的实施例6的合成吸附剂(F)的HCP浓度低,杂质的除去性能优异。另外,细孔的半径小的实施例4的合成吸附剂(D)的抗体的回收率高,能够有效地生产抗体。
[实施例7]
利用研钵将市售的合成吸附剂(商品名“Sepabeads 825L”,三菱化学株式会社制造,进行了多孔化处理的苯乙烯-二乙烯基苯共聚物)粉碎,在浸渍于20体积%乙醇水溶液中后,使用过滤膜(Merck公司制造,孔径5μm)进行抽滤,得到合成吸附剂(H)。对合成吸附剂(H)测定而得的微分细孔容积的最大值为0.131mL/g。
作为单克隆抗体(免疫球蛋白G,分子量:150000)的培养液,使用使来自中国仓鼠卵巢的细胞(CHO细胞)生产抗体后除去CHO细胞并进行澄清化后的培养液上清液。使用5mol/L的乙酸水溶液,将pH7.4的培养液调整为pH4.5。
接着,将合成吸附剂(H)1.5g和所制备的培养液30mL量取至容器中,在25℃振荡8小时,进行混合。然后,利用离心分离和过滤膜(Merck公司制造,孔径0.22μm)来除去合成吸附剂,得到合成吸附剂的分批的处理液。
利用1mol/L的盐酸将所得到的合成吸附剂的分批的处理液调整为pH3.5,在25℃静置2小时。然后,利用1mol/L的Tris溶液调整为pH5.0,进行过滤,得到病毒灭活液。
利用MilliQ水将所得到的病毒灭活液稀释至4倍,在利用20mmol/L的Tris盐酸缓冲液(pH8.5)的平衡化缓冲液进行了平衡化的阴离子交换色谱柱(商品名“ChromSpeedQ103”,三菱化学株式会社制造)中添加稀释后的病毒灭活液。添加结束后,向柱中通入5柱体积的平衡化缓冲液。将柱非吸附级分作为ChromSpeed Q103溶出液。
利用5mol/L的乙酸将所得到的ChromSpeed Q103溶出液调整为pH4.5,在利用20mmol/L的乙酸缓冲液(pH4.5)进行了平衡化的阳离子交换色谱柱(商品名“ChromSpeedS103”,三菱化学公司制造)中添加所制备的溶出液。添加结束后,将吸附级分溶出至洗脱液(溶解有500mmol/L的食盐的20mmol/L的乙酸缓冲液,pH4.5)中,将溶出液作为三阶段纯化样品。
对培养液上清液和所得到的合成吸附剂处理液、三阶段纯化处理液利用下述方法测定HCP和抗体浓度。
对于来自宿主细胞的蛋白质浓度(HCP浓度)[ng/mL],使用试剂盒(试剂盒名“ELISA试剂盒F550(CHO宿主细胞蛋白质(Host Cell Protein),第三代(3RdGeneration))”,通过ELISA法进行测定。
对于抗体浓度[HCP/Mab ppm],使用亲和色谱柱(商品名“POROS A20μm柱”,ThermoFisher Scientific公司制造),通过高效液相色谱HPLC进行测定。
对于抗体的回收率[%],使用亲和色谱柱(商品名“POROS A20μm柱”,ThermoFisher Scientific公司制造),通过高效液相色谱HPLC进行测定。溶剂使用磷酸缓冲生理盐水(pH7.4),洗脱液使用20mmol/L的磷酸缓冲液(pH2.8,食盐浓度150mmol/L)。
[比较例2]
作为单克隆抗体(免疫球蛋白G,分子量:150000)的培养液,使用使来自中国仓鼠卵巢的细胞(CHO细胞)生产抗体后除去CHO细胞并进行澄清化后的培养液上清液。使用5mol/L的乙酸水溶液,将pH7.4的培养液调整为pH4.5。
将所制备的培养液30mL添加于亲和色谱柱(商品名“MabSelect SuRetm LX”、GEHealthcare Life Sciences公司制造,包含作为亲和吸附剂的吸附剂(I)的柱)中,在吸附的状态下利用20mmol/L的磷酸缓冲盐水进行清洗。利用溶解有150mmol/L的食盐的100mmol/L的乙酸进行脱附,得到吸附剂的分批的处理液。
利用MilliQ水将所得到的吸附剂的分批的处理液稀释至10倍后,利用1mol/L的盐酸调整为pH3.5,在25℃静置2小时。然后,利用1mol/L的Tris溶液调整为pH5.0,并进行过滤,得到病毒灭活液。
在利用20mmol/L的Tris盐酸缓冲液(pH8.5)的平衡化缓冲液进行了平衡化的阴离子交换色谱柱(商品名“ChromSpeed Q103”,三菱化学株式会社制造)中添加所得到的病毒灭活液。添加结束后,向柱中通入5柱体积的平衡化缓冲液。将柱非吸附级分作为ChromSpeed Q103溶出液。
从所得到的ChromSpeed Q103溶出液中,将利用与实施例7同样的操作在阳离子交换色谱柱中得到的溶出液作为三阶段纯化样品,与实施例7同样地对培养液上清液、合成吸附剂处理液以及三阶段纯化处理液分别进行分析。
将上述实施例7和比较例2的结果示于表4。
[表4]
由表4可知,与比较例2中使用的亲和吸附剂即吸附剂(I)相比,实施例7中制造的合成吸附剂(H)在第一阶段的吸附剂处理中,虽然如吸附剂(I)那样没有抗体的吸附脱附、清洗工序,但能够粗纯化至抗体浓度为3位数ppm水平,回收率也为80%以上。在三阶段纯化的最终样品中,显示与使用亲和吸附剂即吸附剂(I)的工序同等的HCP除去率、抗体回收率。
[实施例8]
(培养液上清液的制备)
作为单克隆抗体(免疫球蛋白G,分子量:150000)的培养液,使用使来自中国仓鼠卵巢的细胞(CHO细胞)生产抗体后除去CHO细胞并进行澄清化后的培养液上清液1(抗体浓度为2.4mg/mL,导电率为20mS/cm)。
(脱盐处理)
在4mL的上述培养液上清液1中分别添加阴离子交换树脂(Diaion(注册商标)“UBA120OH”,三菱化学株式会社制造)0.4g和阳离子交换树脂(Diaion(注册商标)“UBK10H”,三菱化学株式会社制造)0.4g。然后,使用0.1mol/L的氢氧化钠水溶液将pH7.4的上述培养液上清液1调整为pH4.5。
然后,利用过滤膜(Merck公司制造,孔径0.22μm)除去阴离子交换树脂和阳离子交换树脂,得到脱盐处理液(导电率3mS/cm)。
(利用合成吸附剂进行的纯化)
接着,将合成吸附剂(B)0.3g和所制备的脱盐处理液4mL量取至容器中,在25℃振荡8小时。然后,利用离心分离和过滤膜(Merck公司制造,孔径0.22μm)来除去合成吸附剂(B),得到基于合成吸附剂(B)的处理液。将处理液的评价结果示于表5。
[实施例9]
除了使用将单克隆抗体的抗体浓度调整为15mg/mL的培养液上清液2代替培养液上清液1以外,利用与实施例8相同的工序进行处理,将所得到的处理液的评价结果示于表5。
[实施例10]
除了不进行脱盐处理以外,利用与实施例8相同的工序进行处理,将所得到的处理液的评价结果示于表5。
[实施例11]
除了不进行脱盐处理以外,利用与实施例9相同的工序进行处理,将所得到的处理液的评价结果示于表5。
[表5]
由表5可知,相对于实施例10和实施例11中实施的无脱盐处理的工艺,进行了脱盐处理的实施例8和实施例9的HCP浓度、LRV、抗体回收率优异。特别是培养液中的抗体浓度高时,HCP浓度、LRV、抗体回收率显著优异。
[实施例12]
除了对培养液上清液进行实施例8中记载的脱盐处理、使用合成吸附剂(B)代替合成吸附剂(H)、不稀释病毒灭活液以外,进行与实施例7相同的处理。
将评价结果与实施例7和比较例2的结果一起示于表6。
[表6]
由表6可知,与实施例7相比,在实施例12中通过将脱盐工序(工序(A))组入至工艺中,能够省略阴离子交换色谱前利用MilliQ水进行的四倍稀释,三阶段纯化后的抗体回收率也为81%这样的较高的值。另外,在三阶段纯化的最终样品中,显示与使用亲和吸附剂即吸附剂(I)的工序同等的HCP除去率。
使用特定的方式对本公开进行了详细说明,但对于本领域技术人员而言显而易见的是能够在不脱离本公开的意图和范围的情况下进行各种变更。
本申请基于2021年6月10日提出申请的日本申请2021-097149,其整体通过引用而被援用。
- 蛋白A亲和层析法纯化抗体的方法以及抗体的纯化方法
- Fc结合性蛋白质、该蛋白质的制造方法和使用了该蛋白质的抗体吸附剂、以及使用了该吸附剂的抗体的纯化方法及识别方法
- Fc结合性蛋白质、该蛋白质的制造方法和使用了该蛋白质的抗体吸附剂、以及使用了该吸附剂的抗体的纯化方法及识别方法