用于生成基因组坐标的置信度分类的机器学习模型
文献发布时间:2024-04-18 19:59:31
相关申请的交叉引用
本申请要求2021年6月29日提交的标题为“MACHINE-LEARNING MODEL FORGENERATING CONFIDENCE CLASSIFICATIONS FOR GENOMIC COORDINATES”的美国临时申请第63/216,382号的权益和优先权,该申请的内容特此通过引用整体并入。
背景技术
近年来,生物技术公司和研究机构已经改进了用于核苷酸测序和鉴别含有与规范或参考基因组不同的核碱基的样品的变体检出的硬件和软件。例如,一些现有的核酸测序平台通过使用常规桑格测序或通过使用边合成边测序(SBS)确定核酸序列的个体核碱基。使用SBS时,现有平台可以监测并行合成的数千、数万或更多核酸聚合物,以从更大的碱基检出数据集检测更准确的碱基检出。例如,SBS平台中的照相机可以捕获来自掺入此类寡核苷酸中的核碱基的经照射荧光标签的图像。在捕获此类图像后,现有SBS平台向具有测序-数据-分析软件的计算设备发送碱基检出数据(或图像数据)以确定核酸聚合物的核碱基序列(例如,核酸聚合物的外显子区域)并且使用变体检出程序来鉴别样品的核酸序列内的任何单核苷酸变体(SNV)、插入缺失、或其他变体。
尽管在测序和变体检出方面有这些最新进展,但是现有的测序-数据-分析软件常常包括鉴别核苷酸变体的变体检出程序,而不管(或没有指示)核苷酸变体在序列或基因组内的位置。因为变体检出的位置的背景可影响检出的可靠性-某些基因组区域更可能展现出可预测序列而其他基因组区域更可能展现出变异-所以核苷酸变体的位置可影响将变体鉴别为真阳性或假阳性的概率。进一步就这一点而言,正确鉴别给定基因组区域的变体的概率可根据特定测序方法或设备而不同。在没有用于分析基因组区域的准确度并将变体检出与此类区域相关联的内置机制的情况下-特别是对于特定的测序流水线,临床医生常常使用其他测序方法(例如,桑格补充SBS测序)或补充验证试验以正交验证变体检出。
根据变体检出的基因组区域,对于特定变体的变体检出范围可以介于无关紧要或关键之间。因为现有的变体检出程序常常不能将变体检出与基因组区域或位置的准确度概率相关联,然而,临床医生对变体检出的准确度的置信度有限。例如,鉴别血红蛋白β(HBB)基因中的特定单核苷酸多态性(SNP)的变体检出可具有重要含义。当变体检出程序鉴别染色体11上rs344处的SNP时,变体检出程序可正确鉴别镰状细胞贫血的遗传原因或遗漏该疾病的原因。作为另一个示例,正确或不正确地鉴别血红蛋白亚基α1(HbA1)或血红蛋白亚基α2(HbA2)基因的一个或多个拷贝的缺失的变体检出可引起正确地鉴别遗传性血液病的遗传原因或完全遗漏基因缺失。因此,对基因上的此类SNP或其他变体的变体检出可能是关键的,但常常缺乏对常规变体检出程序从中鉴别变体的区域的准确度概率的基于经验的指示。
尽管核碱基检出的基因组区域中存在变异且变体检出具有潜在重要性,但是现有的核酸测序平台和测序数据分析软件(以及下文中的现有测序系统)缺乏凭经验证明的在基因组内鉴别更高或更低准确度的区域的可报告范围的方式。此类现有测序系统同样缺乏凭经验证明的区分此类可报告范围内不同变体类型的方式。现有测序系统还缺乏对于特定测序流水线而言凭经验证明的鉴别可报告范围或区分那些范围内的变体类型的方式。
常规地,临床医生和生物技术机构可依赖于不限于特定测序流水线的参考基因组的特征。研究人员已经鉴别了参考基因组中更高或更低准确度的区域的可报告范围,包括由瓶中基因组联盟(Genome in a Bottle Consortium,GIAB)和全球基因组健康联盟(Global Alliance for Genomic Health,GA4GH)鉴别的参考基因组的高置信度区域。但是来自GIAB和GA4GH的这些现有的可报告范围将可报告范围限制到排除了困难基因组区域的基准基因组区域,其中大约79%-84%的人类基因组处于基准基因组区域内;未能区分区域的不同类型的准确度等级;并且不通过变体类型区分可报告范围(例如,SNV对比插入缺失)。仅约79%-84%的参考基因组映射到基准区域并且未按变体检出类型在可报告范围中进行区分,常规的可报告范围留下参考基因组的很大一部分,没有对检测准确度的指示并且没有特定变体检出类型是否会影响检测准确度的指示。
即使具有这些常规的可报告范围,临床医生也需要关于参考基因组的特征如何转化为特定测序流水线的专门知识,以例如说明核苷酸样品制备的变化(例如,PCR或更长的读段)、不同的测序设备或不同的测序数据分析软件。实际上,尽管参考基因组有可报告范围,现有测序系统也不能鉴别测序流水线特定的或从经验数据推导出的可报告范围。
除了来自GIAB和GA4GH的常规可报告范围以外,Illumina公司与研究机构合作开发基准基因组集合中高置信度变体检出的目录。通过生成具有三代谱系的人的全基因组序列数据并在每个基因组中检出变体,该团队开发了与这些人中的遗传模式一致的具有470万个SNV和70万个小插入缺失(1-50个碱基对)的目录的Platinum Genomes。虽然铂基因组中的变体检出真集可用于验证和测量所策划样品中变体检出的性能,但铂基因组和来自GIAB的其他真集排除了含有随机和系统误差的有问题的基因组区域。铂基因组或其他真集也不能说明变体检出中的样品特定误差。因为问题区域被排除而不管问题的根本原因,并且此类时间密集的编目难以(如果不是不可能的话)标度,所以高置信度变体检出的目录证明了确定每个基因组坐标处变体检出的准确度和可靠性的不切实际的方法。
发明内容
本公开描述了可以训练基因组位置分类模型,以按照可以在基因组坐标或区域处准确地鉴定核碱基的程度来对此类基因组坐标或基因组区域分类或评分的方法、非暂态性计算机可读介质和系统的实施方案。例如,所公开的系统可以确定多样化样品核酸序列的测序指标和特定核碱基检出周围的上下文核酸子序列中的一者或两者。通过利用基因组坐标的基准真值分类,在一些情况下,所公开的系统训练基因组位置分类模型以将来自测序指标和上下文核酸子序列中的一者或两者的数据与此类基因组坐标或区域的置信度分类相关联。训练了此类模型后,所公开的系统同样可以将基因组位置分类模型应用于来自测序指标或上下文核酸子序列的数据,以确定个体基因组坐标或区域的个体置信度分类。此类坐标特定或区域特定置信度分类可进一步打包到新增文件或新文件类型中-即,具有基因组坐标或区域的置信度分类的数字文件(例如,以补充变体检出)。
除了训练新类型的机器学习模型外,所公开的系统还可以应用该模型以利用凭经验训练的置信度分类来补充或上下文化变体检出。在样品序列中的基因组坐标(或区域)处检测到检出变体之后,例如,所公开的系统可以从对应于变体检出的基因组坐标或区域的数字文件中鉴别坐标特定置信度分类或区域特定置信度分类。基于所鉴别的坐标特定置信度分类或区域特定置信度分类,所公开的系统可以生成对应于变体检出的基因组坐标或区域的置信度分类的指示符,用于在图形用户界面上显示。所公开的系统可以相应地促进计算设备上的图形或文本指示符,说明基因组坐标或区域处的变体检出的置信度分类。
通过训练如本文所述的基因组位置分类模型,所公开的系统创建首创机器学习模型以生成基因组坐标或区域的可报告置信度分类范围。与依赖于与参考基因组相关而与来自测序流水线的经验数据不相关的置信区域的现有解决方案不同,所公开的基因组位置分类模型可以凭经验训练和定制以生成用于特定测序流水线的置信度分类。因为基因组位置分类模型从凭经验训练的过程生成置信度分类,所以来自基因组位置分类模型的坐标或区域特定置信度分类为变体检出或其他核碱基检出给出情境和新发现的准确度。
附图说明
详细描述参考下面简要描述的附图。
图1示出了根据一个或多个实施方案,包括基因组分类系统在内的测序系统的方框图。
图2示出了根据一个或多个实施方案,训练机器学习模型以确定基因组坐标的置信度分类的基因组分类系统的概况。
图3示出了根据一个或多个实施方案,确定关于参考基因组的测序指标的基因组分类系统的概况。
图4示出了根据一个或多个实施方案,其中基因组分类系统调整或准备用于输入到基因组位置分类模型中的测序指标的过程的概况。
图5示出了根据一个或多个实施方案,在核碱基检出周围的上下文核酸子序列。
图6A示出了根据一个或多个实施方案,基因组分类系统训练机器学习模型以基于测序指标和上下文核酸子序列中的一者或两者确定基因组坐标的置信度分类。
图6B示出了根据一个或多个实施方案,基因组分类系统应用基因组位置分类模型的训练版本以基于测序指标和上下文核酸子序列中的一者或两者确定基因组坐标的置信度分类。
图6C示出了根据一个或多个实施方案,测序系统或基因组分类系统从基因组位置分类模型鉴别和显示对应于变体检出的基因组坐标的置信度分类。
图6D-图6H示出了根据一个或多个实施方案,基因组分类系统基于来自基因组样品的样品核酸序列的测序指标及用于基于基因组样品混合物检出反映癌症或镶嵌现象的特定变体类型的再检出率或精确率中的一者或两者来确定基准真值分类。
图7A-图7G示出了根据一个或多个实施方案,指示基因组位置分类模型的信息性测序指标和测序指标导出数据的图表。
图8示出了根据一个或多个实施方案,描绘基因组位置分类模型基于测序指标正确确定基因组坐标的置信度分类的准确度的图表。
图9示出了根据一个或多个实施方案,描绘基因组位置分类模型基于上下文核酸子序列正确确定对应于不同核苷酸变体的基因组坐标的置信度分类的准确度的图表。
图10A-图10B示出了根据一个或多个实施方案,描绘基因组位置分类模型基于测序指标和上下文核酸子序列两者正确确定对应于不同核苷酸变体的基因组坐标的置信度分类的准确度的图表。
图11A-图11B示出了根据一个或多个实施方案,用于训练机器学习模型以确定基因组坐标的置信度分类的一系列动作的流程图。
图12示出了根据一个或多个实施方案,用于从数字文件生成变体核碱基检出的基因组坐标的置信度分类的指示符的一系列操作的流程图。
图13示出了用于实施本公开的一个或多个实施方案的示例性计算设备的方框图。
具体实施方式
本公开描述了基因组分类系统的实施方案,该基因组分类系统训练基因组位置分类模型以确定基因组坐标(或基因组区域)的标记或评分,这些标记或评分指示可以在基因组坐标或区域处准确地鉴别核碱基的程度或范围。为了准备基因组位置分类模型的输入,基因组分类系统确定样品核酸序列的测序指标和特定核碱基检出周围的上下文核酸子序列中的一者或两者。在一些情况下,基因组分类系统使用特定测序和生物信息学流水线来确定此类指标和上下文核酸子序列。因此,基于从测序指标和上下文核酸子序列中的一者或两者导出或准备的数据,并且通过利用基因组坐标的基准真值分类,基因组分类系统训练基因组位置分类模型以确定基因组坐标的置信度分类。
在某些实施方式中,基因组分类系统还通过基因组位置分类模型提供来自于对应于样品的测序指标或上下文核酸子序列的数据来确定基因组坐标(或区域)的置信度分类。基因组分类系统还将此类坐标特定置信度分类或区域特定置信度分类编码到至少一个数字文件中,该数字文件包含特定基因组坐标或基因组区域的置信度分类。例如,该数字文件可以包括基因组坐标和/或基因组区域的注释或其他数据指示符。
除了训练基因组位置分类模型之外或独立于训练基因组位置分类模型,该基因组分类系统还可基于核碱基检出(例如,不变检出或变体检出)的特定基因组坐标或区域来确定对这些检出的置信度分类。使用来自测序设备的数据,例如,基因组分类系统确定样品核酸序列中特定基因组坐标(或特定区域)处的变体核碱基检出或不变核碱基检出。此类核碱基检出可以使用与用于训练数据以训练基因组位置分类模型相同的测序和生物信息学流水线来确定。基因组分类系统然后可以鉴别对应于核碱基检出的基因组坐标或区域的置信度分类(例如,通过访问由经过训练的基因组位置分类模型生成的数字文件内的置信度分类数据)。通过鉴别置信度分类,基因组分类系统生成变体核碱基检出或不变核碱基检出的基因组坐标或区域的置信度分类的指示符用于在图形用户界面中显示。
如前面段落中所述,在一些情况下,基因组分类系统使用单个测序流水线来确定构成测序指标基础的核碱基检出、上下文核酸子序列或变体核碱基检出。例如,基因组分类系统可以使用具有相同核酸序列提取方法(例如,提取试剂盒)、相同测序设备和相同序列分析软件的单个测序流水线。此类序列分析软件可包括将序列读段与参考基因组比对的比对软件和鉴别变体核碱基检出的变体检出程序软件,使得单个测序流水线使用相同的比对软件和/或变体检出程序。通过使用单个测序流水线,在某些实施方式中,基因组分类系统可以训练并应用基因组位置分类模型,该基因组位置分类模型确定该测序流水线特有的置信度分类并且增加用于通过该流水线的变体检出或其他核碱基检出的那些分类的准确度。
为了准备数据以输入用于训练或应用基因组位置分类模型,在一些实施方案中,基因组分类系统确定测序指标,测序指标包括以下中的一项或多项:(i)用于定量样品核酸序列与示例核酸序列(例如,参考基因组或来自祖先单倍型的核酸序列)的基因组坐标的比对的比对指标,(ii)用于定量样品核酸序列在示例核酸序列的基因组坐标处的核碱基检出的深度的深度指标,或(iii)用于定量样品核酸序列在示例核酸序列的基因组坐标处的核碱基检出的质量的检出数据质量指标。例如,基因组分类系统确定映射质量指标、软剪切指标或度量样品序列与参考基因组的比对的其他比对指标。作为另一个示例,基因组位置分类系统确定前向-反向深度指标(或其他此类深度指标)或变体核碱基检出的可检出性(callability)指标(或其他此类检出数据质量指标)。
作为使用此类测序指标作为基因组位置分类模型的数据输入的补充或替代,在某些情况下,基因组分类系统确定在特定基因组坐标处的核碱基检出周围的上下文核酸子序列。例如,在一些实施方案中,基因组分类系统将来自参考基因组(或来自祖先单倍型序列)的核碱基鉴别为上下文核酸子序列,该核碱基位于任何不变核碱基检出或变体核碱基检出(诸如SNV、插入缺失、结构变异或拷贝数变异(CNV))的上游和下游。为了说明,基因组分类系统可将参考基因组或祖先单倍型序列上游的五十个核碱基和位于特定基因组坐标处的SNV下游的五十个核碱基鉴别为上下文核酸子序列。
不管基因组分类系统是否使用从测序指标或上下文核酸子序列或两者导出的数据,基因组分类系统都准备数据作为用于训练基因组位置分类模型的输入。在一些情况下,基因组分类系统通过确定基因组坐标的预计置信度分类并将预计分类与反映基因组坐标处的孟德尔遗传模式或核碱基检出的重复一致性的基准真值分类进行比较来训练基因组位置分类模型。通过使用损失函数来比较针对特定基因组坐标的预计置信度分类与基准真值分类,基因组分类系统可以迭代地调整基因组位置分类模型的参数以更准确地确定置信度分类。
如以上所表明的,基因组位置分类模型可以以各种形式(包括标记或评分)输出置信度分类。基因组分类系统可以确定置信度水平的等级,包括例如高置信度分类、中等置信度分类或低置信度分类,这些等级指示在给定基因组坐标处核碱基检出可信赖的程度。另外或另选地,基因组分类系统可以从指示在给定基因组坐标处核碱基检出可信赖的程度的评分范围确定置信度评分。
在训练和确定置信度分类之后,基因组分类系统可以生成或注释一个或多个数字文件以包括基因组坐标特定的置信度分类。仅举一个例子,在一些情况下,基因组分类系统生成浏览器可扩展数据(BED)文件的修改版本,该修改版本包含针对基因组坐标处的每个核碱基检出的注释,该注释指示该基因组坐标的对应置信度分类。在一些情况下,基因组分类系统根据置信度分类类型生成包含针对基因组坐标的注释的BED文件,诸如具有针对具有高置信度分类的基因组坐标的注释的BED文件,具有针对具有中等置信度分类的基因组坐标的注释的BED文件,以及具有针对具有低置信度分类的基因组坐标的注释的BED文件。基因组分类系统同样可以生成Wiggle(WIG)格式、序列比对/映射的二进制版本(BAM)格式、变体检出文件(VCF)格式、微阵列格式或其他数字文件格式的具有置信度分类的数字文件。在从数字文件中鉴别核苷酸检出变体的相关置信度分类后,基因组分类系统同样可以提供分类指示符以在图形用户界面上显示。此类指示符可以是例如高置信度分类、中等置信度分类或低置信度分类的图形指示符(例如,颜色编码的图形指示符)。
如以上所表明的,相对于常规核酸测序系统和相应的测序数据分析软件,基因组分类系统提供了几个技术益处和技术改进。例如,基因组分类系统引入首创机器学习模型,该模型经独特训练以进行新应用-生成在此处确定核苷酸变体检出或其他核碱基的特定基因组坐标的置信度分类。不同于主要依赖于参考基因组特征的常规变体检出程序或常规可报告范围,该基因组分类系统使用经验数据来训练基因组位置分类模型以生成坐标特定置信度分类或区域特定置信度分类,以核碱基检出的置信度分类的经验可报告范围为结果。可报告范围可包括多种易于理解的标记,诸如高置信度、中等置信度或低置信度分类-与参考基因组的整体常规分类不同。进一步与依赖于针对参考基因组开发的置信度区域的现有测序系统的通用型(one-size-fits-all)方法大不相同,在一些实施方案中,基因组分类系统可以定制基因组位置分类模型的置信度分类以适合单个测序流水线,从而在个体基因组坐标水平上增加特定测序设备(和相应流水线组件)的核碱基检出的置信度分类的准确度。
除了引入首创机器学习模型之外,与现有测序系统相比,基因组分类系统提高在基因组上特定基因组坐标处确定核碱基检出的置信度水平的准确度和广度。例如,基因组分类系统增加测序系统准确鉴别基因组坐标处的变体的精确度、再检出率和一致性。在一些实施方式中,对于约90.3%的参考基因组,测序系统在通过所公开的基因组位置分类模型以高置信度分类标记的基因组坐标处以大约99.9%的精确度、99.9%的再检出率和99.9%的一致性准确地鉴别SNV。本公开报告了以下关于精确度、再检出率和一致性的额外统计资料。与所公开的基因组分类系统的准确度和宽度大不相同,GIAB或GA4GH对于参考基因组的常规可报告范围(具有单一分类)限于参考基因组的约79%-84%。此外,PlatinumGenomes排除了基因组分类现在可以以异常的精确度、再检出率和一致性进行分类的有问题的基因组区域。
除了提高的准确度之外,在某些实施方案中,基因组分类系统通过可靠地确定特定基因组坐标处不同变体类型的置信度分类来提高优于常规方法的灵活性。如上所述,由GIAB和GA4GH开发的常规可报告范围不区分变体类型。相比之下,在一些实施方式中,基因组分类系统确定变体类型(例如,反映癌症或镶嵌现象的SNV、插入缺失、变体核碱基检出)特定的基因组坐标的置信度分类。例如,基因组位置分类模型可以生成基因组坐标的不同置信度分类,在这些基因组坐标处检测到单核苷酸变体、核碱基插入、核碱基缺失、结构变体的一部分或CNV的一部分。因此,来自基因组位置分类模型的置信度分类可以指示可在特定基因组坐标处准确地确定单核苷酸变体的特定置信度程度-与对于核碱基插入、核碱基缺失、结构变异的一部分或CNV的一部分可能不同的置信度分类大不相同。
与提高的准确度或灵活性无关,在一些情况下,基因组分类系统生成新文件类型或新增文件类型,这些文件类型引入特定基因组坐标或区域的特定置信度分类-与常规基因组文件不同。作为背景,常规BED文件通常包括染色体名称的字段(例如,chrom=chr3,chrY)、染色体的核碱基或特征的起始位置(例如,第一个碱基编号chromStart=0)和特征结束位置(例如,chromEnd=100)。在一些情况下,BED文件还包括用于标识特定基因和标识检测到的变体的字段。与WIG文件、BAM文件、VSF文件或微阵列文件一样,常规BED文件没有针对特定基因组坐标的置信度分类的字段或注释。相比之下,基因组分类系统生成BED、BAM、WIG、VCF、微阵列或其他数字文件格式的新的数字文件,该数字文件具有针对特定基因组坐标或区域的置信度分类的注释或其他指示符。如上所述,在一些情况下,基因组分类系统根据不同置信度分类类型生成各自包含基因组坐标的注释的不同数字文件(例如,针对高置信度分类、中等置信度分类、低置信度分类中的每一种的不同数字文件)。通过引入新的置信度分类指标,基因组分类系统可以针对特定基因组坐标或区域处的多种不同变体-核碱基检出以标记或评分形式提供特定置信度分类。
如前面的描述所示,本公开描述了基因组分类系统的各种特征和优点。如本公开中所用,例如,术语“样品核酸序列”或“样品序列”是指从样品生物体分离或提取的核苷酸序列(或此类分离或提取的序列的拷贝)。具体而言,样品核酸序列包括从样品生物体分离或提取并由含氮杂环碱基组成的核酸聚合物的片段。例如,样品核酸序列可包括脱氧核糖核酸(DNA)、核糖核酸(RNA)的片段,或者核酸的其他聚合形式或下文所述核酸的嵌合或杂合形式。更具体地,在一些情况下,样品核酸序列是在由试剂盒制备或分离并且由测序设备接收的样品中发现的。
如本文进一步所用,术语“核碱基检出”是指用以添加到用于测序循环的寡核苷酸内的特定核碱基的指定或确定。具体地,核碱基检出指示已掺入核苷酸样品玻片上的寡核苷酸内的核苷酸类型的指定或确定。在一些情况下,核碱基检出包括核碱基至强度值的指定或确定,该强度值由添加到核苷酸样品玻片(例如,在流动池的孔中)的寡核苷酸的荧光标记核苷酸产生。另选地,核碱基检出包括核碱基至色谱峰或电流变化的指定或确定,该色谱峰或电流变化由穿过核苷酸样品玻片的纳米孔的核苷酸产生。通过使用核碱基检出,测序系统确定核酸聚合物的序列。例如,单个核碱基检出可包括DNA的腺嘌呤检出、胞嘧啶检出、鸟嘌呤检出或胸腺嘧啶检出(缩写为A、C、G、T)或RNA的尿嘧啶检出(而不是胸腺嘧啶检出)(缩写为U)。
如上所述,在一些实施方案中,基因组分类系统确定用于将样品核酸序列与示例核酸序列(例如,参考基因组或来自祖先单倍型的核酸序列)的测序指标。如本文所用,术语“测序指标”是指定量测量或评分,指示个体核碱基检出(或核碱基检出的序列)相对于示例核酸序列的基因组坐标或基因组区域比对、比较或定量的程度。具体而言,测序指标可以包括对样品核酸序列与示例核酸序列的基因组坐标的比对程度进行定量的比对指标,诸如缺失大小指标或映射质量指标。此外,测序指标可以包括定量样品核酸序列在示例核酸序列的基因组坐标处的核碱基检出的深度的深度指标,诸如前向-反向深度指标或归一化深度指标。测序指标还可以包括定量核碱基检出的质量或准确度的检出数据质量指标,诸如核碱基检出质量指标、可检出性指标或体细胞质量指标。在一些实施方案中,从测序指标推导或准备的数据可以输入到基因组位置分类模型中。本公开还描述了测序指标且提供下文参考图3的附加示例。
如上所述,在一些实施方案中,基因组分类系统可确定在基因组坐标处的核碱基检出周围的上下文核酸子序列。如本文所用,术语“上下文核酸子序列”是指来自示例核酸序列的一系列核碱基,这一系列核碱基在样品核酸序列中特定核碱基检出的基因组坐标的周围(例如,侧接在该基因组坐标的每一侧或与之相邻)。在一些示例中,上下文核酸子序列是指来自参考序列(或来自祖先单倍型的基因组或序列)的一系列核碱基,这一系列核碱基在样品核酸序列中的核苷酸变体检出或不变检出的周围。具体而言,上下文核酸子序列包括来自示例核酸序列的核碱基,这些核碱基(i)位于样品核酸序列的特定核碱基检出的基因组坐标的上游和下游,并且(ii)处于特定核碱基检出的基因组坐标的基因组坐标阈值数目内。因此,上下文核酸子序列可以包括示例核酸序列(例如,参考基因组)上游的五十个核碱基和位于特定基因组坐标处的SNV下游的五十个核碱基中的核碱基。
正如刚才提到的,基因组分类系统可以从示例核酸序列确定上下文核酸子序列。如本文所用,术语“示例核酸序列”是指来自参考或相关基因组的核苷酸序列,诸如参考基因组或祖先单倍型的序列。具体而言,示例核酸序列包括从样品的祖先(例如,祖先单倍型)遗传的核酸序列的片段或数字核酸序列(例如,参考基因组)的片段。在一些情况下,祖先单倍型序列来自样品的亲本或祖亲本。
如本文进一步所用,术语“基因组坐标”是指基因组(例如,生物体的基因组或参考基因组)内核碱基的特定定位或位置。在一些情况下,基因组坐标包括基因组的特定染色体的标识符和特定染色体内核碱基位置的标识符。例如,一个或多个基因组坐标可以包括染色体的编号、名称或其他标识符(例如,chr1或chrX)以及一个或多个特定位置,诸如在染色体的标识符之后的编号位置(例如,chr1:1234570或chr1:1234570-1234870)。此外,在某些实施方式中,基因组坐标是指参考基因组的来源(例如,线粒体DNA参考基因组的mt或SARS-CoV-2病毒的参考基因组的SARS-CoV-2)和参考基因组的来源内核碱基的位置(例如,mt:16568或SARS-CoV-2:29001)。相比之下,在某些情况下,基因组坐标是指参考基因组内核碱基的位置,而不提及染色体或来源(例如,29727)。
如上所述,“基因组区域”是指基因组坐标的范围。与基因组坐标一样,在某些实施方案中,基因组区域可以通过染色体的标识符和一个或多个特定位置,诸如染色体标识符之后的编号位置来鉴别(例如,chr1:1234570-1234870)。
如上所述,基因组坐标包括参考基因组内的位置。此类位置可以处于特定参考基因组内。如本文所用,术语“参考基因组”是指作为生物体基因的代表性示例而组装的数字核酸序列。无论序列长度如何,在一些情况下,参考基因组表示数字核酸序列中由科学家确定为代表特定物种的生物的示例基因集合或核酸序列集合。例如,线性人类参考基因组可以是GRCh38或来自基因组参考联盟的参考基因组的其他版本。作为另一个示例,参考基因组可包括参考图基因组,其包括线性参考基因组和表示来自祖先单倍型的核酸序列的路径,诸如Illumina DRAGEN图参考基因组hg19。
如本文所用,术语“基因组位置分类模型”是指经训练以生成基因组坐标或基因组区域的置信度分类的机器学习模型。因此,基因组位置分类模型可包括统计机器学习模型或经训练以生成此类置信度分类的神经网络。在一些情况下,例如,基因组位置分类模型采取逻辑回归模型、随机森林分类器或卷积神经网络(CNN)的形式。但是可以训练或使用其他机器学习模型。
正如刚才所表明的,基因组位置分类模型可以是基因组位置分类神经网络。神经网络包括互相连接的人工神经元的模型(例如,分层组织),这些人工神经元基于提供给神经网络的多个输入进行通信并学习近似复杂函数并生成输出(例如,生成的数字图像)。在一些情况下,神经网络是指实施深度学习技术以对数据中的高级抽象建模的算法(或算法集合)。
不管形式如何,基因组位置分类模型都生成置信度分类。如本文所用,术语“置信度分类”是指指示可在基因组坐标或基因组区域确定或检测核碱基的置信度或可靠性的标记、评分或指标。具体而言,置信度分类包括对于特定基因组坐标或在特定基因组区域内可以准确地检出核碱基的程度进行分类的标记、评分或指标。例如,在某些实施方式中,置信度分类包括标识基因组坐标的高置信度分类、中等置信度分类或低置信度分类的标记。另外或另选地,置信度分类包括指示可以在基因组坐标处准确地确定核碱基的概率或可能性的评分。
以下段落就描绘示例实施方案和实现方式的说明性附图来描述基因组分类系统。例如,图1示出根据一个或多个实施方案的系统环境(或“环境”)100的示意图,基因组分类106在其中操作。如所示,环境100包括经由网络112连接到用户客户端设备108和测序设备114的一个或多个服务器设备102。虽然图1显示了基因组分类系统106的一个实施方案,但本公开描述了以下替代实施方案和配置。
如图1中所示,服务器设备102、用户客户端设备108和测序设备114经由网络112连接。因此,环境100的每个部件可经由网络112通信。网络112包括计算设备可在其上通信的任何合适的网络。下文就图13更详细地讨论示例性网络。
如图1中所示,测序设备114包括用于对核酸聚合物进行测序的设备。在一些实施方案中,测序设备114分析从样品中提取的核酸片段或寡核苷酸以利用(本文所述的)计算机实现的方法和系统在测序设备114上直接或间接生成数据。更具体地,测序设备114在核苷酸样品载玻片(例如,流动池)内接收并且分析从样品中提取的核酸序列。在一个或多个实施方案中,测序设备114利用SBS以对核酸聚合物测序。作为跨网络112进行通信的补充或替代,在一些实施方案中,测序设备114绕过网络112并且直接与用户客户端设备108通信。
如图1进一步所示,服务器设备102可生成、接收、分析、存储和传输数字数据,诸如用于确定核碱基检出或测序核酸聚合物的数据。如图1所示,测序设备114可以发送(并且服务器设备102可以接收)来自测序设备114的检出数据116。服务器设备102还可与用户客户端设备108通信。具体而言,服务器设备102可以向用户客户端设备108发送包含基因组坐标的置信度分类的数字文件118。如图1所示,在一些实施方案中,服务器设备102发送各自包含不同置信度分类的单独数字文件(例如,针对高置信度分类、中等置信度分类、低置信度分类中的每一者的不同数字文件)。在一些情况下,数字文件118(和/或其他数字文件)还包括核碱基检出、误差数据和其他信息。
在一些实施方案中,服务器设备102包括分布式服务器集合,其中服务器设备102包括跨网络112分布并且位于相同或不同物理位置的许多服务器设备。进一步地,服务器设备102可包括内容服务器、应用程序服务器、通信服务器、网络托管服务器或另一类型的服务器。
如图1中进一步所示,服务器设备102可包括测序系统104。通常,测序系统104分析从测序设备114接收的检出数据116,以确定核酸聚合物的核碱基序列。例如,测序系统104可接收来自测序设备114的原始数据并且确定核酸片段的核碱基序列。在一些实施方案中,测序系统104确定DNA和/或RNA片段或寡核苷酸中核碱基的序列。除了处理和确定核酸聚合物的序列之外,测序系统104还生成包含置信度分类的数字文件118,并且可以将数字文件118发送给用户客户端设备108。
正如刚才提到的,并且如图1中所示,基因组分类系统106分析来自测序设备114的检出数据116以确定样品核酸序列的核碱基检出。在一些实施方案中,基因组分类系统106确定此类样品核酸序列的测序指标和特定核碱基检出周围的上下文核酸子序列中的一者或两者。基于从测序指标和上下文核酸子序列中的一者或两者导出或准备的数据-以及基因组坐标的基准真值分类-基因组分类系统106训练基因组位置分类模型以确定基因组坐标的置信度分类。基因组分类系统106还通过向基因组位置分类模型提供从(i)对应于样品的测序指标集合或(ii)对应于样品的上下文核酸子序列制备的数据作为输入来确定针对基因组坐标(或区域)集合的置信度分类集合。基于这些输入,例如,基因组分类系统106使用基因组位置分类模型来确定参考基因组的每个基因组坐标的置信度分类。如上所述,基因组分类系统106还生成包含基因组坐标或区域的集合的置信度分类的数字文件。
如图1中进一步所示和指示,用户客户端设备108可生成、存储、接收和发送数字数据。具体地,用户客户端设备108可从测序设备114接收检出数据116。此外,用户客户端设备108可以与服务器设备102通信以接收包含核碱基检出和/或置信度分类的数字文件118。用户客户端设备108可以相应地在图形用户界面内向与用户客户端设备108相关联的用户呈现基因组坐标的置信度分类-有时连同核苷酸变体检出或核苷酸不变检出一起呈现。
图1中示出的用户客户端设备108可包括各种类型的客户端设备。例如,在一些实施方案中,用户客户端设备108包括非移动设备,诸如台式计算机或服务器,或其他类型的客户端设备。在又其他实施方案中,用户客户端设备108包括移动设备,诸如便携式电脑、平板电脑、移动电话或智能电话。关于用户客户端设备108的附加细节在下文关于图13论述。
如图1中进一步所示,用户客户端设备108包括测序应用程序110。测序应用程序110可以是在用户客户端客户端设备108上存储和执行的网络应用程序或本机应用程序(例如,移动应用程序、桌面应用程序)。测序应用程序110可以接收来自基因组分类系统106的数据并呈现来自数字文件118的数据(例如,通过按基因组坐标呈现特定置信度分类)以在用户客户端设备108处显示。此外,测序应用程序110可以指示用户客户端设备108显示变体核碱基检出或不变核碱基检出的基因组坐标的置信度分类的指示符。
如图1中进一步所示,基因组分类系统106可作为测序应用程序110的一部分位于用户客户端设备108上或位于测序设备114上。因此,在一些实施方案中,基因组分类系统106通过(例如,完全或部分地位于)在用户客户端设备108上实施。在又其他实施方案中,基因组分类系统106由环境100的一个或多个其他部件,诸如测序设备114实施。具体地,基因组分类系统106可以多种不同的方式跨服务器设备102、网络112、用户客户端设备108和测序设备114实施。
尽管图1示出经由网络112进行通信的环境100的部件,但是在某些实施方式中,环境100的部件还可以绕过网络直接与彼此通信。例如,并且如前所述,在一些实施方式中,用户客户端设备108直接与测序设备114通信。另外,在一些实施方案中,用户客户端设备108直接与基因组分类系统106通信。而且,基因组分类系统106可访问容纳在服务器设备102上或由该服务器设备访问的一个或多个数据库,或者环境100中的其他地方。
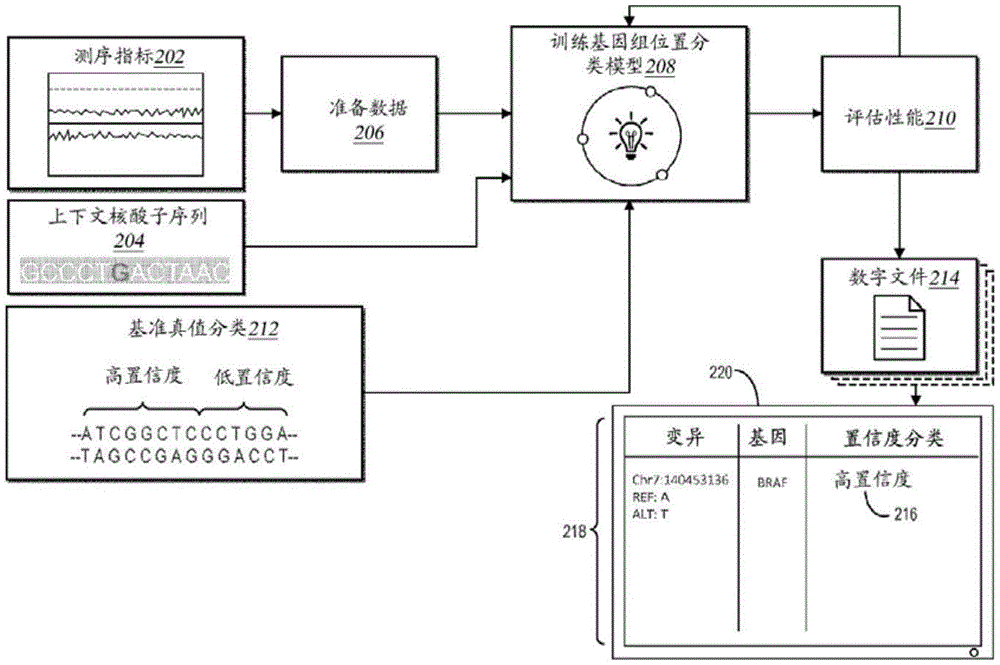
如上所示,基因组分类系统106训练基因组位置分类模型以确定基因组坐标或基因组区域的置信度分类。图2示出了基因组分类系统106使用测序指标和上下文核酸子序列中的一者或两者来训练基因组位置分类模型208的概况。如下文进一步描述的,基因组分类系统106确定样品核酸序列的测序指标202和上下文核酸子序列204中的一者或两者。基于从测序指标202和上下文核酸子序列204中的一者或多者导出或准备的数据,基因组分类系统106训练基因组位置分类模型208以生成基因组坐标的置信度分类。在训练和测试基因组位置分类模型208之后,基因组分类系统106生成包含特定基因组坐标的置信度分类的数字文件214,并且可以使得计算设备220显示来自数字文件214的此类置信度分类。
如图2所示,例如,基因组分类系统106任选地确定用于将样品核酸序列与示例核酸序列(例如,参考基因组或来自祖先单倍型的核酸序列)的基因组坐标进行比较的测序指标202。为准备确定测序指标202,在一些情况下,测序系统104或基因组分类系统106接收检出数据并且确定从多样化样品群组提取的核酸序列的核碱基检出。在一些情况下,例如,基因组分类系统106使用从跨不同群体的30-150个样品确定的核碱基检出和核酸序列。为了提取和确定每个样品核酸序列的核碱基检出,在某些实施方式中,基因组分类系统106使用共用或单个测序流水线,包括用于每个样品的相同核酸序列提取方法、测序设备和序列分析软件。
基于样品核酸序列内的核碱基检出,基因组分类系统106确定测序指标202。如上所示,测序指标202可以包括以下一项或多项:(i)定量样品核酸序列与示例核酸序列(例如,参考基因组或祖先单倍型的核酸序列)比对的程度的比对指标,(ii)定量样品核酸序列在示例核酸序列的基因组坐标处的核碱基检出深度的深度指标,或(iii)定量示例核酸序列的核碱基检出的质量或准确度的检出数据质量指标。当确定比对指标时,例如,基因组分类系统106确定样品核酸序列的缺失熵指标、缺失大小指标、映射质量指标、正插入片段大小指标、负插入片段大小指标、软剪切指标、读段位置指标或读段参考错配指标中的一项或多项。相反,当确定深度指标时,基因组分类系统106确定前向-反向深度指标、归一化深度指标、深度过低指标、深度过高指标或峰值计数指标中的一项或多项。当确定检出数据质量指标时,例如,基因组分类系统106确定样品核酸序列的核碱基检出质量指标、可检出性指标或体细胞质量指标中的一项或多项。下面就图3进一步描述测序指标202。
除了确定测序指标202之外,如图2所示,基因组分类系统106还从测序指标202准备数据206,用于输入到基因组位置分类模型208中。当准备用于输入的数据时,基因组分类系统106可以通过以各种方式汇总或平均测序指标202来从测序指标202提取数据。除了提取之外,在某些情况下,基因组分类系统106还修改测序指标202或从测序指标202提取的数据,以格式化数据用于输入到基因组位置分类模型208中。在提取和修改测序指标202之后或除此之外,在一些实施方案中,基因组分类系统106还将不同类型的测序指标202标准化为相同标度(例如,平均值为0且标准偏差为1)。
如图2进一步所示,作为确定测序指标202的补充或替代,基因组分类系统106从示例核酸序列(例如,参考基因组或祖先单倍型序列)确定上下文核酸子序列204-其在特定基因组坐标处的核碱基检出的周围。对于每个此类上下文核酸子序列,在一些情况下,基因组分类系统106确定参考基因组中在距特定核碱基检出的一个基因组坐标或距特定核碱基检出的多个基因组坐标的阈值坐标距离内的上游和下游核碱基。例如,基因组分类系统106可以确定在距SNV、插入缺失、结构变体、CNV或其他变体的基因组坐标的二十个、五十个、一百个或不同数目的核碱基内的上游和下游核碱基。
如下文进一步解释的,上下文核酸子序列204可以包括或排除对应于特定SNV、插入缺失、结构变体、CNV或所讨论的其他变体类型的基因组坐标的核碱基检出。另外,在某些实施方式中,基因组分类系统106通过例如应用矢量算法将上下文核酸子序列204打包或压缩成用于输入到基因组位置分类模型208中的格式来从上下文核酸子序列204导出或准备数据。
已经确定了从测序指标202和上下文核酸子序列204准备的数据中的一者或两者,基因组分类系统106基于此类数据训练基因组位置分类模型208。例如,基因组分类系统106将从测序指标202和上下文核酸子序列204准备的数据中的一者或两者-连同相应的基因组坐标或区域的指示符-迭代地输入到基因组位置分类模型208中。基于迭代输入,基因组位置分类模型208为每个相应的基因组坐标或基因组区域生成预计置信度分类。
在生成预计置信度分类后,基因组分类系统106在训练迭代中使用预计置信度分类来评估基因组位置分类模型208的性能210。例如,对于相应的基因组坐标或基因组区域,基因组分类系统106将预计置信度分类与来自基准真值分类212的基准真值分类进行比较。在每次训练迭代中,例如,基因组分类系统106执行损失函数以确定基因组坐标的预测置信度分类与该基因组坐标的基准真值分类之间的损失。基于所确定的损失,基因组分类系统106调整基因组位置分类模型208的一个或多个参数以提高基因组位置分类模型208生成预计置信度分类的准确度。通过迭代地执行此类训练迭代,基因组分类系统106训练基因组位置分类模型208以确定置信度分类。
在训练基因组位置分类模型208之后,在一些实施方案中,基因组分类系统106使用基因组位置分类模型208的训练版本以基于测序指标集合和/或上下文核酸子序列集合来确定基因组坐标(或区域)集合的置信度分类集合。在一些实施方案中,基因组分类系统106确定来自不同样品的测序指标集合和/或上下文核酸子序列集合。通过确定每个基因组坐标或区域或对应于参考基因组的基因组坐标或区域的至少一个子集的置信度分类,基因组分类系统106生成坐标特定分类或区域特定分类,指示是否可以在此类基因组坐标或区域准确地检测核碱基。因为确定测序指标202或上下文核酸子序列204的核碱基检出使用单个或限定的测序流水线,所以基因组分类系统106同样可以基于使用相同限定的测序流水线分析的样品核酸序列来确定基因组坐标或区域的置信度分类。
如图2进一步所示,基因组分类系统106生成包含基因组坐标或区域的置信度分类的数字文件214。在一些情况下,数字文件214包括置信度分类作为参考文件,计算设备可以访问该参考文件以鉴别特定基因组坐标或区域的置信度分类。数字文件214(或数字文件集合)可以包括每个基因组坐标的高置信度、中等置信度或低置信度的置信度分类或置信度评分。另外,在一些情况下,基因组分类系统106在数字文件214中检出核碱基以使用不同测序方法进行正交验证,因为核碱基检出位于对应于较低可靠性的置信度分类(例如,低置信度分类或低于置信度评分阈值)的基因组坐标处。
如下文进一步解释的,在某些情况下,数字文件214包括特定基因组坐标的核苷酸变体检出和特定基因组坐标的置信度分类。在此类情况下,数字文件214提供临床医生或患者可信赖核碱基检出(包括核苷酸变体检出)的可靠性的情境。如图2进一步所示,在一些实施方案中,基因组分类系统106生成各自包含不同置信度分类的单独数字文件(例如,针对高置信度分类、中等置信度分类、低置信度分类中的每一者的不同数字文件)。
除了生成数字文件214并且如图2中进一步所示,在一些实施方案中,基因组分类系统106还向计算设备220提供核碱基检出(诸如变体核碱基检出或不变核碱基检出)的基因组坐标的特定置信度分类的置信度指示符216。如图2所示,基因组分类系统106不仅可以将置信度分类整合到数字文件214中,还可以整合到用于在计算设备220的图形用户界面218上报告变体检出或不变检出的数据中。例如,如图2所描绘的,测序系统104或基因组分类系统106提供置信度指示符216,用于连同变体检出的基因组坐标和特定基因的标识符一起在图形用户界面218内显示。测序系统104或基因组分类系统106同样可以提供用于连同相同或相似的基因组坐标和/或基因信息一起在图形用户界面上显示的不变检出的置信度指示符。
如上所述,基因组分类系统106确定用于将样品核酸序列与参考基因组的基因组坐标进行比较的测序指标。根据一个或多个实施方案,图3示出基因组分类系统106确定样品核酸序列的核碱基检出302,将序列核碱基检出与示例核酸序列比对304,并且确定样品核酸序列的测序指标306。如下所述,基因组分类系统106确定核碱基检出,比对样品核酸序列,并确定参考基因组内特定基因组坐标的测序指标。
如图3所示,例如,基因组分类系统106确定样品核酸序列302的核碱基检出。为准备此类核碱基检出,在一些实施方案中,使用提取试剂盒或特定核酸序列提取方法从多样化种族的样品中提取或分离核酸序列。提取后,测序设备114使用SBS测序或桑格测序来合成样品核酸序列的拷贝和反向链,并生成指示掺入增长的核酸序列中的个体核碱基的检出数据。基于检出数据,测序系统104确定核酸序列内的核碱基检出。
在一些实施方案中,单个或限定的流水线处理并确定每个样品的此类核酸序列的核碱基。例如,测序系统104可以使用包括相同核酸序列提取方法(例如,提取试剂盒)、相同测序设备和相同序列分析软件的单个测序流水线。具体而言,单个流水线可以包括,例如,使用用于核酸序列提取方法的Illumina Inc.的TruSeq PCR-Free样品制备试剂盒提取DNA片段;使用用于测序设备的NovaSeq 6000Xp、NextSeq 550、NextSeq 1000或NextSeq 2000进行测序;并且使用用于序列分析软件的Dragen种系流水线确定核碱基检出。
在确定样品核酸序列的核碱基检出后,如图3进一步所示,基因组分类系统106将序列核碱基检出与示例核酸序列304进行比对。例如,测序系统104或基因组分类系统106将特定核酸序列的核碱基(在各种读段上)与参考基因组(例如,线性参考基因组或图参考基因组)的核碱基大致匹配。如图3所示,基因组分类系统106对来自每个样品的核酸序列重复该比对过程。如上所示,在一些情况下,作为将核碱基检出与参考基因组比对的补充或替代,将核碱基序列(例如,来自核苷酸读段)与来自祖先单倍型的一个或多个核酸序列进行比对。一旦大致比对,基因组分类系统106就可鉴别每个样品的参考基因组的特定基因组坐标处的核碱基检出。
如图3所表明的,在一些实施方式中,测序系统104或基因组分类系统106将序列核碱基检出与示例核酸序列304进行比对,并且聚集此类核碱基检出的读段和样品数据作为生成BAM和VCF文件中的一者或两者的一部分。为此,测序系统104或基因组分类系统106为每个样品生成BAM文件和VCF文件,该BAM文件包含比对的样品核酸序列的数据,该VCF文件包含在参考基因组的基因组坐标处的核酸变体检出的数据。
如图3进一步所示,在确定核碱基检出并比对样品核酸序列之后,基因组分类系统106确定样品核酸序列306的测序指标。在一些实施方案中,基因组分类系统106确定样品核酸序列在每个基因组坐标(或每个基因组区域)处的测序指标。如上所示,基因组分类系统106任选地从各种样品的BAM和VCF文件确定测序指标。如以下解释的,基因组分类系统106确定定量基因组坐标处的深度、比对或检出数据质量的一个或多个测序指标。以下段落描述了根据对准、深度和检出数据质量粗略分组的示例测序指标。
正如刚才所示,基因组分类系统106可以确定定量样品核酸序列的核碱基检出与示例核酸序列(例如,参考基因组或祖先单倍型的核酸序列)的基因组坐标的比对的比对指标。为了说明,在一些情况下,基因组分类系统106通过例如确定在基因组坐标处读段的平均或中值映射质量来确定样品核酸序列的映射质量指标。在一些此类实施方案中,基因组分类系统106鉴别或生成基因组坐标处核碱基检出的映射质量(MAPQ)评分,其中MAPQ评分表示-10log10 Pr{映射位置错误},四舍五入至最接近的整数。在平均值或中值映射质量的替代方案中,在一些实施方案中,基因组分类系统106通过确定与基因组坐标或祖先单倍型比对的所有读段的映射质量的全分布来确定样品核酸序列的映射质量指标。作为映射质量指标的补充或替代,基因组分类系统106可以通过例如确定跨越对应于参考基因组或祖先单倍型的基因组坐标的软剪切核碱基的总数来确定样品核酸序列的软剪切指标。因此,在一些情况下,基因组分类系统106确定在读段的任一侧(例如,读段的5引物端或3引物端)的特定基因组坐标处不匹配示例核酸序列(例如,参考基因组或祖先单倍型)并且出于比对的目的被忽略的核碱基的数目。
作为比对指标的另一示例,在一些实施方案中,基因组分类系统106通过例如跨多个读段(例如,与特定基因组坐标重叠的所有读段)或跨多个循环(例如,所有循环)确定在特定基因组坐标处与示例核酸序列(例如,参考基因组或祖先单倍型)的核碱基不匹配的核碱基的总数来确定样品核酸序列的读段-参考错配指标。相比之下,在某些情况下,基因组分类系统106通过例如确定覆盖基因组坐标的核碱基的测序读段内的平均或中值位置来确定样品核酸序列的读段位置指标。
除了上述比对指标之外,基因组分类系统106还可以通过确定对样品核酸序列的基因组坐标处的插入缺失进行定量的插入缺失指标(诸如缺失指标)来确定比对。在一些情况下,基因组分类系统106通过例如确定跨越参考基因组的基因组坐标的缺失的平均大小或中值大小来确定样品核酸序列的缺失大小指标。此外,在某些实施方式中,基因组分类系统106通过例如确定参考基因组的基因组坐标或基因组区域的缺失大小的分布或方差来确定样品核酸序列的缺失熵指标。样品核酸序列中具有单核碱基的一致或重复缺失的基因组坐标或区域(例如,20%的样品包括单核碱基缺失)具有比样品核酸序列中具有不同缺失大小的不同基因组坐标或区域(例如,20%的样品包括单核碱基缺失、5核碱基缺失或10核碱基缺失)更小的缺失熵。
除了作为上述比对指标的示例的缺失指标之外,基因组分类系统106还可以确定对样品核酸序列的基因组坐标处的插入进行定量的插入大小指标。例如,在某些实施方式中,基因组分类系统106通过确定覆盖基因组坐标的读段的平均或中值正插入片段大小来确定样品核酸序列的正插入片段大小指标。此类正插入片段可以包括DNA或RNA片段的未被两个测序读段覆盖的区域。与正插入片段大小指标相反,在一些情况下,基因组分类系统106确定样品核酸序列的负插入片段大小指标。例如,基因组分类系统106确定覆盖基因组坐标的测序读段的平均或中值负插入片段大小作为负插入片段大小指标。此类负插入片段可以包括两个测序读段之间的重叠。
作为比对指标的补充或替代,基因组分类系统106可以确定定量样品核酸序列的基因组坐标处的核碱基检出深度的深度指标。深度指标可以例如定量已经确定并在基因组坐标处比对的核碱基检出的数目。在某些实施方式中,基因组分类系统106通过确定基因组坐标处的前向链和反向链上的深度来确定样品核酸序列的前向-反向深度指标。另外或另选地,基因组分类系统106通过例如确定基因组坐标处归一化标度上的深度来确定样品核酸序列的归一化深度指标。在一些此类情况下,基因组分类系统106使用归一化深度为1指二倍体且归一化深度为0.5指单倍体的标度。
除了前向-反向深度指标或归一化深度指标之外,在一些情况下,基因组分类系统106还确定样品核酸序列的深度过低指标或深度过高指标。例如,基因组分类系统106可通过定量在基因组坐标或基因组区域处低于预期深度或阈值深度覆盖率的核碱基检出的数目来确定深度过低指标。在一些情况下,基因组分类系统106将基因组坐标处的平均深度覆盖率乘以-1,加1,并且设置最小值为0。例如,如果基因组坐标具有0.75的平均深度覆盖率,则基因组分类系统106将确定基因组坐标的深度过低指标为0.25。相比之下,基因组分类系统106可通过定量在基因组坐标或基因组区域处高于预期深度或阈值深度覆盖率的核碱基检出的数目来确定深度过高指标。
如上所述,在一些实施方式中,基因组分类系统106通过例如确定跨基因组样品(例如,多样化基因组样品群组)的基因组坐标或区域的深度分布并且从该分布鉴别深度覆盖率的局部最大值来确定峰值计数指标。在某些实现方式中,基因组分类系统106使用高斯核(Gaussian kernel)将基因组区域的深度过高指标平滑化到深度覆盖的分布中,并且将来自SciPy.org的信号处理分包的寻找峰值函数应用于该分布,以鉴别深度覆盖率的局部最大值。
不依赖深度指标,基因组分类系统106可以确定定量样品核酸序列在基因组坐标处的核碱基检出质量的检出数据质量指标。在某些实施方案中,例如,基因组分类系统106通过确定在示例核酸序列(例如,参考基因组或祖先单倍型的核酸序列)的基因组坐标处满足阈值质量评分(例如,Q20)的核碱基检出的百分比或子集来确定核碱基检出质量指标。为了说明,质量评分(或Q评分)可以指示基因组坐标处不正确核碱基检出的概率对于Q20评分等于1/100,对于Q30评分等于1/1,000,对于Q40评分等于1/10,000,等等。
作为核碱基检出质量指标的补充或替代,在一些实施方案中,基因组分类系统106通过例如确定指示基因组坐标处的正确核苷酸变体检出或核碱基检出的评分来确定样品核酸序列的可检出性指标。在一些情况下,可检出性指标表示具有通过的基因型检出的非N参考位置的分数或百分比,如由Illumina,Inc.实施的。此外,在一些实施方式中,基因组分类系统106使用一个版本的基因组分析工具包(GATK)来确定可检出性指标。
除了核碱基检出质量指标或可检出性指标之外,在一些实施方案中,基因组分类系统106通过例如确定估计确定肿瘤样品中异常读段数目的概率的评分来确定样品核酸序列的体细胞质量指标。例如,体细胞质量指标可以表示使用费雪精确检验(Fisher ExactTest)确定肿瘤样品中异常读段的给定(或更极端)数目—肿瘤和正常BAM文件中的异常读段和正常读段的给定计数的概率的估计值。在一些情况下,基因组分类系统106使用Phred算法来确定体细胞质量指标并且将体细胞质量指标表示为范围从0到60的Phred标度评分,诸如质量评分(或Q评分)。此类质量评分可以等于-10log10(变体概率是体细胞的)。
如以上所表明的,在确定测序指标之后,基因组分类系统106可以从测序指标准备数据以输入到基因组位置分类模型中。根据一个或多个实施方案,图4示出基因组分类系统106通过以下方式从测序指标准备数据404:(i)从测序指标提取数据406,(ii)转换测序指标或指标提取值408,并且(iii)重新工程化或重新组织测序指标或指标提取值410。如统一流形逼近与投影(UMAP)图402a和402b所示以及如以下进一步解释的,数据准备有效地整理基因组位置分类模型的数据,如由来自Platinum Genomes编目的区域的铂碱基和非铂碱基所测量的。如本文所用,术语“铂碱基”或“真集碱基”表示来自由Illumina,Inc.开发的Platinum Genomes的限定置信区域的核碱基。具体而言,铂碱基(或真集碱基)表示来自具有限定的孟德尔遗传模式和一致的纯合遗传中的一种或两种的基因组坐标的核碱基。
如图4所描绘的,例如,基因组分类系统106从测序指标406提取数据以准备用于输入到基因组位置分类模型中的数据。通过从测序指标中提取数据或特征,基因组分类系统106可以概括来自基因组位置分类模型不能以其他方式鉴别或学习的测序指标的信息。例如,在一些实施方案中,基因组分类系统106通过确定以下各项中的一项或多项从测序指标提取数据:(i)某些测序指标的滚动平均值以提供基因组坐标的测序指标的局部汇总,(ii)某些测序指标的掩蔽滚动平均值以提供无基因组坐标的测序指标的局部汇总,或(iii)来自评估给定测序指标的特定假设的统计检验的统计测量。
正如刚才提到的,基因组分类系统106可以进行各种统计检验以从某些测序指标中提取数据用于输入到基因组位置分类模型中。在一些情况下,例如,基因组分类系统106对深度指标(例如,前向-反向深度指标、归一化深度指标)进行柯尔莫戈洛夫-斯米尔诺夫(KS)检验以确定深度是否跨样品群体正态分布。在一些情况下,KS检验根据经验分布函数量化来自每个样品的样品核酸序列的深度之间的距离。作为统计检验的进一步示例,在某些实施方案中,基因组分类系统106对深度指标(例如,前向-反向深度指标)进行二项式检验以确定深度是否在前向链和反向链上平均分布。在某些情形下,二项式检验确定与进入前向链和反向链的类别的深度的预期分布的偏差的统计显著性。
除了作为统计检验的KS检验或二项式检验之外(或作为替代),基因组分类系统106对检出数据质量指标(例如,核碱基检出质量指标)和/或其他测序指标进行二项比例检验以确定前向链和反向链上的读段是否具有相同百分比的满足质量评分阈值(例如,Q20评分)的质量评分。在一些情况下,二项式检验确定在前向链和反向链上的读段具有相同百分比的至少Q20评分的概率的二项式分布。相比之下,在某些实施方式中,基因组分类系统106进行贝茨分布检验(Bates distribution test)以确定来自参考基因组的基因组坐标的平均起始位置是否在样品核酸序列的读段的中途。例如,贝茨分布检验可以确定在读段中途的平均起始位置的平均数的概率分布。
除了从测序指标中提取数据之外,如图4进一步所示,基因组分类系统106转换测序指标或指标提取值408以准备用于输入到基因组位置分类模型中的数据。通过将测序指标(或从测序指标中提取的数据)转换为新的形式或标度,基因组分类系统106可以重新标定某些测序指标以避免过度训练或不必要地训练基因组位置分类模型。例如,在一些实施方案中,基因组分类系统106通过以下一种或多种方式来转换测序指标(或从测序指标提取值的数据):(i)将包括计数或总数的测序指标归一化以将此类计数或总数除以覆盖率,(ii)将所有或一些测序指标和/或从测序指标提取值的数据归一化到相同标度,(iii)确定测序指标的平均值或局部平均值,或(iv)对于测序指标确定来自基因组样品的原始寡核苷酸的前向链对比反向链上的读段的份数或分数。相比之下,基因组分类系统106任选地不转换某些测序指标,例如通过不转换映射质量指标、读段位置指标、缺失大小指标、深度指标、深度过低指标、深度过高指标、正插入片段大小指标、负插入片段大小指标和核碱基检出质量指标。
为了说明特定转换,在一些实施方案中,基因组分类系统106覆盖率通过将跨越基因组坐标的软剪切核碱基的总数转化成基于来自样品的读段总数的百分比来归一化软剪切指标。作为进一步转换示例,在某些情况下,基因组分类系统106将深度指标归一化成为在标准偏差内的值,诸如平均值为0且标准偏差为1。此外,基因组分类系统106有时通过确定在基因组坐标或基因组区域处与参考基因组的核碱基不匹配的核碱基的平均数目来确定读段-参考错配指标的局部平均值。作为另一个转换示例,在一些实施方案中,基因组分类系统106针对核碱基检出质量指标或深度指标确定来自基因组样品的原始寡核苷酸的前向链对比反向链上的读段的份数或分数。通过确定用于测序指标的前向链对比反向链的分数,基因组分类系统106可以生成前向分数指标,诸如前向分数-核碱基检出质量指标或前向分数-深度指标。
在从测序指标中提取数据并转换测序指标之后,在一些实施方案中,基因组分类系统106重新工程化或重新组织测序指标或指标提取值410以准备用于输入到基因组位置分类模型中的数据。通过重新工程化或重新组织某些测序指标或指标提取值,基因组分类系统106可将某些测序指标或指标提取值打包成基因组位置分类模型可以处理的格式。例如,基因组分类系统106可以通过以下方式重新工程化或重新组织测序指标或指标提取值:(i)应用线性标度函数来标度某些测序指标或指标提取值;(ii)从某些测序指标中剪切概率值(p值);(iii)确定某些测序指标或指标提取值的绝对值;(iv)离散化某些测序指标以将此类指标从连续值改变为值的类别;(v)用其他值替换某些测序指标或指标提取值(例如,以避免零值);或(vi)通过对限定范围之外的值进行对数转换来平滑剪切某些测序指标以最小化离群值效应。相比之下,基因组分类系统106任选地不重新工程化或重新组织某些测序指标,诸如映射质量指标、软剪切指标、核碱基检出质量指标、缺失熵指标、深度指标、读段参考错配指标和峰值计数指标。
为了说明特定重新工程化或重新组织测序指标,在一些实施方案中,基因组分类系统106应用线性标度函数以通过例如使用y=(a*x)+b的线性函数对值进行标度来对某些测序指标或指标提取值进行标度,其中“x”表示测序指标或指标提取值的原始值,“y”表示测序指标或指标提取值的标度值,并且“a”和“b”表示标度的不同变量。在某些情况下,基因组分类系统106将线性标度函数应用于读段位置指标、深度过低指标、深度过高指标和前向分数指标的值。作为重新工程化或重新组织测序指标的进一步示例,在一些情况下,基因组分类系统106对于读段位置指标和前向分数指标用0.5值替换0.0值,和/或对于对核碱基检出质量指标的二项式比例检验用1.0e-100替换0.0值。此外,基因组分类系统106有时确定读段位置指标和前向分数指标的绝对值。
作为替换值或确定用于重新工程化或重新组织某些测序指标的绝对值的补充(或替代),在一些实施方案中,基因组分类系统106对数平滑剪切缺失大小指标、深度指标和深度过高指标,以有效地创建缺失大小剪切指标、深度剪切指标和深度过高剪切指标。例如,基因组分类系统106对数平滑剪切缺失大小指标、归一化深度指标和高于值5的深度过高指标,而不修改这些测序指标的其他值。例如,对于值1.5,基因组分类系统106不会修改该值并且保持输入到基因组位置分类模型中的对应测序指标的原始值。但是对于值9,基因组分类系统106使用5+log(9–5+1)的对数公式转换值9以输出并且使用值~5.7。
除了平滑剪切之外或代替平滑剪切,在某些情况下,基因组分类系统106剪切来自对深度指标的KS检验、对深度指标的二项式检验、对检出数据质量指标的二项式比例检验、或对读段位置指标的贝茨分布检验的p值。对于此类统计检验中的每个值,例如,基因组分类系统106对高于5.0的Phred标度p值进行对数平滑化以避免过度训练基因组位置分类模型。例如,基因组分类系统106将对40的Phred标度p值进行对数平滑化以变为~6.5。
为了进一步说明测序指标的特定重新工程或重新组织,在一些实施方案中,基因组分类系统106将来自正插入片段大小指标和负插入片段大小指标的连续值离散化为值的类别。例如,基因组分类系统106将不同大小的正插入或负插入离散化为三个类别:在第一类中低于200个核碱基插入,在第二类别中200至800个核碱基插入,在第三类别中高于800个核碱基插入。
如下面进一步解释的,在一些实施方案中,基因组分类系统106将从测序指标提取、转换和重新标度的数据输入到基因组位置分类模型中用于训练或应用。例如,基因组分类系统106聚集来自每个基因组坐标的测序指标的重新标度的数据,并且将重新标度的测序指标数据连同基因组坐标标识符一起迭代输入到基因组位置分类模型中。
通过从如上所示的测序指标准备数据,基因组分类系统106有效地转换测序指标(或来自测序指标的推导值)以向基因组位置分类模型指示基因组坐标的相对更高或更低的可靠性。为了正交检验此类数据准备的有效性,研究人员执行UMAP算法以(i)在数据准备之前在UMAP图402a中根据测序指标可视化特定基因组坐标处的核碱基,并且(ii)在数据准备之后在UMAP图402b中根据测序指标可视化特定基因组坐标处的核碱基,如图4所示。如UMAP图402a和402b所指示,数据准备有效地将来自根据Platinum Genomes具有验证的变体检出(这里,在铂碱基处)的基因组区域的核碱基检出与来自根据Platinum Genomes没有验证的变体检出(这里,在非铂碱基处)的基因组区域的核碱基检出分开。注意,UMAP图402a和402b不表示基因组位置分类模型的组成部分或数据准备的组成部分,而仅可视化数据准备的正交检验。
作为确定测序指标的补充或替代,在一些实施方案中,基因组分类系统106从在核碱基检出周围的示例核酸序列(例如,参考基因组、祖先单倍型)确定上下文核酸子序列,作为基因组位置分类模型的输入。根据一个或多个实施方案,图5示出了基因组分类系统106确定对应于核碱基检出502的上下文核酸子序列504作为此类输入的示例。
如图5所示,基因组分类系统106鉴别特定基因组坐标的核碱基检出502。在一些情况下,基因组分类系统106从VCF文件鉴别在基因组坐标处的变体核苷酸检出或不变核苷酸检出。基于基因组坐标,基因组分类系统106进一步鉴别来自参考基因组的一系列核碱基,这一系列核碱基位于核碱基检出502的基因组坐标的上游和下游,并且在距核碱基检出502的基因组坐标的基因组坐标的阈值数目内。如图5所描绘的,基因组分类系统106将来自示例核酸序列的这一系列上游和下游核碱基鉴别为核碱基检出502的上下文核酸子序列504。在鉴别后,在一些实施方案中,基因组分类系统106还通过应用矢量算法(例如,Nucl2Vec,独热矢量(one-hot vector))准备上下文核酸子序列504以将上下文核酸子序列504编码到矢量中用于输入到基因组位置分类模型中。
当从示例核酸序列鉴别上下文核酸子序列时,基因组分类系统106可以使用多种阈值数目的基因组坐标。例如,上下文核酸子序列可以包括在距特定核碱基检出的基因组坐标10个、50个、100个、400个或任何其他数目的基因组坐标内的参考基因组核碱基。如下文进一步描述的,在一些情况下,基因组分类系统106提高了基因组位置分类模型确定基因组坐标的置信度分类的准确度,因为对于上下文核酸子序列而言,核碱基的基因组坐标的阈值数目增加。
除了基因组坐标的阈值数目变化之外,在一些实施方案中,基因组分类系统106使用多种不同变体检出类型作为从中确定阈值数目的基因组坐标的核碱基检出。如图5所描绘的,例如,基因组分类系统106鉴别核碱基检出502的SNV。然而,在一些实施方案中,基因组分类系统106将插入缺失、结构变异或CNV的一个基因组坐标(或多个基因组坐标)鉴别为从中确定构成上下文核酸子序列的基因组坐标的阈值数目内的核碱基的参考点。
为了鉴别核苷酸变体检出作为确定上下文核酸子序列的基础,在一些情况下,基因组分类系统106使用来自VCF文件的变体检出。仅举一个例子,基因组分类系统106可以从来自HapMap计划的NA12878(或其他样品)的VCF文件的一致性数据中鉴别变体检出。在一种此类情况下,基因组分类系统106确定来自NA12878的96个重复的变体检出,作为确定用于输入到基因组位置分类模型和训练的上下文核酸子序列的基础。
在确定测序指标和上下文核酸子序列并准备用于输入的数据之后,基因组分类系统106训练并应用基因组位置分类模型。根据一个或多个实施方案,图6A-图6C示出了基因组分类系统106训练和应用基因组位置分类模型608以确定基因组坐标(或区域)的置信度分类,并且随后提供对应于核碱基检出的置信度分类的置信度指示符以在计算设备上显示。如图6A所示,基因组分类系统106进行多个训练迭代,其中基因组分类系统106(i)基于测序指标和上下文核酸子序列中的一者或两者来确定预测置信度分类,并且(ii)将此类预测置信度分类与基准真值分类进行比较。在训练之后,如图6B中所示,基因组分类系统106应用基因组位置分类模型608的训练版本来确定基因组坐标(或区域)集合的置信度分类集合并且生成包含置信度分类集合的数字文件。基于所生成的数字文件,如图6C所示,基因组分类系统106提供核碱基检出的基因组坐标(或区域)的置信度分类以在图形用户界面上显示。
为简单起见,本发明描述初始训练迭代,随后是如图6A中所描绘的后续训练迭代的总结。例如,在图6A描绘的初始训练迭代中,基因组分类系统106将从测序指标602和对应于特定基因组坐标的基因组坐标标识符604的上下文核酸子序列606中的一者或两者推导出或准备的数据输入到基因组位置分类模型608中。
正如刚才所表明的和图6A中所描绘的,在一些实施方案中,基因组分类系统106输入从基因组坐标标识符604的基因组坐标特定的测序指标602准备的数据,而不输入基因组坐标的相应上下文核酸子序列。在一些此类实施方案中,输入包括来自KS检验、二项式检验、二项式比例检验或贝茨分布检验中的一种或多种的数据。相比之下,在某些实施方式中,基因组分类系统106输入基因组坐标标识符604的基因组坐标特定的上下文核酸子序列606,而不输入相应的测序指标。可选地,基因组分类系统106输入从测序指标602和上下文核酸子序列606两者推导出或准备的数据。
如以上所表明的,基因组分类系统106以多种格式将此类数据输入到基因组位置分类模型608中。例如,在一些实施方案中,基因组分类系统106将来自基因组坐标的测序指标602的重新标度的数据聚集到包含基因组坐标标识符604的每个重新标度的测序指标的矢量或矩阵中。在一些情况下,基因组分类系统106将来自对应于基因组坐标标识符604的基因组坐标的测序指标602的重新标度的数据与上下文核酸子序列606一起聚集到输入矢量或矩阵中。相比之下,在某些实施方式中,基因组分类系统106将来自对应于基因组坐标标识符604的基因组坐标的测序指标602的重新标度的数据以及上下文核酸子序列606中的核碱基的每个基因组坐标的重新标度的测序指标与上下文核酸子序列606一起聚集到输入矢量或矩阵中。
为了说明,在一些实施方案中,基因组分类系统106将从测序指标602推导出或准备的数据作为数值阵列的集合输入到基因组位置分类模型608中。例如,基因组分类系统106将从测序指标602推导出或准备的数据存储在分级信息格式5(HDF5)文件中,并将该数据作为数值阵列(例如,一维Python NumPy数组)的集合输入到基因组位置分类模型608中。
为了进一步说明,在某些实施方式中,基因组分类系统106将从测序指标606和上下文核酸子序列606两者推导出或准备的数据作为矩阵输入(到基因组位置分类模型608中),其中上下文核酸子序列602的大小或长度为第一维度,个体测序指标的数目和/或来自个体测序指标的推导值为第二维度。例如,上下文核酸子序列606的大小或长度的第一维度可包括上下文核酸子序列606中核碱基的数目加1(例如,核碱基检出每侧25个碱基为51个维度,核碱基检出每侧50个碱基为101个维度)。相比之下,个体测序指标数目的第二维度可以包括表示每个个体测序指标、来自测序指标的推导值以及上下文核酸子序列(例如,占据5个位置的独热编码的上下文核酸子序列)的矢量化表示的维数。
此外,将对应于多个核碱基检出的上下文核酸子序列的多个示例输入到基因组位置分类模型608中时,在一些情况下,基因组分类系统106输入三维张量。此类张量可以包括表示示例数目的第一维度、表示上下文核酸子序列的大小或长度的第二维度、以及针对个体测序指标和/或来自个体测序指标的推导值的数目的第三维度。
将从上下文核酸子序列606推导出或准备的数据输入到基因组位置分类模型608中时,在一些情况下,基因组分类系统106输入从DNA或RNA单链推导的数据。例如,基因组分类系统106输入来自示例核酸序列(例如,祖先单倍型)的正义链或负义链的上下文核酸子序列的矢量化形式。在一些实施方案中,基因组分类系统106单独输入来自于从示例核酸序列(例如,祖先单倍型)确定的上下文核酸子序列的正义链和负义链的上下文核酸子序列的矢量化形式,并且确定对应于正义链和负义链中的每一者的置信度分类。
在输入从测序指标602和上下文核酸子序列606中的一者或两者推导出或制备的数据之后,基因组分类系统106执行基因组位置分类模型608。如上所指示,基因组位置分类模型608可以采取各种形式。基因组位置分类模型608可以是例如统计机器学习模型或神经网络。在一些情况下,仅举几个例子,基因组位置分类模型采取逻辑回归模型、随机森林分类器、CNN或长短时记忆(LSTM)网络的形式。
例如,在一些实施方案中,基因组位置分类模型608采取包括2个卷积层和1个完全连接层的CNN的形式。相比之下,在某些情况下,基因组位置分类模型608采取包括8、12、20个卷积层和1个完全连接层的CNN的形式。另选地,基因组位置分类模型608采取改良初始网络(Inception Network)的形式,其在每一层(例如,conv3、conv5、conv7、conv9)中包括连接在一起的多个卷积层,其中每个卷积层来源于相同的前一层。
在接收用于初始训练迭代的输入数据后,如图6A中进一步所示,基因组位置分类模型608确定对应于基因组坐标标识符604的基因组坐标的预测置信度分类610。在一些实施方案中,例如,预测置信度分类610包括指示可以在对应于基因组坐标标识符604的基因组坐标处准确地确定核碱基的高置信度分类、中等置信度分类或低置信度分类的标记。相比之下,在某些实现方式中,预测置信度分类610包括指示可以在对应于基因组坐标标识符604的基因组坐标处以高置信度确定核碱基的概率或可能性的评分。基于此类概率或可能性评分,在一些情况下,基因组分类系统106确定高置信度分类、中等置信度分类或低置信度分类。
如以上所指示,在某些实施方式中,基因组分类系统106确定变体类型特定的基因组坐标的置信度分类。因此,当确定预测置信度分类610时,基因组分类系统106可以确定SNP特定的基因组坐标、各种大小的插入(例如,短插入、中等插入或长插入)、各种大小的缺失(例如,短缺失、中等缺失或长缺失)、各种大小的结构变异或各种大小的CNV的预测变体置信度分类。另外或可选地,基因组分类系统106可以确定体细胞核碱基变体或种系核碱基变体,诸如反映癌症或体细胞镶嵌现象的体细胞核碱基变体或反映种系镶嵌现象的种系核碱基变体特定的基因组坐标的预测变体置信度分类。为了训练基因组位置分类模型608以生成变体类型特定的变体置信度分类,如以下所解释的,基因组分类系统106使用相应变体类型特定的基准真值分类。
如图6A中进一步所示,在确定预测置信度分类610之后,基因组分类系统106针对对应于基因组坐标标识符604的基因组坐标将预测置信度分类610与基准真值分类614进行比较。例如,在一些实现方式中,基因组分类系统106使用损失函数612来比较预测置信度分类610与基准真值分类614(并且确定之间的任何差异)。如下文所解释的,在一些情况下,基准真值分类614反映在对应于基因组坐标标识符604的基因组坐标处的孟德尔遗传模式或核碱基检出的重复一致性。如图6A进一步所示,基因组分类系统106利用损失函数612从预测置信度分类610和基准真值分类614确定损失616。
取决于基因组位置分类模型608的形式,基因组分类系统106可针对损失函数612使用多种损失函数。在某些实施方案中,例如,基因组分类系统106使用逻辑损失(例如,对于逻辑回归模型)、基尼不纯度(Gini impurity)或信息增益(例如,对于随机森林分类器)、或交叉熵损失函数或最小平方误差函数(例如,对于CNN、LSTM)。
如以上所指示,基因组分类系统106可以使用多种基础或基准来鉴别基准真值分类。在一些实施方案中,例如,当基因组坐标对应于具有以下特征中的一个(或任何组合)的核苷酸变体检出时,基因组分类系统106用高置信度的基准真值分类来标记该基因组坐标:孟德尔遗传模式、一致纯合遗传(例如,其中相同等位基因来自两个亲本的基因组坐标)、或在基因组坐标处展示核苷酸变体检出的重复的阈值数目(或阈值份数)。例如,当重复的阈值数目(或阈值份数)等于或超过展示核苷酸变体检出的样品核酸序列的56%(例如,96个样品中的54个)时,基因组分类系统106可以用高置信度的基准真值分类来标记基因组坐标。在一个另外的示例性实施方案中,当基因组坐标对应于来自Platinum Genomes的铂碱基或真集碱基时,基因组分类系统106用高置信度的基准真值分类来标记该基因组坐标,并且当基因组坐标不对应于来自Platinum Genomes的铂碱基或真集碱基时用低置信度的基准真值分类来标记该基因组坐标。
相比之下,在一些情况下,当基因组坐标对应于具有以下特征中的一个(或任何组合)的核苷酸变体检出时,基因组分类系统106用低置信度的基准真值分类来标记基因组坐标:非孟德尔遗传模式、失败或不一致的纯合遗传、或在基因组坐标处展示核苷酸变体检出的重复的阈值数目(或阈值份数)。例如,当重复的阈值数目(或阈值份数)等于或低于展示核苷酸变体检出的样品核酸序列的15%(例如,96个样品中的14个)时,基因组分类系统106可以用低置信度的基准真值分类来标记基因组坐标。
在一些实施方案中,基因组分类系统106任选地使用用于中等置信度的标记。例如,当基因组坐标对应于具有以下至多两项的核苷酸变体检出时,基因组分类系统106用中等置信度的基准真值分类来标记该基因组坐标:孟德尔遗传模式、一致纯合遗传(例如,相同等位基因来自两个亲本的基因的基因组坐标部分)和跨技术重复的再现性。但是基因组分类系统106还可以使用用于高置信度分类和低置信度分类的标记作为基准真值分类—没有中等置信度分类。
如以上所指示的,在一些情况下,基因组分类系统106用针对特定类型的核苷酸变体检出的基准真值分类来标记基因组坐标。例如,基因组分类系统106用针对以下一种或多种的基准真值分类来标记基因组坐标:SNP、各种大小的插入、各种大小的缺失、各种大小的结构变异、各种大小的CNV、反映癌症或体细胞镶嵌现象的体细胞核碱基变体、或反映种系镶嵌现象的种系核碱基变体。此类体细胞镶嵌现象可以包括癌细胞或具有镶嵌变异的健康细胞中的一种或两种镶嵌现象。在某些实现方式中,基因组分类系统106基于在基因组坐标处展示核苷酸变体检出的重复的阈值数目(或阈值份数),用核苷酸变体检出类型特定的基准真值分类来标记基因组坐标。
如下表1中所示,研究人员鉴别了用于鉴别基因组坐标处的特定类型的核苷酸变体检出(例如,SNP、缺失、插入)的阈值重复计数,作为用高置信度或低置信度的基准真值分类来标记基因组坐标的基础。具体而言,研究人员基于来自96个总样品在给定基因组坐标处的特定类型的核苷酸变体检出的技术重复计数,确定检测特定类型的核苷酸变体检出的随机假阳性率的阳性预测值(PPV)。通过将重复计数与PPV进行比较,研究人员确定表1中报告的最小重复计数,在该最小重复计数下,核苷酸变体检出的随机假阳性率满足目标阈值,诸如对于高置信度的基准真值分类,在基因组坐标处小于0.05%的随机假阳性核苷酸变体检出率的目标阈值。
表1
如表1中所报道,短缺失跨越1-5个核碱基,中等缺失跨越5-15个核碱基,长缺失跨越超过15个核碱基并且可以包括(或短于)50个核碱基的缺失,短插入跨越1-5个核碱基,中等插入跨越5-15个核碱基,并且长插入跨越超过15个核碱基并且可以包括(或短于)50个核碱基的插入。研究人员分别对于SNP、短缺失、中等缺失、长缺失、短插入、中等插入和长插入在总共96个样品中确定54、64、63、70、63、80和47的最小重复计数作为用高置信度的基准真值分类来标记基因组坐标的阈值。如表1中所示,用高于刚才列出的相应最小重复计数的高置信度的基准真值分类来标记基因组坐标的最小重复计数,分别对应于SNP、短缺失、中等缺失、长缺失、短插入、中等插入和长插入的变体检出再现性的95.07%、95.22%、93.83%、94.14%、95.25%、97.39%和81.92%的平均置信度。换句话说,表1中的平均高置信度再现性指示设定高置信度阈值的变体的最小重复次数。表1还报告了根据一个或多个实施方案,对于SNP、缺失和插入,基因组分类系统106用高置信度或低置信度的基准真值分类进行标记的位点(例如,基因组坐标或基因组区域)的数目。
作为标记的替代,在一些实施方案中,基因组分类系统106为基因组坐标分配反映置信度评分的基准真值分类,该置信度评分具有关于基因组坐标是否对应于具有孟德尔遗传模式、一致纯合遗传或跨技术重复的再现性中的一种或多种的核苷酸变体检出的权重。例如,在一些实施方案中,基因组坐标的此类置信度评分表示孟德尔遗传模式的一个值点乘以第一权重、一致纯合遗传的一个值点乘以第二权重以及跨技术重复的再现性的一个值点乘以第三权重的总和或乘积。
基于来自损失函数612的确定的损失616,基因组分类系统106随后调整基因组位置分类模型608的参数。通过调整参数,基因组分类系统106增加基因组位置分类模型608在训练迭代中准确地确定预测的置信度分类的准确度。在初始训练迭代和参数调整之后,如图6A所示,基因组分类系统106还基于从不同基因组坐标的测序指标和上下文核酸子序列中的一者或两者推导出或准备的数据来确定不同基因组坐标的预测置信度分类。在一些情况下,基因组分类系统106执行训练迭代,直到基因组位置分类模型608的参数(例如,值或权重)跨训练迭代没有显著改变或以其他方式符合收敛标准为止。
尽管图6A描绘了生成针对基因组坐标的预测置信度分类的训练迭代,但是在一些实施方案中,基因组分类系统106同样输入数据并且确定针对基因组区域的置信度分类。在此类实施方案的训练迭代中,基因组分类系统106输入基因组区域的基因组区域标识符和从基因组区域内每个基因组坐标的测序指标和上下文核酸子序列中的一者或两者推导出或准备的数据。基因组分类系统106还使用基因组位置分类模型608来基于此类基因组区域特定输入确定基因组区域的预测置信度分类。基因组分类系统106同样使用损失函数来比较基因组区域的预测置信度分类和基因组区域的基准真值分类,并且基于从损失函数确定的损失来调整基因组位置分类模型608的参数。
在训练基因组位置分类模型608之后,并且如图6B中所描绘的,基因组分类系统106应用基因组位置分类模型608的训练版本来确定基因组坐标集合的置信度分类集合并且生成包含置信度分类集合的数字文件。类似于上述训练过程,如图6B所示,基因组分类系统106基于从测序指标和对应于特定基因组坐标的上下文核酸子序列中的一者或两者推导出或准备的数据,确定在基因组坐标之后的基因组坐标的置信度分类。为简单起见,本公开描述用以确定单个置信度分类的初始应用迭代或初始过程,随后是图6B中所描绘的后续应用迭代的汇总。
例如,在图6B描绘的初始应用迭代中,基因组分类系统106将从测序指标618和对应于特定基因组坐标的基因组坐标标识符620的上下文核酸子序列622中的一者或两者推导出或准备的数据输入到基因组位置分类模型608的训练版本中。当训练时,基因组分类系统106可以输入从基因组坐标特定的测序指标618和/或对应于基因组坐标标识符620的基因组坐标特定的上下文核酸子序列622准备的数据的任何组合。基因组分类系统106同样可以通过使用与上述相同格式的输入矢量或输入矩阵来输入从测序指标618和/或上下文核酸子序列622准备的数据。输入到基因组位置分类模型608的训练版本中的上下文核酸子序列622同样可以是DNA或RNA单链(例如,正义链或负义链)。然而,在一些实施方案中,基因组分类系统106使用与用于训练的测序指标和上下文核酸子序列不同的测序指标集合和/或不同的上下文核酸子序列集合(和相应的核碱基检出)来应用基因组位置分类模型608的训练版本。
如图6B中进一步所示在初始应用迭代中,基因组位置分类模型608的训练版本确定对应于基因组坐标标识符620的基因组坐标的置信度分类624。与上述训练一致,置信度分类624可包括(i)可在对应于基因组坐标标识符620的基因组坐标处准确地确定核碱基的高置信度分类、中等置信度分类或低置信度分类的标记,或者另选地,(ii)指示可在对应于基因组坐标标识符620的基因组坐标处以高置信度确定核碱基的概率或可能性的评分。基于用于训练基因组位置分类模型608的基准真值分类的类型,置信度分类624同样可以特定于一种类型的核苷酸变体检出,诸如特定于SNP、各种大小的插入、各种大小的缺失、各种大小的结构变异、各种大小的CNV、反映癌症或体细胞镶嵌现象的体细胞核碱基变体、或反映种系镶嵌现象的种系核碱基变体中的一种或多种。
在初始应用迭代之后,基因组分类系统106还基于从不同基因组坐标的测序指标和上下文核酸子序列中的一者或两者推导出或准备的数据来确定不同基因组坐标的置信度分类。当完成此类应用迭代时,如图6B中所示,基因组分类系统106基于从测序指标和上下文核酸子序列的集合推导出或准备的数据确定基因组坐标集合的置信度分类集合。在一些情况下,该组置信度分类集合包含参考基因组中每个基因组坐标的置信度分类。相比之下,在某些实现方式中,该置信度分类集合包含参考基因组中的一些(但不是全部)基因组坐标的置信度分类。
如图6B中进一步所示,基因组分类系统106还生成包含置信度分类628的数字文件626。如图6B所描绘的,置信度分类628包括由图6B中的基因组位置分类模型608生成的针对该基因组坐标集合的置信度分类集合。与置信度分类624一样并且取决于用于训练基因组位置分类模型608的基准真值分类的类型,置信度分类628同样可以特定于一种类型的核苷酸变体检出,诸如特定于SNP、各种大小的插入、各种大小的缺失、结构变异、CNV、反映癌症或体细胞镶嵌现象的体细胞核碱基变体、或反映种系镶嵌现象的种系核碱基变体中的一种或多种。
为了生成或修改数字文件626,在某些实现方式中,基因组分类系统106生成或修改BED文件以包括用于每个基因组坐标的注释,该注释包括相应的置信度分类。相比之下,在一些实施方案中,基因组分类系统106生成或修改WIG文件、BAM文件、VCF文件、微阵列文件或其他合适的数字文件类型以包括置信度分类628。如图6B进一步所示,在一些实施方案中,基因组分类系统106可以从预测置信度分类生成各自包含不同置信度分类类型的单独数字文件(例如,针对高置信度分类、中等置信度分类、低置信度分类中的每一者的不同数字文件)。
尽管图6B描绘了生成针对基因组坐标的置信度分类的应用迭代,但是在一些实施方案中,基因组分类系统106同样输入数据并且确定针对基因组区域的置信度分类。在此类实施方案的应用迭代中,基因组分类系统106输入基因组区域的基因组区域标识符和从基因组区域内每个基因组坐标的测序指标和上下文核酸子序列中的一者或两者推导出或准备的数据。基因组分类系统106还使用基因组位置分类模型608来基于此类基因组区域特定输入确定基因组区域的置信度分类。
在生成数字文件626(例如,单独数字文件的一部分)之后,在一些情况下,基因组分类系统106使用数字文件626来提供核碱基检出的基因组坐标(或区域)的特定置信度分类以在图形用户界面上显示。根据一个或多个实施方案,图6C示出了测序系统104或基因组分类系统106,从基因组位置分类模型608鉴别并显示对应于核苷酸变体检出的特定基因组坐标的特定置信度分类。
如图6C所指示,例如,测序设备630在测序期间将核碱基掺入样品核酸序列中并捕获指示所掺入的核碱基的相应图像(或其他数据)。基于图像或其他数据,测序系统104或基因组分类系统106在样品核酸序列内的基因组坐标处检测到变体核碱基检出632a、632b和632n。在一些实施方案中,变体核碱基检出632a-632n表示SNV、核碱基插入、核碱基缺失、结构变化、CNV。另外或另选地,在某些实施方式中,变体核碱基检出632a-632n表示反映癌症或体细胞镶嵌现象的体细胞核碱基变体或反映种系镶嵌现象的种系核碱基变体。变体核碱基检出632a-632n同样可以由遗传修饰或表观遗传修饰引起。
如图6C中进一步描绘的,基因组分类系统106将变体核碱基检出632a-632n与来自数字文件626(或来自多个数字文件之一)的置信度分类628中的一个或多个整合。例如,在一些情况下,基因组分类系统106将变体核碱基检出632a-632n编码到数字文件626中,将变体核碱基检出632a-632n与来自数字文件626(或来自多个数字文件之一)的置信度分类628进行比较,或从数字文件626中检索置信度分类628以整合在变体核碱基检出632a-632n的单独数字文件(例如,VCF文件)内。另外或另选地,在某些实施方式中,数字文件626包括对应于置信度分类的基因组坐标的查找表,诸如不同变体类型的不同查找表,其中基因组坐标包括相应的置信度分类。不管此类整合是如何进行的,基因组分类系统106从针对变体核碱基检出632a-632n的特定基因组坐标的置信度分类628中鉴别特定置信度分类。
除了包括变体核碱基检出632a-632n之外,在一些情况下,基因组分类系统106使用不同的测序方法在建议用于正交验证的数字文件214中鉴别变体核碱基检出或非变体核碱基检出。例如,当变体核碱基检出位于对应于特定类型变体的较低可靠性的置信度分类(例如,低置信度分类或低于置信度评分阈值)的基因组坐标处时,基因组分类系统106在数字文件214中包括此类变体核碱基检出的标识符以建议正交验证。通过使用某些置信度分类作为置信度阈值,基因组分类系统106可以标记单个测序流水线不能以足够置信度确定的特定变体核碱基检出或非变体核碱基检出。
在从数字文件626鉴别此类置信度分类之后,如图6C进一步所示,基因组分类系统106向计算设备636提供变体核碱基检出632a-632n的基因组坐标的特定置信度分类的置信度指示符。例如,如图6C中所描绘的,测序系统104或基因组分类系统106提供置信度分类的置信度指示符638a和638b,用于连同变体核碱基检出632a和632b的基因组坐标以及相应基因的标识符一起在计算设备636的图形用户界面634内显示。通过提供置信度指示符638a和638b,基因组分类系统106向临床医生、试验受试者或其他人提供指示某些基因的变体核碱基检出632a和632b的可靠性的关键信息。
如以上所表明的,在一些实施方案中,基因组分类系统106训练或应用基因组位置分类模型以确定反映癌症或体细胞镶嵌现象的体细胞核碱基变体特定的或种系核碱基变体特定的置信度分类。为了训练此类基因组位置分类模型,在一些实施方案中,基因组分类系统106确定来自不同基因组样品的核酸序列子集,该子集模拟来自一种类型的癌症或镶嵌现象的核碱基变体。基因组分类系统106还相对于参考基因组的基因组坐标确定样品核酸序列的某些测序指标。基于这些测序指标,基因组分类系统106生成特定基因组坐标和特定变体核碱基检出(诸如反映镶嵌现象的体细胞核碱基变体或种系核碱基变体)两者特定的基准真值分类。如上所述,使用基准真值分类,基因组分类系统106可进一步训练基因组位置分类模型以确定基因组坐标和该类型的变体核碱基检出两者特定的置信度分类。
根据一个或多个实施方案,图6D-图6H示出基因组分类系统106基于以下的一项或两项确定基准真值分类:(i)来自基因组样品的样品核酸序列的某些测序指标(例如,如以上解释的多样化基因组样品群组)和(ii)反映癌症或镶嵌现象的基因组样品的混合物的变体检出数据(例如,用于检出基因组样品混合物的反映癌症或镶嵌现象的特定类型的变体的再检出率或精确率)。如图6D所描绘的,基因组分类系统106从雄性和雌性基因组样品的组合确定样品核酸序列的子集(例如,百分比),这些子集一起模拟具有癌症或镶嵌现象的基因组样品的变体等位基因频率。如图6E所示,基因组分类系统106确定在样品核酸序列的深度指标、映射质量指标或核碱基检出质量指标中的一项或多项中表现出正常行为的基因组坐标,作为确定高置信度基因组坐标的基准真值分类的基础。如图6F-图6H中进一步描绘的,基因组分类系统106进一步基于以下的一项或两项来确定基准真值分类:来自样品核酸序列的核碱基检出的体细胞质量指标以及用于基于基因组样品的混合物来确定变体核碱基检出的特定类型的再检出率或精确率。
如图6D所示,例如,基因组分类系统106确定来自不同基因组样品的形成混合基因组的样品核酸序列的子集。当相应的样品核酸序列子集混合在一起时,混合基因组模拟具有癌症或镶嵌现象的基因组样品。为了模拟此类具有癌症或镶嵌现象的基因组样品,例如,基因组分类系统106确定来自第一基因组样品639a的样品核酸序列640a的百分比和来自第二基因组样品639b的样品核酸序列640b的百分比,当混合在一起时,模拟表现出癌症或镶嵌现象特征的基因组样品的变体等位基因频率。作为确定样品核酸序列640a和640b的子集的一部分,基因组分类系统106对于第一基因组样品639a和第二基因组样品639b从Platinum Genomes的真集碱基估计不同子集混合物(或百分比混合物)的变体等位基因频率。
根据一些实施方案,基因组分类系统106使用来自混合基因组,而不是单个天然存在的基因组的样品核酸序列,因为测序系统往往不能一致地或准确地检测来自天然存在的基因组的序列中反映癌症或镶嵌现象的核碱基变体。例如,转移的肿瘤可突变一些体细胞类型的DNA中的核碱基,但不突变其他体细胞类型。事实上,一些肿瘤可以影响特定细胞类型的所有细胞,诸如在血液中扩散的白血病,使得仅有肿瘤的样品是专门可用并且使得获得对照样品是不切实际或不可能的。在不同的活检组织样品中或在不同的活检时间,从具有癌症的天然存在的基因组提取的DNA可以具有显著不同的核碱基等位基因频率,使得天然存在的基因组的样品是估计由一些癌症引起的变体等位基因频率不可预测的样品。为了避免癌细胞或健康细胞的DNA中核碱基变体的不可预测的可变性,在一些实施方式中,基因组分类系统106确定模拟反映癌症的变体的混合基因组。
与癌症引起的变体大不相同,样品DNA中天然存在的镶嵌现象可表现出在测序期间难以检测的不常见的变体-不管该镶嵌现象是由肿瘤、基因遗传、复制错误还是一些其他因素引起的。虽然一个人可能具有小百分比的表现出镶嵌现象的DNA,但是许多现有的测序系统不能检测反映镶嵌现象的常见核碱基变体,除非该测序系统对来自具有该类型的镶嵌现象的大得多的样品组的寡核苷酸进行测序。为了产生训练基因组样品而不发现表现出镶嵌现象的稀有样品组,在某些实施方案中,基因组分类系统106确定混合基因组以模拟反映体细胞镶嵌现象或种系镶嵌现象的变体。
图6D示出了基因组分类系统106的示例,其确定一个此类混合基因组的样品核酸序列的子集并确定相应的变体等位基因频率。如图6D中所描绘的,基因组分类系统106确定混合基因组的杂合等位基因和纯合等位基因两者的SNP的变体等位基因频率。根据样品核酸序列子集640a(此处为60%)和样品核酸序列子集640b(此处为40%)所反映的百分比,基因组分类系统106通过参考来自Platinum Genomes的第一基因组样品639a(例如,NA12877)和第二基因组样品639b(例如,NA12878)的真集碱基来确定或预测相关变体等位基因频率。虽然图6D描述了来自混合基因组的SNP的变体等位基因频率,但是基因组分类系统106可以确定混合基因组和其他特定变体类型(诸如插入、缺失、结构变异或CNV)的变体等位基因频率。
例如,如图6D中呈现的等位基因频率表642中所示,基因组分类系统106确定来自第二基因组样品639b的独特纯合等位基因和独特杂合等位基因在混合基因组中分别以0.4和0.2的变体等位基因频率出现。如进一步所示,基因组分类系统106确定来自第一基因组样品639a的独特纯合等位基因和独特杂合等位基因在混合基因组中分别以0.6和0.3的变体等位基因频率出现。相比之下,基因组分类系统106确定根据第二基因组样品639b和第一基因组样品639a中的相应等位基因接合性,作为纯合-纯合组合、杂合-纯合组合、纯合-杂合组合和杂合-杂合组合存在于60%和40%混合基因组中的常见等位基因分别以1.0、0.8、0.7和0.5的变体等位基因频率出现。
为了选择代表具有癌或镶嵌现象的基因组样品的合适混合物基因组,基因组分类系统106可以从给定混合物基因组中基因组样品的各种组合(和百分比)的真集碱基确定变体等位基因频率。除了图6D中描绘的60%和40%混合基因组中存在的变异等位基因频率之外,在一些实施方案中,基因组分类系统106确定其他可能的混合基因组的变体等位基因频率以模拟具有癌症或镶嵌现象的基因组样品。例如,基因组分类系统106确定来自第一基因组样品639a的30%样品核酸序列和来自第二基因组样品639b的70%样品核酸序列将分别以0.7和0.3的变体等位基因频率产生来自第一基因组样品639a和来自第二基因组样品639b的独特纯合等位基因,以及来自第一基因组样品639a和来自第二基因组样品639b的独特杂合等位基因,变体等位基因频率分别为0.35和0.15。相比之下,基因组分类系统106确定或预测在此类30%和70%混合基因组中作为纯合-纯合组合、杂合-纯合组合、纯合-杂合组合和杂合-杂合组合存在的常见等位基因—根据相同的30%和70%混合—将分别产生1.0、0.85、0.65和0.5的变体等位基因频率。
除了确定来自第一基因组样品639a和第二基因组样品639b的各种混合基因组之外,在某些实施方式中,基因组分类系统106从不同样品基因组的组合确定变体等位基因频率以鉴别模拟具有癌症或镶嵌现象的基因组样品的合适的混合基因组。通过确定多种混合基因组的变体等位基因频率,基因组分类系统106可以选择更接近地(或最接近地)模拟靶类型或癌症或镶嵌现象的变体等位基因频率的混合物基因组。
如以上所指示,基因组分类系统106可以部分地基于某些测序指标生成反映癌症或镶嵌现象的体细胞-核碱基变体特定的或种系核碱基变体特定的基准真值分类。如图6E所示,在一些实施方案中,基因组分类系统106通过以下方式以高置信度分类(或其他置信度分类)对基因组坐标进行分选或标记:(i)确定来自基因组样品(例如,如上文所解释的多样化基因组样品群组)的样品核酸序列跨基因组坐标的测序指标分布644,并且(ii)鉴别具有落入正态分布的目标部分内的某些测序指标的基因组坐标。在所描绘的示例中,当基因组坐标表现出深度指标、映射质量指标和核碱基检出质量指标在三个测序指标中的每一个的正态分布的标准偏差内时,基因组分类系统106鉴别在高置信度区域652内的基因组坐标。如下文所讨论的,表现出正常深度指标、映射质量指标和核碱基检出质量指标并且因此是高置信度区域652的一部分的基因组坐标也表现出用于基于基因组样品的混合物确定变体核碱基检出的更佳精确度。
如图6E所示,基因组分类系统106确定来自基因组样品(例如,多样化基因组样品群组)的样品核酸序列在参考基因组的基因组坐标处的测序指标分布644。为了确定此类分布,基因组分类系统106确定来自多样化群组的测序基因组样品的测序指标,并且根据不同的基因组坐标确定测序指标的分布。例如,在某些情况下,基因组分类系统106确定基因组样品的核碱基检出(例如,通过使用DRAGEN体细胞流水线中的仅肿瘤分析)并且确定基因组样品的确定的序列的测序指标。在一些实施方案中,基因组分类系统106确定样品核酸序列相对于每个基因组坐标的深度指标、映射质量指标和核碱基检出质量指标。相比之下,在某些实施方式中,基因组分类系统106确定上述任何测序指标中的一个或多个,包括但不限于上述比对指标、深度指标或检出数据质量指标中的一个或多个中的任一个。
如图6E进一步所示,基因组分类系统106基于一个或多个测序指标分布644来鉴别正常基因组坐标646和离群值基因组坐标648。例如,基因组分类系统106针对深度指标、映射质量指标、核碱基检出质量指标和/或上述跨基因组坐标的其他测序指标中的每一个,将贝叶斯-高斯混合模型与全基因组分布拟合。基因组分类系统106随后使用算法来修剪或移除对每个测序指标的全基因组分布与贝叶斯-高斯混合模型的适当拟合没有贡献或贡献很少的组成部分(例如,测序指标子集)。基于每个测序指标的拟合分布,基因组分类系统106设置p值阈值以根据每个特定测序指标定义或鉴别落入拟合分布内的正常基因组坐标646和落入拟合分布外的离群基因组坐标648。因此,基因组坐标对于一个测序指标可以是正常基因组坐标646中的一个,但是对于另一个测序指标可以是离群基因组坐标648中的一个。
在鉴别正常基因组坐标646和离群基因组坐标648之后,基因组分类系统106进一步鉴别表现出正常深度指标、映射质量指标和核碱基检出质量指标的基因组坐标作为高置信度区域652的一部分。如重叠可视化650所示,基因组分类系统106确定落入深度指标、映射质量指标和核碱基检出质量指标中的每一项的分布(例如,拟合分布)内的基因组坐标。所鉴别的基因组坐标形成高置信度区域652,并且包括参考基因组的89.9%-不包括其他区域的空位。落在深度指标、映射质量指标和核碱基检出质量指标中任一项的分布外的基因组坐标形成低置信度区域654。如图6E所示,在某些实施方案中,基因组分类系统106对于反映癌症的体细胞核碱基变体用高置信度的基准真值分类来标记在高置信度区域652内的基因组坐标。
如以上所表明的,表现出正常深度指标、映射质量指标和核碱基检出质量指标的基因组坐标也表现出确定变体核碱基检出的准确度或精确度。为了测试可靠性并进一步区分基准真值分类,在一些实施方案中,基因组分类系统106确定混合物基因组的核碱基检出,并将核碱基检出与形成来自Platinum Genomes的混合物基因组的基因组样品所独有的真集碱基进行比较。通过将混合物基因组的变体检出与相应真集碱基进行比较,基因组分类系统106可以在相应的基因组坐标处鉴别真阳性变体。
因为模拟癌症或镶嵌现象的混合基因组中的变体如此少,所以在一些实施方式中,基因组分类系统106使用正态-正态扣除方法来鉴别在基因组坐标处确定的假阳性变体。具体而言,基因组分类系统106通过在来自Illumina,Inc.的肿瘤/正常数据分析中将一个重复作为肿瘤样品并且将另一个重复作为正常样品处理来确定来自混合物的相同基因组样品(例如,NA12877)的两个重复的核碱基检出,并且比较来自两个重复的核碱基检出以鉴别假阳性变体。当执行此类分析时,例如,基因组分类系统106可以使用由Illumina,Inc.,“Evaluating Somatic Variant Calling in Tumor/Normal Studies”(2015),获自https://www.illumina.com/content/dam/illumina-marketing/documents/products/whitepapers/whitepaper_wgs_tn_somatic_variant_calling.pdf描述的肿瘤/正常数据分析,其内容特此通过引用并入。通过测量基因组坐标或基因组区域处的假阳性变体的密度,基因组分类系统106可以鉴别在确定具有癌症或镶嵌现象的给定基因组样品的核碱基变体检出中最不可能产生错误的基因组坐标或区域。根据一个或多个实施方案,图6F示出假阳性密度图656,描绘在不同读段深度处在来自图6E的高置信度区域652和低置信度区域654内确定的假阳性的密度。
除了确定假阳性变体的密度之外,在一些实施方案中,基因组分类系统106确定来自混合基因组的样品核酸序列的核碱基检出的体细胞质量指标,并且确定如按照体细胞质量指标阈值分隔的来自图6E的低置信度区域654的部分内的假阳性变体的密度。如下文进一步解释的,在一些情况下,基因组分类系统106使用体细胞质量指标阈值来区分低置信度区域654或高置信度区域652中的基因组坐标的基准真值分类的不同等级。根据一个或多个实施方案,图6F还示出了假阳性密度图656,描绘了在不同的体细胞质量指标阈值和不同的读段深度处在来自图6E的低置信度区域654的不同等级内确定的假阳性的密度。
如图6F的假阳性密度图656中所示,基因组分类系统106确定不同读段深度处的高置信度区域和低置信度区域的基因组坐标处每百万碱基假阳性变体的密度(Mb)。基因组分类系统106还根据不同的体细胞质量指标阈值(即,值为17.5、20和25的体细胞质量指标)来确定低置信度区域中假阳性变体的密度。对于基因组坐标处的读段深度100,基因组分类系统106确定高置信度区域中的基因组坐标刚好高于0.1/Mb的假阳性密度、体细胞质量指标介于17.5至20之间的低置信度区域中的基因组坐标高于1.6/Mb的假阳性密度、体细胞质量指标介于20至25之间的低置信度区域中的基因组坐标高于0.8/Mb的假阳性密度、以及体细胞质量指标高于25的低置信度区域中的基因组坐标高于0.2/Mb的假阳性密度。对于给定基因组坐标处的读段深度75,基因组分类系统106确定高置信度区域中的基因组坐标刚好低于0.1/Mb的假阳性密度、体细胞质量指标介于17.5至20之间的低置信度区域中的基因组坐标高于1.1/Mb的假阳性密度、体细胞质量指标介于20至25之间的低置信度区域中的基因组坐标高于0.7/Mb的假阳性密度、以及体细胞质量指标高于25的低置信度区域中的基因组坐标大约为0.3/Mb的假阳性密度。
如假阳性密度图656所指示,假阳性变体的密度随着低置信度区域中基因组坐标的体细胞质量指标的降低而增加。相反,当体细胞质量指标阈值增加时,假阳性变体的密度降低,而假阴性变体的密度增加。因为假阳性变体的密度是体细胞变体检出程序的准确度的反向指示符,所以假阳性密度图656显示基因组分类系统106根据假阳性变体确定体细胞变体检出的准确度随着低置信度区域中基因组坐标的体细胞质量指标的降低而增加。
通过使用体细胞质量指标阈值,在某些实现方式中,基因组分类系统106可以相应地区分低置信度区域内的基因组坐标的基准真值分类。例如,在一些情况下,基因组分类系统106可以在相应的体细胞质量指标低于25时用低置信度分类来标记来自低置信度区域的基因组坐标,并且在相应的体细胞质量指标超过25时用中等置信度分类来标记来自低置信度区域的基因组坐标。相比之下,基因组分类系统106可以在相应的体细胞质量指标低于25时用较低置信度评分对来自低置信度区域的基因组坐标进行评分,并且在相应的体细胞质量指标超过25时用较高置信度评分对来自低置信度区域的基因组坐标进行评分。如刚刚阐述的,用于区分基准真值分类的阈值25仅仅是示例。在另外的实施方案中,基因组分类系统106对于体细胞质量指标使用一个或多个不同的阈值(例如,15、20、30)。
如图6F的假阳性密度图656进一步所指示,在一些实施方案中,基因组分类系统106可以对低置信度区域使用不同且更严格的体细胞质量指标阈值,以在通常被常规系统认为是低质量的基因组区域中鉴别更可靠的基因组区域。常规变体检出程序通常使用体细胞变体检出质量的阈值。当候选核碱基检出具有低于阈值的质量时,常规变体检出程序过滤出相应的核碱基检出(例如,标记为不通过)。当阈值体细胞质量指标增加时,变体检出程序过滤出更多的核碱基检出,这导致假阳性变体减少但假阴性变体增加。通常,选择变体检出程序所使用的体细胞质量指标的阈值以实现假阳性变体和假阴性变体的最佳平衡。然而,通过使用上述体细胞质量指标阈值来过滤核碱基检出,基因组分类系统106可以显著减少假阳性变体而不会过度不利于再检出,如下文进一步所示。
如上所指示,在某些实施方式中,基因组分类系统106确定用于确定在特定基因组坐标处的变体核碱基检出的再检出率,并且部分地基于再检出率生成基准真值分类。例如,在某些情况下,基因组分类系统106确定对基因组样品的混合物的体细胞变体检出,并且将该体细胞变体检出与针对来自该混合物的相应基因组样品的真集(例如,来自PlatinumGenomes)进行比较以确定再检出率。在一些实施方案中,基因组分类系统106通过确定正确确定的真阳性核碱基检出变体的数目除以所有真阳性核碱基检出变体的数目来确定再检出率。基因组分类系统106可相应地确定并使用此类再检出率来鉴别(i)反映癌症或镶嵌现象的体细胞核碱基变体或(ii)反映镶嵌现象的种系核碱基变体特定的基准真值分类。
根据一个或多个实施方案,图6G示出了再检出图658a和658b,描绘了基因组分类系统106确定不同基因组区域内的基因组坐标处和不同变体等位基因频率下反映癌症的体细胞核碱基变体的再检出率。具体而言,再检出图658a和658b分别示出跨不同变体等位基因频率,针对根据17.5、20和25的体细胞质量指标阈值分隔的高置信度区域内和低置信度区域内的基因组坐标在100读段深度和75读段深度处的再检出率。
如分别针对在给定基因组坐标处读段深度100和75的再检出图658a和658b所指示的,基因组分类系统106确定用于确定在各个基因组坐标处和跨各种变体等位基因频率反映癌症的体细胞变体的再检出率。如再检出图658a和658b所示,高置信度区域内的基因组坐标表现出比任何分隔的低置信度区域更高的跨变体等位基因频率的再检出率。因为变体等位基因频率为0.05至0.2的核碱基变体在相对较少的读段中在给定基因组坐标处存在,所以测序系统缺乏足够的读段(甚至在基因组坐标的100和75的读段深度下)以在较高变体等位基因频率下表现出的接近1.0的再检出率确定高置信度区域中的相应核碱基变体检出。
如在再检出图658a和658b中进一步所示,在体细胞质量指标为25的低置信度区域、体细胞质量指标阈值为20的低置信度区域、以及体细胞质量指标阈值为17.5的低置信度区域中的每一个区域中的基因组坐标表现出跨变体等位基因频率越来越好的再检出率。换句话说,随着对于基因组坐标而言用于过滤的体细胞质量指标阈值增加,对于基因组坐标而言用于确定反映癌症的体细胞变体的再检出率降低。注意,体细胞质量指标阈值与再检出率之间的这种关系不代表体细胞质量指标增加。随着体细胞质量指标的增加,用于确定体细胞变体的再检出率应该同样增加,并且体细胞变体检出较不易于出现假阴性变体和假阳性变体。
通过使用体细胞质量指标阈值和再检出率两者,在某些实现方式中,基因组分类系统106可以相应地区分低置信度区域内的基因组坐标的基准真值分类。例如,在一些情况下,基因组分类系统106在相应的体细胞质量指标低于25(或一些其他体细胞质量指标阈值)时用低置信度分类来标记来自低置信度区域的基因组坐标。相反,基因组分类系统106在相应的体细胞质量指标超过25(或一些其他体细胞质量指标阈值)时用中等置信度分类来标记来自低置信度区域的基因组坐标。相比之下,基因组分类系统106可以在相应的体细胞质量指标高于或低于25时用较低(或较高)置信度评分来对来自低置信度区域的基因组坐标进行评分。
相比之下,在一些实施方案中,基因组分类系统106可以基于具有不同体细胞质量指标阈值的基因组坐标的F评分来区分低置信度区域中的基因组坐标的基准真值分类。例如,基因组分类系统106可以基于再检出率和精确率两者来确定用于确定低置信度区域中的基因组坐标处的变体核碱基检出的F评分。在一些实施方案中,基因组分类系统106通过确定正确确定的真阳性核碱基检出变体的数目除以所有确定的核碱基检出变体的数目来确定精确率。在一些情况下,基因组分类系统106通过确定精确率和再检出率的调和平均值来确定F
如以上进一步所指示,在某些实施方式中,基因组分类系统106确定用于确定在特定基因组坐标处的变体核碱基检出的精确率和再检出率中的一者或两者,并且部分地基于精确率和再检出率中的一者或两者生成基准真值分类。例如,在某些情况下,基因组分类系统106确定基因组样品的混合物的体细胞变体检出(例如,当确定模拟癌症的体细胞变体检出时通过使用肿瘤/正常DRAGEN体细胞流水线,或者当确定模拟镶嵌现象的体细胞变体检出时在DRAGEN体细胞流水线中使用仅瘤分析)。基因组分类系统106随后针对来自混合物的相应基因组样品将体细胞变体检出与真集(例如,Platinum Genomes)进行比较以确定精确率和再检出率。基因组分类系统106可相应地确定并使用此类精确率或再检出率来鉴别(i)反映癌症或镶嵌现象的体细胞核碱基变体或(ii)反映镶嵌现象的种系核碱基变体特定的基准真值分类。
根据一个或多个实施方案,图6H示出了再检出图660a和660b,描绘了基因组分类系统106确定反映不同基因组区域内的基因组坐标处和不同变体等位基因频率下反映镶嵌现象的变体核碱基检出的精确度。图6H还示出了再检出图662a和662b,描绘了基因组分类系统106确定不同基因组区域内的基因组坐标处和不同变体等位基因频率下反映镶嵌现象的核碱基变体的再检出率。
如分别针对在给定基因组坐标处读段深度100和75的精确度图660a和660b所指示的,基因组分类系统106确定用于确定在各个基因组坐标处和跨各种变体等位基因频率反映镶嵌现象的核碱基变体的精确率。如精确度图660a和660b所示,高置信度区域内的基因组坐标通常表现出比低置信度区域的基因组坐标更高的跨变体等位基因频率的精确率。在精确度图660a和660b中以变体等位基因频率0.15开始,低置信度区域内的基因组坐标表现出与高置信度区域内的基因组坐标几乎相同的接近1.000的精确率。
如分别针对在给定基因组坐标处读段深度100和75的再检出图662a和662b所指示的,基因组分类系统106确定用于确定在各个基因组坐标处和跨各种变体等位基因频率反映镶嵌现象的核碱基变体的再检出率。如再检出图662a和662b所示,高置信度区域内的基因组坐标一致地表现出比低置信度区域的基因组坐标更高的跨变体等位基因频率的再检出率。
如以上所指示的,在给定的基因组坐标处,变体等位基因频率为0.05至0.15的核碱基变体在相对较少的核苷酸读段中存在。因此,测序系统缺乏足够的读段(甚至在基因组坐标的读段深度为100和75时)来确定相应的核碱基变体检出,在更高的变体等位基因频率下表现出接近1.0的精确率或接近1.0的再检出率。
除了确定精确率和再检出率之外,在某些实施方式中,基因组分类系统106还基于精确率和再检出率确定用于确定基因组坐标处的变体核碱基检出的F评分。如以上所指示的,在一些情况下,基因组分类系统106通过确定精确率和再检出率的调和平均值来确定F
基于再检出率和精确率中的一项或两项,在某些实施方式中,基因组分类系统106区分高置信度区域和低置信度区域内的基因组坐标的基准真值分类。例如,在一些情况下,基因组分类系统106用高置信度分类来标记高置信度区域中的基因组坐标,这部分是因为高置信度区域中的基因组坐标表现出更好的再检出率和精确率。相比之下,在一些情况下,基因组分类系统106用低置信度分类(或中等置信度分类)来标记低置信度区域中的基因组坐标,这是因为低置信度区域表现出较低的再检出率和精确率。
不管基因组分类系统106如何确定或标记此类基准真值分类,在某些情况下,基因组分类系统106训练基因组位置分类模型608以针对反映癌症或体细胞镶嵌现象的体细胞核碱基变体或针对反映种系镶嵌现象的种系核碱基变体,基于此类确定的基准真值分类确定基因组坐标的变体置信度分类,如图6A中所描绘。因此,基因组分类系统106同样可以利用基因组位置分类模型608的训练版本来确定既针对基因组坐标集合并且是反映癌症或体细胞镶嵌现象的体细胞核碱基变体或是反映种系镶嵌现象的种系核碱基变体特定的变体置信度分类,如图6B中所描绘。因此,基因组分类系统106还可从基因组位置分类模型608的训练版本鉴别并显示变体置信度分类,这些变体置信度分类对应于针对反映癌症或体细胞镶嵌的核碱基变体或针对反映种系镶嵌现象的种系核碱基变体的变体检出的基因组坐标,如图6C中所描绘。
如以上所指示,为了评估基因组位置分类模型的不同实施方案的性能,研究人员测量由基因组分类系统106的置信度分类所证明的变量和各种准确度指标。以下段落描述如图7-10B中所描绘的那些测量中的一些测量。根据一个或多个实施方案,例如,图7A-图7G描绘了图700a-图700g,指示当从逻辑回归模型训练时通知用于特定变体类型的基因组位置分类模型的测序指标和测序指标推导的输入数据。具体而言,图700a-图700g示出了由基因组位置分类模型用于前二十三个测序指标和测序指标推导的输入数据的逻辑回归系数,以基于不同的核碱基检出变体类型确定基因组坐标的高置信度分类或低置信度分类。
如图7A和图7B中所示,例如,图700a和图700b显示分别使用对应于1-5个核碱基长度的短缺失(对于图700a)或1-5个核碱基长度的短插入(对于图700b)的基准真值分类训练的基因组位置分类模型的逻辑回归系数。图7A和图7B显示使用短缺失或短插入训练的逻辑回归模型用与其他数据输入相比具有最大量值的系数对映射质量指标(MAPQ)或标准化深度进行加权,以确定基因组坐标或基因组区域的高置信度分类或低置信度分类。
具体而言,图7A中的图700a显示针对短缺失训练的逻辑回归模型使用超过-1.5的系数和超过1.5的系数用于映射质量指标,以分别确定基因组坐标或基因组区域的高置信度分类和低置信度分类。图7B中的图700b显示针对短插入训练的逻辑回归模型使用超过-1.5的系数和超过1.5的系数用于标准化深度指标,以分别确定基因组坐标或基因组区域的高置信度分类和低置信度分类。此类标准化深度指标经受标准偏差并且可以包括前向-反向深度指标或归一化深度指标。
相比之下,图7A中的图700a显示针对短缺失训练的逻辑回归模型使用0.0的系数和接近0.0的系数—在量值上低于针对短缺失的其他数据输入—用于前向分数指标和读段参考错配指标的局部均值(局部_均值_错配),以确定基因组坐标的高置信度分类和低置信度分类。图7B中的图700b显示针对短插入训练的逻辑回归模型使用接近0.0的系数—在量值上低于针对短插入的其他数据输入—用于较高的负插入片段大小指标,以确定基因组坐标的高置信度分类和低置信度分类。
如图7C和图7D所示,图700c和图700d显示分别使用对应于5-15个核碱基长度的中等缺失(对于图700c)或5-15个核碱基长度的中等插入(对于图700d)的基准真值分类训练的基因组位置分类模型的逻辑回归系数。图700c和图700d都显示逻辑回归模型用与其他数据输入相比具有最大量值的系数对映射质量指标(MAPQ)进行加权,以确定基因组坐标或基因组区域的高置信度分类或低置信度分类。
具体而言,图7C中的图700c显示针对中等缺失训练的逻辑回归模型使用量值接近-0.8的系数和量值接近0.8的系数用于映射质量指标,以分别确定基因组坐标的高置信度分类和低置信度分类。类似地,图7D中的图700d显示针对中等插入训练的逻辑回归模型使用量值超过-0.75的系数和量值超过0.75的系数用于映射质量指标,以分别确定基因组坐标的高置信度分类和低置信度分类。
相比之下,图7C中的图700c显示针对中等缺失训练的逻辑回归模型使用0.0的系数—在量值上低于针对中等缺失的其他数据输入—用于二项比例检验和贝茨分布检验两者,以分别确定基因组坐标的高置信度分类和低置信度分类。图7D中的图700d显示针对中等插入训练的逻辑回归模型使用0.0和接近0.0的系数—在量值上低于针对中等插入的其他数据输入—用于前向分数指标和较高的负插入片段大小指标,以分别确定基因组坐标的高置信度分类和低置信度分类。
如图7E和图7F所示,图700e和图700f显示分别使用对应于多于15个核碱基长度的长缺失(对于图700e)或多于15个核碱基长度的长插入(对于图700f)的基准真值分类训练的基因组位置分类模型的逻辑回归系数。图7E和图7F显示使用长缺失或长插入训练的逻辑回归模型用与其他数据输入相比具有最大量值的系数对映射质量指标(MAPQ)或深度剪切指标进行加权,以确定基因组坐标或基因组区域的高置信度分类或低置信度分类。
具体而言,图7E中的图700e显示针对长缺失训练的逻辑回归模型使用超过-0.4和超过0.4的系数用于映射质量指标(MAPQ),以分别确定基因组坐标或基因组区域的高置信度分类和低置信度分类。图7F中的图700f显示针对长插入训练的逻辑回归模型使用量值超过-0.4和量值超过0.4的系数用于深度剪切指标,以分别确定基因组坐标或基因组区域的高置信度分类和低置信度基因组学分类。
相比之下,图7E中的图700e显示针对长缺失训练的逻辑回归模型使用0.0的系数—低于针对长缺失的其他数据输入—用于峰值计数指标和读段位置指标两者,以确定基因组坐标的高置信度分类和低置信度分类。图7F中的图700f显示针对长插入训练的逻辑回归模型使用接近0.0的系数和0.0的系数—在量值上低于针对长插入的其他数据输入—用于读段参考错配指标的局部均值(局部_均值_错配)和二项式比例检验,以确定基因组坐标的高置信度分类和低置信度分类。
如图7G所示,图700g示出了使用对应于SNP的基准真值分类训练的基因组位置分类模型的逻辑回归系数。如图7G所示,图700g显示针对SNP训练的逻辑回归模型使用超过-2.0的系数和超过2.0的系数—高于针对SNP的其他数据输入—用于映射质量指标(MAPQ),以分别确定基因组坐标或基因组区域的高置信度分类和低置信度分类。相比之下,图700g显示针对SNP训练的逻辑回归模型使用低于针对SNP的其他数据输入的系数用于缺失熵指标,以确定基因组坐标或基因组区域的高置信度分类和低置信度分类。
为了进一步评估作为基因组位置分类模型基于测序指标训练的逻辑回归模型的性能,研究人员确定此类基因组位置分类模型正确确定置信度分类的比率。根据一个或多个实施方案,图8示出了具有接收者操作特征(ROC)曲线的图800,这些曲线定义了作为基因组位置分类模型训练的对数回归模型正确地(i)将在基因组坐标处的高置信度分类或低置信度分类确定为真阳性或假阳性并且(ii)对于具有常见缺失的基因组坐标将置信度分类确定为真阳性和假阳性的比率的曲线下面积(AUC)。如图8中所示,基因组分类系统106将从测序指标推导出或准备的数据输入到基因组位置分类模型中以确定基因组坐标的置信度分类。
如图800所示,作为基因组位置分类模型训练的逻辑回归模型基于与基准真值分类的比较以99.34%的AUC正确地将高置信度分类确定为基因组坐标的真阳性或假阳性。如图800进一步指示,此类基因组位置分类模型基于与基准真值分类的比较以97.39%的AUC正确地将低置信度分类确定为基因组坐标的真阳性或假阳性。最后,此类基因组位置分类模型基于与参考基因组的比较,正确地将置信度分类确定为发生常见缺失的基因组坐标的真阳性或假阳性,AUC为97.32%。
除了确定图8中描绘的图800的ROC曲线之外,研究人员还评估了变体检出程序可以在通过作为基因组位置分类模型训练的逻辑回归模型分类的基因组坐标处鉴别SNV和插入缺失的精确度、再检出性和一致性(或再现性)。各种检验证明,作为基因组位置分类模型训练的逻辑回归模型正确地对人类基因组中具有高置信度坐标(或区域)的较大一部分进行分类,在所述高置信度坐标(或区域)处,相比于通过GIAB鉴别的那些,可以正确地鉴别SNV和插入缺失。实际上,此类基因组位置分类模型可以鉴别GIAB鉴别为在困难区域内的具有高置信度分类的某些基因组坐标(或区域)。例如,下面的表2证明基因组分类系统106提高了现有测序系统鉴别可以在特定基因组坐标处确定核碱基的置信度的准确度。
表2
/>
如表2所示,作为基因组位置分类模型训练的逻辑回归模型正确地分类90.3%非N常染色体人类基因组的基因组坐标。相比之下,GIAB已经鉴别了在仅79-84%的非N常染色体人类基因组中没有困难地准确确定变体的基因组区域。如表2进一步所指示,基于使用SNV数据确定的基准真值分类,此类逻辑回归模型以大约99.9%的精确度、99.9%的再检出率和99.9%的一致性将基因组坐标准确分类。类似地,基于使用插入缺失数据确定的基准真值分类,此类逻辑回归模型以大约99.0%的精确度、99.5%的再检出率和98.5%的一致性将基因组坐标准确分类。在通过此类逻辑回归模型用中等置信度分类或低置信度分类标记的基因组坐标处—或包含常见缺失的基因组区域处—此类逻辑回归模型基于从SNV或插入缺失推导出的基准真值数据以表2中进一步报告的较低精确度、再检出率和一致性率对基因组坐标进行分类。
为了评估作为基因组位置分类模型基于上下文核酸子序列训练的CNN的性能,研究人员确定此类基因组位置分类模型正确确定置信度分类的比率。根据一个或多个实施方案,图9示出了图900a,其中ROC曲线定义了作为基因组位置分类模型训练的CNN的AUC,该基因组位置分类模型基于从插入缺失数据推导出的基准真值分类来确定基因组坐标的置信度分类。图9还示出了图900b,其中ROC曲线定义了作为基因组位置分类模型训练的CNN的AUC,该基因组位置分类模型基于从单核苷酸多态性(SNP)的数据推导出的基准真值分类确定基因组坐标的置信度分类。如图9所示,为了确定基因组坐标的置信度分类,基因组分类系统106将从上下文核酸子序列推导出或准备的数据输入到作为基因组位置分类模型训练的CNN中。
作为概述,图900a和图900b证明作为基因组位置分类模型训练的CNN基于从插入缺失或SNP推导出的基准真值数据以介于77.9%至91.7%之间的AUC将基因组坐标的置信度分类正确地确定为真阳性或假阳性—这取决于输入到基因组位置分类模型中的上下文核酸子序列的长度。具体而言,如图900a所指示,针对插入缺失训练的基因组位置分类模型基于21个碱基对、101个碱基对、151个碱基对、301个碱基对和801个碱基对的上下文核酸子序列分别以AUC 81.4%、87.4%、87.6%、88.2%和87.9%将基因组坐标的置信度分类正确地确定为真阳性或假阳性。如图900b所指示,针对SNP训练的基因组位置分类模型基于21个碱基对、101个碱基对、151个碱基对、301个碱基对和801个碱基对的上下文核酸子序列分别以AUC 77.9%、88.8%、90.0%、91.2%和91.7%将基因组坐标的置信度分类正确地确定为真阳性或假阳性。因此,对于插入缺失和SNP两者,对于对于置信度分类而言当上下文核酸子序列的长度增加时,作为基因组位置分类模型训练的CNN更准确地确定基因组坐标的置信度分类。
为了测试作为基因组位置分类模型基于测序指标和上下文核酸子序列两者训练的CNN的性能,研究人员还确定此类基因组位置分类模型使用测试或保留数据集正确确定置信度分类的比率。根据一个或多个实施方案,图10A和图10B示出了图1002a-1002b、直方图1004a-1004b和混淆矩阵1006a-1006b,描绘了此类基因组位置分类模型基于从插入缺失和SNP数据推导出的基准真值分类来正确地确定特定基因组坐标的置信度分类的比率和置信度。如图10A和图10B所示,为了确定基因组坐标的置信度分类,基因组分类系统106将从测序指标和上下文核酸子序列两者推导出(或准备)的数据输入到作为基因组位置分类模型训练的CNN中。
如图10A中的图1002a所指示,作为基因组位置分类模型针对插入缺失训练的CNN基于101个碱基对的上下文核酸子序列以97.8%的AUC正确地将基因组坐标的高置信度分类确定为真阳性或假阳性。如图10B中的图1002b所示,作为基因组位置分类模型针对SNP训练的CNN基于101个碱基对的上下文核酸子序列以99.7%的AUC正确地将基因组坐标的置信度分类确定为真阳性或假阳性。因此,图1002a和图1002b证明,当使用测序指标和上下文核酸子序列两者作为输入时,如图10A和图10B所示的作为基因组位置分类模型训练的CNN可以以异常高的比率正确地确定特定基因组坐标的置信度分类。
现在转回到图10A中针对插入缺失的直方图1004a。如直方图1004a所指示,针对插入缺失作为基因组位置分类模型训练的CNN在超过80,000次预测中在基因组坐标处以大约1.0的置信度正确地将置信度分类确定为真阳性。换句话说,基于101个碱基对的上下文核酸子序列,此类基因组位置分类模型在检测到真阳性插入缺失的基因组坐标处以高置信度确定分类。如直方图1004a进一步所指示,针对插入缺失作为基因组位置分类模型训练的CNN在超过80,000次预测中在基因组坐标处以大约0.0的置信度正确地将置信度分类确定为假阳性。换句话说,基于101个碱基对的上下文核酸子序列,此类基因组位置分类模型在检测到假阳性插入缺失的基因组坐标处以低置信度确定分类。
现在转回到图10B中针对SNP的直方图1004b。如直方图1004b所指示,针对SNP作为基因组位置分类模型训练的CNN在近800,000次预测中在基因组坐标处以大约1.0的置信度正确地将置信度分类确定为真阳性。换句话说,基于101个碱基对的上下文核酸子序列,基因组位置分类模型在检测到真阳性SNP的基因组坐标处以高置信度确定分类。如直方图1004b所进一步指示,针对SNP作为基因组位置分类模型训练的CNN在超过700,000次预测中在基因组坐标处以大约0.0的置信度正确地将置信度分类确定为假阳性。换句话说,基于101个碱基对的上下文核酸子序列,基因组位置分类模型在检测到假阳性SNP的基因组坐标处以低置信度确定分类。
现在转回到图10A和图10B中的混淆矩阵1006a和1006b。如图10A中的混淆矩阵1006a所描绘的,针对插入缺失作为基因组位置分类模型训练的CNN从基因组坐标处的总预测以92.322%的比率正确地将置信度分类确定为真阳性(例如,高置信度分类)或真阴性(例如,低置信度分类)。相比之下,此类CNN测序系统从基因组坐标处的总预测仅以7.678%的比率错误地将置信度分类确定为真阳性或真阴性。如图10B中的混淆矩阵1006b所描绘的,针对SNP作为基因组位置分类模型训练的CNN从基因组坐标处的总预测以97.409%的比率正确地将置信度分类确定为真阳性或真阴性。相比之下,此类CNN从基因组坐标处的总预测仅以2.591%的比率错误地将置信度分类确定为真阳性或真阴性。
现在转向图11A,该图示出了根据一个或多个实施方案,训练机器学习模型以确定基因组坐标的置信度分类的一系列动作1100a的流程图。虽然图11A示出根据一个实施方案的动作,替代实施方案可以省略、添加、重新排序和/或修改图11A中所示的任何动作。图11A的动作可作为方法的一部分来执行。另选地,非暂态计算机可读存储介质可包括当由一个或多个处理器执行时导致计算设备执行图11A描绘的动作的指令。在又一些实施方案中,一种系统包括至少一个处理器和包括指令的非暂态计算机可读介质,这些指令在由一个或多个处理器执行时,使得该系统执行图11A的动作。
如图11A所示,动作1100a包括确定测序指标或上下文核酸子序列中的一种或多种的动作1102。具体而言,在一些实施方案中,动作1102包括确定用于将样品核酸序列与示例核酸序列的基因组坐标进行比较的测序指标。在一些情况下,动作1102包括从示例核酸序列确定样品核酸序列中变体核碱基检出周围在来自参考基因组的基因组坐标的基因组坐标处的上下文核酸子序列。在一个或多个实施方案中,使用包括核酸序列提取方法、测序设备和序列分析软件的单个测序流水线确定样品核酸序列。相关地,在某些实施方案中,示例核酸序列包含参考基因组或祖先单倍型的核酸序列。
如以上所指示,在一些情况下,确定测序指标包括确定以下的一项或多项:用于定量样品核酸序列与示例核酸序列的基因组坐标的比对的比对指标;用于定量所述样品核酸序列在所述示例核酸序列的所述基因组坐标处的核碱基检出深度的深度指标;或用于定量所述样品核酸序列在所述示例核酸序列的所述基因组坐标处的所述核碱基检出的质量的检出数据质量指标。
相关地,在某些实施方式中,确定比对指标包括确定样品核酸序列的缺失大小指标、映射质量指标、正插入大小指标、负插入大小指标、软剪切指标、读段位置指标或读段参考错配指标中的一项或多项;确定深度指标包括确定前向-反向深度指标或归一化深度指标中的一项或多项;或确定检出数据质量指标包括确定样品核酸序列的核碱基检出质量指标或可检出性指标中的一项或多项。
如图11A中进一步所示,动作1100a包括训练基因组位置分类模型以基于测序指标或上下文核酸子序列中的一项或多项来确定基因组坐标的置信度分类的动作1104。具体而言,在一些实施方案中,动作1104包括训练基因组位置分类模型以基于所述测序指标和特定基因组坐标的基准真值分类来确定所述基因组坐标的置信度分类。此外,在一些情况下,动作1104包括训练基因组位置分类模型以基于所述基因组坐标的所述上下文核酸子序列和基准真值分类来确定所述基因组坐标的置信度分类。
如以上所表明的,在某些实施方案中,训练基因组位置分类模型以确定置信度分类包括训练统计机器学习模型或神经网络以确定置信度分类。相关地,在一个或多个实施方案中,训练基因组位置分类模型以确定置信度分类包括训练逻辑回归模型、随机森林分类器或卷积神经网络以确定所述置信度分类。
此外,在以下情形下,置信度分类指示在所述特定基因组坐标处可以准确确定核碱基的程度。相关地,在一些情况下,确定置信度分类包括确定基因组坐标处单核苷酸变体、核碱基插入、核碱基缺失、结构变异的一部分或拷贝数变异的一部分的置信度分类。
如以上进一步表明的,在一个或多个实施方案中,训练基因组位置分类模型以确定置信度分类包括:对于基因组坐标,将预计置信度分类与反映基因组坐标处的孟德尔遗传模式或核碱基检出的重复一致性的基准真值分类进行比较;根据所述预计置信度分类与所述基准真值分类的比较来确定损失;以及基于所确定的损失调整所述基因组位置分类模型的参数。
如图11A中进一步所示,动作1100a包括确定基因组坐标集合的置信度分类集合的动作1106。具体而言,在某些实施方式中,动作1106包括利用基因组位置分类模型,基于一个或多个样品核酸序列的测序指标集合确定基因组坐标集合的置信度分类集合。在一些情况下,动作1106包括利用基因组位置分类模型,基于上下文核酸子序列确定基因组坐标的置信度分类。
例如,在一个或多个实施方式中,从置信度分类集合确定置信度分类包括确定针对包含遗传修饰或表观遗传修饰的基因组坐标的置信度分类。相关地,在一些实施方案中,从置信度分类集合确定置信度分类包括确定基因组坐标处单核苷酸变体、核碱基插入、核碱基缺失或结构变异的一部分的置信度分类。
此外,在一些情况下,从置信度分类集合确定置信度分类包括确定基因组坐标的高置信度分类、中等置信度分类或低置信度分类中的至少一种。另外或可选地,从该置信度分类集合确定置信度分类包括确定在指示可以在基因组坐标处准确地确定核碱基的程度的置信度评分范围内的置信度评分。
如图11A中进一步所示,动作1100a包括生成包含该置信度分类集合的至少一个数字文件的动作1108。具体而言,在某些实施方式中,动作1108包括生成包含该基因组坐标集合的置信度分类集合的至少一个数字文件。类似地,在一些实施方案中,动作1108包括生成包含变体核碱基检出的基因组坐标的置信度分类的数字文件。
除了动作1102-1108之外,在某些实现方式中,动作1100a包括从示例核酸序列确定变体核碱基检出周围的上下文核酸子序列;以及训练基因组位置分类模型以基于以下项确定变体核碱基检出的基因组坐标的置信度分类:上下文核酸子序列;对应于所述上下文核酸子序列的基因组坐标子集的测序指标子集;以及对应于所述上下文核酸子序列的基因组坐标子集的基准真值分类子集。
现在转向图11B,该图示出了根据一个或多个实施方案,训练机器学习模型以确定基因组坐标的变体置信度分类的一系列动作1100b的流程图。虽然图11B示出根据一个实施方案的动作,替代实施方案可以省略、添加、重新排序和/或修改图11B中所示的任何动作。图11B的动作可作为方法的一部分来执行。另选地,非暂态计算机可读存储介质可包括当由一个或多个处理器执行时导致计算设备执行图11B描绘的动作的指令。在又一些实施方案中,一种系统包括至少一个处理器和包括指令的非暂态计算机可读介质,这些指令在由一个或多个处理器执行时,使得该系统执行图11B的动作。
如图11B所示,动作1100b包括确定来自基因组样品混合物的样品核酸序列的测序指标的动作1110。具体而言,在一些实施方案中,动作1110包括确定用于将来自基因组样品的样品核酸序列与示例核酸序列的基因组坐标进行比较的测序指标。例如,在一些情况下,确定测序指标包括确定样品核酸序列的映射质量指标、前向-反向深度指标和核碱基检出质量指标。在一个或多个实施方案中,使用包括核酸序列提取方法、测序设备和序列分析软件的单个测序流水线确定样品核酸序列。
如图11B中进一步所示,动作1100b包括针对变体核碱基检出基于一个或多个测序指标生成基因组坐标的基准真值分类的动作1112。例如,动物1112可以包括对于特定的变体核碱基检出,基于基因组样品混合物的测序指标或变体检出数据中的一项或多项生成特定基因组坐标的基准真值分类。作为进一步的示例,动作1112可以包括基于包括样品核酸序列的映射质量指标、前向-反向深度指标和核碱基检出质量指标的测序指标中的一项或多项生成基准真值分类。
如以上所表明的,在某些实施方案中,对于特定的变体核碱基检出,基于基因组样品混合物的变体检出数据生成特定基因组坐标的基准真值分类包括确定用于确定来自基因组样品混合物的一个或多个样品核酸序列在特定基因组坐标处的变体核碱基检出集合的精确率或再检出率中的一项或多项;以及基于用于确定变体核碱基检出集合的精确率或再检出率中的一项或多项生成基准真值分类。此外,在一些实施方式中,对于特定的变体核碱基检出,基于基因组样品混合物的变体检出数据生成特定基因组坐标的基准真值分类包括确定来自基因组样品混合物的一个或多个样品核酸序列的变体核碱基检出集合的变体等位基因频率;确定用于确定来自所述基因组样品混合物的一个或多个样品核酸序列在所述特定基因组坐标处并且在来自所述变体等位基因频率的不同变体等位基因频率下不同变体核碱基检出的精确率或再检出率中的一项或多项;以及基于用于确定在所述不同变体等位基因频率下的不同变体核碱基检出的所述精确率或所述再检出率中的一项或多项生成所述基准真值分类。
相关地,在一些情况下,对于特定的变体核碱基检出,基于基因组样品混合物的变体检出数据生成特定基因组坐标的基准真值分类包括确定来自基因组样品混合物的一个或多个样品核酸序列的核碱基检出的体细胞质量指标;生成用于区分所述特定基因组坐标的不同基准真值分类的体细胞质量指标阈值;以及根据所述体细胞质量指标阈值生成所述特定基因组坐标的分层基准真值分类。在一些此类情况下,生成分级基准真值分类包括根据体细胞质量指标阈值仅生成分级基准真值分类的子集。
此外,在一些实施方案中,对于特定的变体核碱基检出,基于基因组样品混合物的变体检出数据生成特定基因组坐标的基准真值分类包括确定来自基因组样品混合物的一个或多个样品核酸序列的变体核碱基检出集合的变体等位基因频率;确定用于确定来自基因组样品混合物的该一个或多个样品核酸序列在特定基因组坐标处并且在来自这些变体等位基因频率的不同变体等位基因频率下变体核碱基检出的子集的精确率和再检出率;基于所述精确率和所述再检出率确定用于确定在所述特定基因组坐标处的不同变体核碱基检出的F评分;以及进一步基于用于确定所述不同变体核碱基检出的所述F评分生成所述基准真值分类。
除了动作1110和1112之外,在一些实施方案中,动作1100b还包括从一个或多个示例核酸序列确定一个或多个样品核酸序列中在一个或多个基因组坐标处的变体核碱基检出周围的上下文核酸子序列。在某些实施方式中,该一个或多个示例核酸序列包含参考基因组或祖先单倍型的核酸序列。
如图11B中进一步所示,动作1100b包括训练基因组位置分类模型以基于基准真值分类确定基因组坐标的变体置信度分类的动作1114。具体而言,在一些实施方案中,动作1114包括训练基因组位置分类模型以基于测序指标和基准真值分类来针对变体核碱基检出确定基因组坐标的变体置信度分类。此外,在一些情况下,动作1114包括训练基因组位置分类模型以基于上下文核酸子序列和基准真值分类来针对变体核碱基检出确定基因组坐标的变体置信度分类。
如以上所表明的,在某些实施方案中,变体置信度分类指示可以在基因组坐标处准确地确定反映癌症或体细胞镶嵌现象的体细胞核碱基变体的程度。相比之下,在一些情况下,变体置信度分类指示在基因组坐标处可以准确地确定反映种系镶嵌现象的种系核碱基变体的程度。
如图11B中进一步所示,动作1100b包括确定基因组坐标集合的变体置信度分类集合的动作1116。具体而言,在某些实施方式中,动作1116包括利用基因组位置分类模型,基于一个或多个样品核酸序列的测序指标集合确定基因组坐标集合的变体置信度分类集合。在一些情况下,动作1116包括利用基因组位置分类模型,基于相应的变体核碱基检出集合周围的上下文核酸子序列集合来确定基因组坐标集合的变体置信度分类集合。例如,确定该测序指标集合可以包括确定来自一个或多个基因组样品的该一个或多个样品核酸序列的测序指标集合。
作为进一步的示例,在一些情况下,动作1116包括通过基于体细胞核碱基变体周围反映癌症或体细胞镶嵌现象的上下文核酸子序列确定基因组坐标的变体置信度分类,从变体置信度分类集合中确定变体置信度分类。相比之下,在某些情况下,动作1116包括通过基于种系核碱基变体周围反映种系镶嵌现象的上下文核酸子序列确定基因组坐标的变体置信度分类,从变体置信度分类集合中确定变体置信度分类。此外,在一个或多个实施方案中,动作1116包括通过确定在指示可以在基因组坐标处准确地确定核碱基变体的程度的变体置信度评分范围内的变体置信度评分来从变体置信度分类集合中确定变体置信度分类。
除了动作1110-1116之外,在某些实施方式中,动作1100b包括通过确定来自第一基因组样品的第一核酸序列子集和来自第二基因组样品的第二核酸序列子集的组合来确定基因组样品混合物,第一核酸序列子集和第二核酸序列子集一起模拟具有癌症或镶嵌现象的基因组样品的变体等位基因频率。类似地,在一些情况下,动作1100b包括通过确定来自第一天然存在的基因组样品的第一核酸序列百分比和来自第二天然存在的基因组样品的第二核酸序列百分比的组合来确定基因组样品混合物,第一核酸序列百分比和第二核酸序列百分比一起模拟具有癌症或镶嵌现象的基因组样品的变体等位基因频率。
现在转到图12,该图示出了根据一个或多个实施方案,用于从数字文件生成变体核碱基检出的基因组坐标的置信度分类的指示符的一系列动作1200的流程图。虽然图12示出根据一个实施方案的动作,替代实施方案可以省略、添加、重新排序和/或修改图12中所示的任何动作。图12的动作可作为方法的一部分来执行。另选地,非暂态计算机可读存储介质可包括当由一个或多个处理器执行时导致计算设备执行图12描绘的动作的指令。在又一些实施方案中,一种系统包括至少一个处理器和包括指令的非暂态计算机可读介质,这些指令在由一个或多个处理器执行时可以使得该系统执行图12的动作。
如图12所示,动作1200包括检测基因组坐标处变体核碱基检出的动作1202。具体而言,在一些实施方案中,动作1202包括检测样品核酸序列内的基因组坐标处的变体核碱基检出。如上所述,在一些情况下,检测基因组坐标处的变体核碱基检出包括检测单核苷酸变体、核碱基插入、核碱基缺失或结构变异的一部分。
如图12中进一步所示,动作1200包括根据基因组位置分类模型鉴别基因组坐标的置信度分类的动作1204。具体而言,在一些实施方案中,动作1204包括根据基因组位置分类模型从数字文件鉴别基因组坐标的置信度分类。
如以上所表明的,在某些实施方案中,鉴别基因组坐标的置信度分类包括从数字文件中鉴别指示可以在基因组坐标处准确地确定核碱基的程度的置信度分类。此外,在一些实施方式中,从数字文件中鉴别置信度分类包括从数字文件内针对基因组坐标的注释或评分鉴别置信度分类。相关地,在一个或多个实施方案中,从数字文件中鉴别置信度分类包括鉴别基因组坐标的高置信度分类、中间置信度分类或低置信度分类中的至少一种。
如图12中进一步所示,动作1200包括生成置信度分类的指示符的动作1206。具体而言,在某些实现方式中,动作1206包括生成变体核碱基检出的基因组坐标的置信度分类的指示符以用于在图形用户界面内显示。
本文所述的方法可与多种核酸测序技术结合使用。特别适用的技术是其中核酸附接到阵列中的固定位置处使得其相对位置不改变并且其中该阵列被重复成像的那些技术。在不同颜色通道(例如,与用于将一种核苷酸碱基类型与另一种核苷酸碱基类型区分开的不同标记吻合)中获得图像的实施方案特别适用。在一些实施方案中,确定靶核酸(即,核酸聚合物)的核苷酸序列的过程可以是自动化过程。优选的实施方案包括边合成边测序(SBS)技术。
SBS技术通常包括通过针对模板链反复加入核苷酸进行的新生核酸链的酶促延伸。在传统的SBS方法中,可在每次递送中在存在聚合酶的情况下将单个核苷酸单体提供给靶核苷酸。然而,在本文所述的方法中,可在递送中存在聚合酶的情况下向靶核酸提供多于一种类型的核苷酸单体。
SBS可利用具有终止子部分的核苷酸单体或缺少任何终止子部分的核苷酸单体。使用缺少终止子的核苷酸单体的方法包括例如焦磷酸测序和使用γ-磷酸标记的核苷酸的测序,如下文进一步详细描述的。在使用缺少终止子的核苷酸单体的方法中,在每个循环中加入的核苷酸的数目通常是可变的,并且该数目取决于模板序列和核苷酸递送的方式。对于利用具有终止子部分的核苷酸单体的SBS技术,终止子在使用的测序条件下可为有效不可逆的,如利用双脱氧核苷酸的传统桑格测序的情况,或者终止子可为可逆的,如由Solexa(现为Illumina,Inc.)开发的测序方法的情况。
SBS技术可利用具有标记部分的核苷酸单体或缺少标记部分的核苷酸单体。因此,可基于以下项来检测掺入事件:标记的特性,诸如标记的荧光;核苷酸单体的特性,诸如分子量或电荷;掺入核苷酸的副产物,诸如焦磷酸盐的释放;等等。在测序试剂中存在两种或更多种不同的核苷酸的实施方案中,不同的核苷酸可以是彼此可区分的,或者另选地,两种或更多种不同的标记在所使用的检测技术下可以是不可区分的。例如,测序试剂中存在的不同核苷酸可具有不同的标记,并且它们可使用适当的光学器件进行区分,如由Solexa(现为Illumina,Inc.)开发的测序方法所例示。
优选的实施方案包括焦磷酸测序技术。焦磷酸测序检测当将特定的核苷酸掺入新生链中时无机焦磷酸盐(PPi)的释放(Ronaghi,M.、Karamohamed,S.、Pettersson,B.、Uhlen,M.和Nyren,P.(1996年),“Real-time DNA sequencing using detection ofpyrophosphate release.”,Analytical Biochemistry 242(1),84-9;Ronaghi,M.(2001)“Pyrosequencing sheds light on DNA sequencing.”Genome Res.,11(1),3-11;Ronaghi,M.,Uhlen,M.和Nyren,P.(1998)“A sequencing method based on real-timepyrophosphate.”Science 281(5375),363;美国专利号6,210,891;美国专利号6,258,568和美国专利号6,274,320,这些文献的公开内容全文以引用方式并入本文)。在焦磷酸测序中,释放的PPi可通过被腺苷三磷酸(ATP)硫酸化酶立即转化为ATP成来进行检测,并且通过荧光素酶产生的光子来检测所产生的ATP水平。待测序的核酸可附接到阵列中的特征部,并且可对阵列进行成像以捕获由于在阵列的特征部处掺入核苷酸而产生的化学发光信号。可在用特定核苷酸类型(例如,A、T、C或G)处理阵列后获得图像。在添加每种核苷酸类型后获得的图像将在阵列中哪些特征部被检测到方面不同。图像中的这些差异反映阵列上的特征部的不同序列内容。然而,每个特征部的相对位置将在图像中保持不变。可使用本文所述的方法存储、处理和分析图像。例如,在用每种不同核苷酸类型处理阵列后获得的图像可以与本文针对从用于基于可逆终止子的测序方法的不同检测通道获得的图像所例示的相同方式进行处理。
在另一种示例性类型的SBS中,通过逐步添加可逆终止子核苷酸来完成循环测序,这些可逆终止子核苷酸包含例如可裂解或可光漂白的染料标记,如例如WO 04/018497和美国专利号7,057,026所述,这两份专利的公开内容以引用方式并入本文。该方法由Solexa(现为Illumina Inc.)商业化,并且还在WO 91/06678和WO 07/123,744中有所描述,这些文献中的每一者的公开内容以引用方式并入本文。荧光标记终止子(其中终止可以是可逆的并且荧光标记可被切割)的可用性有利于高效的循环可逆终止(CRT)测序。聚合酶也可共工程化以有效地掺入这些经修饰的核苷酸并从这些经修饰的核苷酸延伸。
优选地,在基于可逆终止子的测序实施方案中,标记在SBS反应条件下基本上不抑制延伸。然而,检测标记可以是可移除的,例如通过裂解或降解移除。可在将标记掺入到阵列化核酸特征部中后捕获图像。在特定实施方案中,每个循环涉及将四种不同的核苷酸类型同时递送到阵列,并且每种核苷酸类型具有在光谱上不同的标记。然后可获得四个图像,每个图像使用对四个不同标记中的一个标记具有选择性的检测通道。另选地,可顺序地添加不同的核苷酸类型,并且可在每个添加步骤之间获得阵列的图像。在此类实施方案中,每个图像将示出已掺入特定类型的核苷酸的核酸特征部。由于每个特征部的不同序列内容,不同特征部存在于或不存在于不同图像中。然而,特征部的相对位置将在图像中保持不变。通过此类可逆终止子-SBS方法获得的图像可如本文所述进行存储、处理和分析。在图像捕获步骤后,可移除标记并且可移除可逆终止子部分以用于核苷酸添加和检测的后续循环。已在特定循环中以及在后续循环之前检测到标记之后移除这些标记可提供减少循环之间的背景信号和串扰的优点。可用的标记和去除方法的示例在下文进行阐述。
在特定实施方案中,一些或所有核苷酸单体可包括可逆终止子。在此类实施方案中,可逆终止子/可裂解荧光团可包括经由3'酯键连接到核糖部分的荧光团(Metzker,Genome Res.15:1767-1776(2005年),该文献以引用方式并入本文)。其他方法已将终止子化学与荧光标记的裂解分开(Ruparel等人,Proc Natl Acad Sci USA 102:5932-7(2005年),该文献全文以引用方式并入本文)。Ruparel等人描述了可逆终止子的发展,这些可逆终止子使用小的3'烯丙基基团来阻断延伸,但是可通过用钯催化剂进行的短时间处理来容易地去阻断。荧光团经由可光裂解的接头附接到碱基,该可光裂解的接头可通过暴露于长波长紫外光30秒来容易地裂解。因此,二硫化物还原或光裂解可用作可裂解的接头。可逆终止的另一种方法是使用天然终止,该天然终止在将大体积染料放置在dNTP上之后接着发生。dNTP上存在带电大体积染料可通过空间位阻和/或静电位阻而充当高效的终止子。除非染料被移除,否则一个掺入事件的存在防止进一步的掺入。染料的裂解移除荧光团并有效地逆转终止。修饰的核苷酸的示例还描述于美国专利号7,427,673和美国专利号7,057,026中,其公开内容全文以引用方式并入本文。
可与本文所述的方法和系统一起利用的附加的示例性SBS系统和方法描述于美国专利申请公布号2007/0166705、美国专利申请公布号2006/0188901、美国专利号7,057,026、美国专利申请公布号2006/0240439、美国专利申请公布号2006/0281109、PCT公布号WO05/065814、美国专利申请公布号2005/0100900、PCT公布号WO 06/064199、PCT公布号WO07/010,251、美国专利申请公布号2012/0270305和美国专利申请公布号2013/0260372中,这些文献的公开内容全文以引用方式并入本文。
一些实施方案可使用少于四种不同标记来使用对四种不同核苷酸的检测。例如,可以利用并入的美国专利申请公布号2013/0079232的材料中所述的方法和系统来执行SBS。作为第一个示例,一对核苷酸类型可在相同波长下检测,但基于对中的一个成员相对于另一个成员的强度差异,或基于对中的一个成员的导致与检测到的该对的另一个成员的信号相比明显的信号出现或消失的变化(例如,通过化学改性、光化学改性或物理改性)来区分。作为第二个示例,四种不同核苷酸类型中的三种能够在特定条件下被检测到,而第四种核苷酸类型缺少在那些条件下可被检测到或在那些条件下被最低限度地检测到的标记(例如,由于背景荧光而导致的最低限度检测等)。可基于其相应信号的存在来确定前三种核苷酸类型掺入到核酸中,并且可基于任何信号的不存在或对任何信号的最低限度检测来确定第四核苷酸类型掺入到核酸中。作为第三示例,一种核苷酸类型可包括在两个不同通道中检测到的标记,而其他核苷酸类型在不超过一个通道中被检测到。上述三种例示性构型不被认为是互相排斥的,并且可以各种组合进行使用。组合所有三个示例的示例性实施方案是基于荧光的SBS方法,该方法使用在第一通道中检测到的第一核苷酸类型(例如,具有当由第一激发波长激发时在第一通道中检测到的标记的dATP),在第二通道中检测到的第二核苷酸类型(例如,具有当由第二激发波长激发时在第二通道中检测到的标记的dCTP),在第一通道和第二通道两者中检测到的第三核苷酸类型(例如,具有当被第一激发波长和/或第二激发波长激发时在两个通道中检测到的至少一个标记的dTTP),以及缺少在任一通道中检测到或最低限度地检测到的标记的第四核苷酸类型(例如,不具有标记的dGTP)。
此外,如并入的美国专利申请公布号2013/0079232的材料中所述,可使用单个通道获得测序数据。在此类所谓的单染料测序方法中,标记第一核苷酸类型,但在生成第一图像之后移除标记,并且仅在生成第一图像之后标记第二核苷酸类型。第三核苷酸类型在第一图像和第二图像中都保留其标记,并且第四核苷酸类型在两个图像中均保持未标记。
一些实施方案可以利用边连接边测序技术。此类技术利用DNA连接酶掺入寡核苷酸并确定此类寡核苷酸的掺入。寡核苷酸通常具有与寡核苷酸杂交的序列中的特定核苷酸的同一性相关的不同标记。与其他SBS方法一样,可在用已标记的测序试剂处理核酸特征部的阵列后获得图像。每个图像将示出已掺入特定类型的标记的核酸特征部。由于每个特征部的不同序列内容,不同特征部存在于或不存在于不同图像中,但特征部的相对位置将在图像中保持不变。通过基于连接的测序方法获得的图像可如本文所述进行存储、处理和分析。可以与本文所述的方法和系统一起使用的示例性SBS系统和方法在美国专利号6,969,488、美国专利号6,172,218和美国专利号6,306,597中有所描述,这些专利的公开内容全文以引用方式并入本文。
一些实施方案可以利用纳米孔测序(Deamer,D.W.和Akeson,M.“Nanopores andnucleic acids:prospects for ultrarapid sequencing.”Trends Biotechnol.18,147-151(2000);Deamer,D.和D.Branton,“Characterization of nucleic acids by nanoporeanalysis”.Acc.Chem.Res.35:817-825(2002);Li,J.、M.Gershow、D.Stein、E.Brandin和J.A.Golovchenko,“DNA molecules and configurations in a solid-state nanoporemicroscope”,Nat.Mater.,2:611-615(2003),这些文献的公开内容全文以引用方式并入本文)。在此类实施方案中,目标核酸穿过纳米孔。纳米孔可为合成孔或生物膜蛋白,诸如α-溶血素。当目标核酸穿过纳米孔时,可以通过测量孔的电导率的波动来识别每个碱基对。(美国专利号7,001,792;Soni,G.V.和Meller,“A.Progress toward ultrafast DNAsequencing using solid-state nanopores.”Clin.Chem.53,1996-2001(2007);Healy,K.,“Nanopore-based single-molecule DNA analysis.”,Nanomed.,2,459-481(2007);Cockroft,S.L.、Chu,J.、Amorin,M.和Ghadiri,M.R.,“A single-molecule nanoporedevice detects DNA polymerase activity with single-nucleotide resolution.”,J.Am.Chem.Soc.130,818-820(2008),这些文献的公开内容全文以引用方式并入本文)。从纳米孔测序获得的数据可如本文所述进行存储、处理和分析。具体地,根据本文所述的光学图像和其他图像的示例性处理,可将数据如同图像那样进行处理。
一些实施方案可利用涉及DNA聚合酶活性的实时监测的方法。可以通过携带荧光团的聚合酶与γ-磷酸标记的核苷酸之间的荧光共振能量转移(FRET)相互作用来检测核苷酸掺入,如例如美国专利号7,329,492和美国专利号7,211,414中所述(这两份专利中的每一者以引用方式并入本文),或者可以用零模波导来检测核苷酸掺入,如例如美国专利号7,315,019中所述(该专利以引用方式并入本文),并且可以使用荧光核苷酸类似物和工程化聚合酶来检测核苷酸掺入,如例如美国专利号7,405,281和美国专利申请公布号2008/0108082中所述(这两份专利中的每一者以引用方式并入本文)。照明可限于表面栓系的聚合酶周围的仄升量级的体积,使得可在低背景下观察到荧光标记的核苷酸的掺入(Levene,M.J.等人,“Zero-mode waveguides for single-molecule analysis at highconcentrations.”,Science 299,682-686(2003);Lundquist,P.M.等人,“Parallelconfocal detection of single molecules in real time.”,Opt.Lett.33,1026-1028(2008);Korlach,J.等人,“Selective aluminum passivation for targetedimmobilization of single DNA polymerase molecules in zero-mode waveguide nanostructures.”,Proc.Natl.Acad.Sci.USA 105,1176-1181(2008),这些文献的公开内容全文以引用方式并入本文)。通过此类方法获得的图像可如本文所述进行存储、处理和分析。
一些SBS实施方案包括检测在核苷酸掺入延伸产物时释放的质子。例如,基于释放质子的检测的测序可使用可从Ion Torrent公司(Guilford,CT,它是Life Technologies子公司)商购获得的电检测器和相关技术或在US 2009/0026082A1、US2009/0127589 A1、US2010/0137143 A1或US 2010/0282617A1中所述的测序方法和系统,这些文献中的每一篇均以引用方式并入本文。本文阐述的使用动力学排阻来扩增靶核酸的方法可以容易地应用于用于检测质子的基板。更具体地,本文阐述的方法可以用于产生用于检测质子的扩增子克隆群体。
上述SBS方法可有利地以多种格式进行,使得同时操纵多个不同的目标核酸。在特定实施方案中,可在共同的反应容器中或在特定基板的表面上处理不同的目标核酸。这允许以多种方式方便地递送测序试剂、移除未反应的试剂和检测掺入事件。在使用表面结合的目标核酸的实施方案中,目标核酸可为阵列格式。在阵列格式中,目标核酸通常可以在空间上可区分的方式结合到表面。目标核酸可通过直接共价附着、附着到小珠或其他粒子或结合到附着到表面的聚合酶或其他分子来结合。阵列可包括在每个位点(也被称为特征部)处的目标核酸的单个拷贝,或者具有相同序列的多个拷贝可存在于每个位点或特征部处。多个拷贝可通过扩增方法(诸如,如下文进一步详细描述的桥式扩增或乳液PCR)产生。
本文所述的方法可使用具有处于多种密度中任一种密度的特征部的阵列,该多种密度包括例如至少约10个特征部/cm
本文阐述的方法的优点是它们并行提供了对多个靶核酸的快速且有效检测。因此,本公开提供了能够使用本领域已知的技术(诸如上文所例示的那些)来制备和检测核酸的整合系统。因此,本公开的整合系统可以包括能够将扩增试剂和/或测序试剂递送到一个或多个固定DNA片段的流体部件,该系统包括诸如泵、阀、贮存器、流体管线等的部件。流通池在整合系统中可以被配置用于和/或用于检测靶核酸。示例性流通池在例如US 2010/0111768 A1和美国序列号13/273,666中有所描述,这两份专利中的每一者以引用方式并入本文。如针对流通池所例示的,整合系统的一个或多个流体部件可以用于扩增方法和检测方法。以核酸测序实施方案为例,整合系统的一个或多个流体部件可以用于本文阐述的扩增方法以及用于在测序方法(诸如上文例示的那些)中递送测序试剂。另选地,整合系统可包括单独的流体系统以执行扩增方法并执行检测方法。能够产生扩增核酸并且还确定核酸序列的整合测序系统的示例包括但不限于MiSeq
上述测序系统对由测序设备接收的样品中存在的核酸聚合物进行测序。如本文所定义,“样品”及其衍生物以其最广泛的意义使用,包括怀疑包含目标的任何标本、培养物等。在一些实施方案中,样品包括DNA、RNA、PNA、LNA、嵌合或杂交形式的核酸。样品可以包括含有一种或多种核酸的任何基于生物、临床、外科、农业、大气或水生动植物的标本。该术语还包括任何分离的核酸样品,诸如基因组DNA、新鲜冷冻或福尔马林固定石蜡包埋的核酸标本。还设想样品的来源可以是:单个个体、来自遗传相关成员的核酸样品的集合、来自遗传不相关成员的核酸样品、来自单个个体的(与之匹配的)核酸样品(诸如肿瘤样品和正常组织样品),或者来自含有两种不同形式的遗传物质(诸如从母体受试者获得的母体DNA和胎儿DNA)的单个来源的样品,或者在含有植物或动物DNA的样品中存在污染性细菌DNA。在一些实施方案中,核酸材料的来源可以包括从新生儿获得的核酸,例如通常用于新生儿筛检的核酸。
该核酸样品可以包括高分子量物质,诸如基因组DNA(gDNA)。该样品可以包括低分子量物质,诸如从FFPE样品或存档的DNA样品获得的核酸分子。在另一实施方案中,低分子量物质包括酶促片段化或机械片段化的DNA。该样品可以包含无细胞循环DNA。在一些实施方案中,该样品可以包括从活检组织、肿瘤、刮取物、拭子、血液、黏液、尿液、血浆、精液、毛发、激光捕获显微解剖、手术切除和其他临床或实验室获得的样品获得的核酸分子。在一些实施方案中,该样品可以是流行病学样品、农业样品、法医学样品或病原性样品。在一些实施方案中,该样品可包括从动物(诸如人类或哺乳动物来源)获得的核酸分子。在另一实施方案中,该样品可包括从非哺乳动物来源(诸如植物、细菌、病毒或真菌)获得的核酸分子。在一些实施方案中,核酸分子的来源可以是存档或灭绝的样品或物种。
另外,本文所公开的方法和组合物可以用于扩增具有低质量核酸分子的核酸样品,诸如来自法医学样品的降解的和/或片段化的基因组DNA。在一个实施方案中,法医学样品可包括从犯罪现场获得的核酸、从失踪人员DNA数据库获得的核酸、从与法医调查相关联的实验室获得的核酸,或者包括由执法机关、一种或多种军事服务或任何此类人员获得的法医学样品。核酸样品可以是经纯化的样品或含有粗DNA的溶胞产物,例如来源于口腔拭子、纸、织物或者其他可以用唾液、血液或其他体液浸渍的基材。因此,在一些实施方案中,该核酸样品可包含少量DNA(诸如基因组DNA),或者DNA的片段化部分。在一些实施方案中,靶序列可存在于一种或多种体液中,其中体液包括但不限于血液、痰、血浆、精液、尿液和血清。在一些实施方案中,靶序列可从受害者的毛发、皮肤、组织样品、尸体解剖或遗骸获得。在一些实施方案中,包含一种或多种靶序列的核酸可从死亡的动物或人获得。在一些实施方案中,靶序列可包括从非人类DNA(诸如微生物、植物或昆虫DNA)获得的核酸。在一些实施方案中,靶序列或扩增的靶序列导向人类身份识别的目的。在一些实施方案中,本公开整体涉及用于识别法医学样品的特性的方法。在一些实施方案中,本公开整体涉及使用本文所公开的一种或多种目标特异性引物或者用本文概述的引物设计标准设计的一种或多种目标特异性引物的人类身份识别方法。在一个实施方案中,含有至少一种靶序列的法医学样品或人类身份识别样品可以使用本文所公开的任何一种或多种目标特异性引物或者使用本文概述的引物标准进行扩增。
基因组分类系统106的部件可包括软件、硬件或两者。例如,基因组分类系统106的部件可包括存储在计算机可读存储介质上并且可由一个或多个计算设备(例如,用户客户端设备108)的处理器执行的一个或多个指令。当由一个或多个处理器执行时,基因组分类系统106的计算机可执行指令可使计算设备执行本文所描述的气泡检测方法。另选地,基因组分类系统106的部件可包括硬件,诸如专用处理设备用以执行某些功能或功能的组。附加地或另选地,基因组分类系统106的部件可包括计算机可执行指令和硬件的组合。
此外,执行本文所描述关于基因组分类系统106的功能的基因组分类系统106的部件可以例如被实施作为独立应用的一部分、作为应用的模块、作为应用的插件、作为可以被其他应用检出的库函数或函数、和/或作为云计算模型。因此,基因组分类系统106的部件可以被实施作为个人计算设备或移动设备上的独立应用的一部分。附加地或另选地,基因组分类系统106的部件可以实施在提供测序服务的任何应用中,包括但不限于IlluminaBaseSpace、Illumina DRAGEN或Illumina TruSight软件。“Illumina”、“BaseSpace”、“DRAGEN”和“TruSight”是Illumina,Inc.公司在美国和/或其他国家的注册商标或商标。
如以下更详细讨论的,本公开的实施方案可以包括或利用包括计算机硬件(诸如例如一个或多个处理器和系统存储器)的专用或通用计算机。本公开范围内的实施方案还包括用于携带或存储计算机可执行指令和/或数据结构的物理和其他计算机可读介质。具体地,本文所述的过程中的一者或多者可以至少部分实施为体现在非暂态计算机可读介质中并且可由一个或多个计算设备(例如,本文所述的介质内容访问设备中的任一者)执行的指令。一般来讲,处理器(例如,微处理器)从非暂态计算机可读介质(例如,存储器等)接收指令,并且执行那些指令,由此执行一个或多个过程,包含本文所述的过程中的一者或多者。
计算机可读介质可以是可由通用或专用计算机系统访问的任何可用介质。存储计算机可执行指令的计算机可读介质是非暂态计算机可读存储介质(设备)。携带计算机可执行指令的计算机可读介质是传输介质。因此,通过示例方式而非限制,本公开的实施方案可包括至少两种明显不同种类的计算机可读介质:非暂态计算机可读存储介质(设备)和传输介质。
非暂态计算机可读存储介质(设备)包括RAM、ROM、EEPROM、CD-ROM、固态驱动器(SSD)(例如,基于RAM)、快闪存储器、相变存储器(PCM)、其他类型的存储器、其他光盘存储装置、磁盘存储装置或其他磁存储设备,或可用于存储呈计算机可执行指令或数据结构形式的期望的程序代码手段并且其可由通用或专用计算机访问的任何其他介质。
“网络”定义为使得能够在计算机系统和/或模块和/或其他电子设备之间传输电子数据的一个或多个数据链路。当通过网络或另一通信连接(硬连线、无线或硬连线或无线的组合)向计算机转移或提供信息时,计算机适当地将该连接视为传输介质。传输介质可包括网络和/或数据链路,该网络和/或数据链路可用于携带呈计算机可执行指令或数据结构形式的期望的程序代码手段,并且其可由通用或专用计算机访问。上述的组合也应当被包括在计算机可读介质的范围内。
此外,在到达各种计算机系统部件后,呈计算机可执行指令或数据结构形式的程序代码手段可从传输介质自动转移到非暂态计算机可读存储介质(设备)(或反之亦然)。例如,通过网络或数据链路接收的计算机可执行指令或数据结构可被缓冲在网络接口模块(例如,NIC)内的RAM中,并且然后最终被转移到计算机系统RAM和/或到计算机系统处的较不易失的计算机存储介质(设备)。因此,应当理解,非暂态计算机可读存储介质(设备)可被包括在也(或甚至主要)利用传输介质的计算机系统部件中。
计算机可执行指令包括例如当在处理器处执行时,使得通用计算机、专用计算机或专用处理设备执行某些功能或功能的组的指令和数据。在一些实施方案中,在通用计算机上执行计算机可执行指令以将通用计算机变成实施本公开的元素的专用计算机。计算机可执行指令可以是例如二进制数、诸如汇编语言的中间格式指令、或者甚至源代码。尽管已经以特定于结构特征和/或方法动作的语言描述了主题内容,但是应当理解,在所附权利要求中定义的主题内容不必限于所描述的特征部或动作。相反,所描述的特征部和动作是作为实施权利要求的示例性形式来公开的。
本领域中的技术人员将理解,本公开可以在具有许多类型的计算机系统配置的网络计算环境中实践,包括个人计算机、台式计算机、便携式电脑、消息处理器、手持式设备、多处理器系统、基于微处理器的或可编程消费电子产品、网络PC、小型计算机、大型计算机、移动电话、PDA、平板电脑、寻呼机、路由器、交换机等。本公开还可以在分布式系统环境中实践,其中通过网络链接(通过硬连线数据链路、无线数据链路或者通过硬连线和无线数据链路的组合)的本地和远程计算机系统两者都执行任务。在分布式系统环境中,程序模块可以位于本地和远程存储器存储设备两者中。
本公开的实施方案还可在云计算环境中实施。在本说明书中,“云计算”定义为用于实现对可配置计算资源的共享池的按需网络访问的模型。例如,可在市场中采用云计算以提供对可配置计算资源的共享池的无处不在并且便利的按需访问。可配置计算资源的共享池可经由虚拟化快速预置并且以低管理努力或服务提供者交互释放,并且然后因此扩展。
云计算模型可由各种特性组成,诸如例如按需自助服务、广泛网络访问、资源池化、快速弹性、可计量服务等。云计算模型还可展示各种服务模型,诸如例如软件即服务(SaaS)、平台即服务(PaaS)和基础设施即服务(IaaS)。云计算模型还可使用不同的部署模型来部署,诸如私有云、社区云、公共云、混合云等。在本说明书和在权利要求书中,“云计算环境”是在其中采用云计算的环境。
图13示出可以被配置为执行上述过程中的一者或多者的计算设备1300的方框图。人们将理解,诸如计算设备1300的一个或多个计算设备可以实施基因组分类系统106和测序系统104。如图13所示,计算设备1300可包括处理器1302、存储器1304、存储设备1306、I/O接口1308和通信接口1310,它们可以通过通信基础设施1312的方式通信地耦合。在某些实施方案中,计算设备1300可包括比图13中示出的部件更少或更多的部件。以下段落更详细地描述图13中所示的计算设备1300的部件。
在一个或多个实施方案中,处理器1302包括用于执行指令的硬件,诸如构成计算机程序的那些指令。作为示例,而非通过限制的方式,为了执行用于动态地修改工作流程的指令,处理器1302可以从内部寄存器、内部高速缓存、存储器1304或存储设备1306检索(或提取)指令,并且解码和执行它们。存储器1304可以是用于存储由处理器执行的数据、元数据和程序的易失性或非易失性存储器。存储设备1306包括用于存储用于执行本文所述的方法的数据或指令的存储装置,诸如硬盘、闪存盘驱动器或其他数字存储设备。
I/O接口1308允许用户向计算设备1300提供输入、从该计算设备接收输出,以及以其他方式向该计算设备转移数据和从该计算设备接收数据。I/O接口1308可以包括鼠标、小键盘或键盘、触摸屏、相机、光学扫描仪、网络接口、调制解调器、其他已知I/O设备或此类I/O接口的组合。I/O接口1308可以包括用于向用户呈现输出的一个或多个设备,包括但不限于图形引擎、显示器(例如,显示屏)、一个或多个输出驱动程序(例如,显示驱动程序)、一个或多个音频扬声器,以及一个或多个音频驱动程序。在某些实施方案中,I/O接口1308被配置为向显示器提供图形数据用于呈现给用户。图形数据可以表示一个或多个图形用户界面和/或可以服务于特定实施的任何其他图形内容。
通信接口1310可包括硬件、软件或两者。在任何情况下,通信接口1310可提供用于计算设备1300与一个或多个其他计算设备或网络之间的通信(诸如例如,基于分组的通信)的一个或多个接口。作为示例,而非通过限制的方式,通信接口1310可以包括用于与以太网或其他基于有线的网络通信的网络接口控制器(NIC)或网络适配器,或用于与无线网络(诸如WI-FI)通信的无线NIC(WNIC)或无线适配器。
附加地,通信接口1310可以促进与各种类型的有线或无线网络的通信。通信接口1310还可以促进使用各种通信协议的通信。通信基础设施1312还可以包括将计算设备1300的部件彼此耦合的硬件、软件或两者。例如,通信接口1310可以使用一个或多个网络和/或协议以使得由特定基础设施连接的多个计算设备能够与彼此通信以执行本文所述的过程的一个或多个方面。为了说明,测序过程可允许多个设备(例如,客户端设备、测序设备和服务器设备)交换诸如测序数据和误差通知的信息。
在前述说明书中,本公开已经参考其特定示例性实施方案进行描述。参考本文所讨论的细节描述了本公开的各种实施方案和方面,并且附图说明各种实施方案。上面的描述和图是对本公开的说明,并且不应被解释为限制本公开。描述了许多特定细节以提供对本公开的各种实施方案的透彻理解。
本公开可以以其它特定形式体现而不脱离其精神或本质特征。所述实施方案在所有方面都应被视为仅为示例性的而非限制性的。例如,本文所描述的方法可以用更少或更多的步骤/动作执行,或者步骤/动作可以以不同的顺序执行。附加地,本文所描述的步骤/动作可以重复或与彼此并行地执行或与相同或类似步骤/动作的不同实例并行地执行。因此,本申请的范围由所附权利要求书而非前述描述来指示。在权利要求的等效含义和范围内的所有改变都将包含在其范围内。
- 一种散料滑动摩擦系数测定装置
- 一种散料滑动摩擦系数测定装置