一种在WES数据中检测单样本SMN基因拷贝数的方法
文献发布时间:2023-06-19 10:03:37
技术领域
本发明涉及生物学与精准医学全基因组变异检测领域,尤其涉及一种WES(WholeExome Sequence,全外显子组测序,简写为WES)数据中检测单样本SMN基因拷贝数的方法。
背景技术
脊髓性肌肉萎缩症(英语:Spinal muscular atrophy,简写为SMA),是一种遗传性神经疾病。它会造成运动神经元退化、肌肉萎缩,肌肉无力,最终造成死亡。SMA是由于人体内被称作为“运动神经元存活1号”基因(SMN1)的缺失或异常(突变)所导致的。SMA主要与两个高度同源(是指这两个基因的序列非常相似)的基因密切相关,即SMN1和SMN2(“运动神经元存活2号”基因),这两个基因主要通过7号外显子和8号外显子上的两个基因位点进行区分。一般来说,大部分正常个体都有2份拷贝的SMN1基因与2份拷贝的SMN2基因,SMN2基因发生外显子7的跳跃,只有少量的全长SMN mRNA,所以如果某个人两份拷贝的SMN1基因都失去功能则一定会患病,只有一份SMN1基因起作用的个体为携带者。在SMN1基因都失去功能的情况下,SMN2基因拷贝数数目,则会影响患者的发病时间与疾病严重程度。
SMA基因检测的方法有以下几类:(1)PCR(Polymerase Chain Reaction,聚合酶链式反应)或一代测序,首先对目标区域进行扩增,然后通过限制性内切酶或一代测序的方法来区分,如果是患者,则在c.840位点缺失SMN1的C峰,只显示SMN2的T纯合峰;正常人或携带者应该为杂合的C/T峰。(2)MLPA,多重连接探针扩增技术(multiplex ligation-dependentprobe amplification,MLPA)于2002年由Schouten等首先报道,是早年几年发展起来的一种针对待检DNA序列进行定性和半定量分析的新技术。该技术高效、特异,在一次反应中可以检测45个核苷酸序列拷贝数的改变,针对c.840C>T位点设计不同的探针序列,对SMN1基因和SMN2基因扩增出不同长度的片段,而峰的高度可以反映拷贝数变异。(3)二代测序:贝勒医学院的Feng等人在Genetics in Medicine杂志上发表了一项利用二代测序检测SMA的研究(pmid:28125085),该研究包括6648例样本。主要的原理是收集同一批次样本进行SMN基因的目标区域捕获测序,统计SMN1与SMN2各个的exon1--exon8的总覆盖度,且提取的是单端reads,然后根据c.840C>T分析SMN1 reads与SMN2 reads比例,再根据SMN1及SMN2reads比例、总覆盖度计算出每个人分别携带几个拷贝的SMN1与SMN2。与MLPA相比,灵敏度大于98%,特异性大于98%。另外该研究还确诊了几个致病点突变位点,并且g.27134T>G与RFLP(Restriction Fragment Length Polymorphism,限制性内切酶片段长度多态性)结果一致,该位点与SMN1 2+0型特殊携带者密切相关。但是对于SMN2拷贝数的灵敏度与特异性未明确描述。
针对上述3种检测方法,每种方法都有各自的不足,如(1)PCR-RFLP或一代测序:这个方法的缺陷是存在酶切不彻底的隐患,且不能区分携带者与正常人,也不能检测SMN2拷贝数,在临床上可以诊断SMN1纯合缺失的患者,其他情况只能作为初筛。(2)MLPA技术该试剂盒不能检测点突变与特殊的SMN1 2+0携带者,且检测通量低。(3)Feng等人的检测方法是基于NGS平台,虽然能够解决特殊的SMN1 2+0携带者变异,但是该方法需要在同批次样本中检测以消除批次效应,如果同批次样本数量不足够多,会对检测结果产生影响。该方法中提取单端reads的比对结果统计覆盖度,会丢失部分有效信息。该方法统计了exon1--exon8的所有外显子上的覆盖度,虽然全面考察了SMN基因的比对情况,但是由于文库制备和测序环节的不确定性,多个外显子间的扩增效率有差异,导致扩增出来的reads会有差异,对检测SMN基因的拷贝数,尤其是exon7和exon8的真实拷贝数有较大影响。
此外,部分开源软件可以在WGS中检测SMN基因拷贝数,但是这些软件要么需要使用WGS数据,要么需要批次的样本,并不能有效解决单样本检测的需求。为了能够快速精确的检测SMN基因拷贝数,尤其是能够满足临床上需要对单个样本的检测,本发明基于NGS平台和WES测序数据,利用大量测试样本构建数据集,预先探究SMN基因不同拷贝数对应的概率值,充分消除样本间的批次效应,增加检测的灵活性和可靠性。
发明内容
本申请通过提供一种在WES数据中检测单样本SMN基因拷贝数的方法,用于解决现有技术中不能精确检测单样本SMN基因拷贝数且不能同时检测出特殊的SMN1 2+0携带者状态的问题。
本申请提供了一种在WES数据中检测单样本SMN基因拷贝数的方法,
S1、收集不同批次WES数据的已知SMN基因实际拷贝数的阴性样本和已知SMN基因实际拷贝数的阳性样本,在全外显子Bed区间内寻找与SMN基因拷贝数高相关性的对照区间;
S2、利用所述对照区间的resds覆盖度校正不同批次的所述阴性样本和阳性样本间的批次效应,定义所述不同批次WES数据的已知SMN基因实际拷贝数的阴性样本和已知SMN基因实际拷贝数的阳性样本为所有样本,计算所述所有样本的SMN1基因的相应拷贝数时的P1值分布范围和SMN2基因的相应拷贝数时的P2值分布范围:①SMN1基因的P1值分布范围是根据样本中SMN1基因的实际拷贝数进行分组的,例如样本SMN1基因的实际拷贝数为0个拷贝的P1值分布范围定义为P1_zero、样本SMN1基因的实际拷贝数为1个拷贝的P1值分布范围定义为P1_one、样本SMN1基因的实际拷贝数为2个拷贝的P1值分布范围定义为P1_two,以此类推。②SMN2基因的P2值分布范围是根据样本中SMN2基因的实际拷贝数进行分组的,例如样本SMN2基因的实际拷贝数为0个拷贝的P2值分布范围定义为P2_zero、样本SMN2基因的实际拷贝数为1个拷贝的P2值分布范围定义为P2_one、样本SMN2基因的实际拷贝数为2个拷贝的P2值分布范围定义为P2_two,以此类推。以下把P1_zero、P1_one、P1_two、P2_zero、P2_one、P2_two等统称为P1值和P2值。
统计所述所有样本中已经验证为是静默携带者的样本的7号内含子的g.27134T>G位点的校正后覆盖度P_silent值分布范围,后期可以根据该覆盖度P_silent值分布范围和单个样本的SMN1基因的拷贝数为2的证据,判断单样本是否为静默携带者;
S3、计算单个测试样本的SMN1基因的7号外显子和8号外显子的p1值、SMN2基因的7号外显子和8号外显子的p2值,根据S2计算所得的P1值和P2值的分布范围判断本步骤中p1值和p2值所对应的SMN1基因和SMN2基因的拷贝数;
统计单个测试样本的7号内含子上的g.27134T>G位点的覆盖度p_silent值;根据所述p_silent值和所述单个测试样本的SMN1基因的拷贝数,判断该单个测试样本状态:;
当p_silent值在S2中计算的P_silent值分布范围内且所述单个测试样本的SMN1基因的拷贝数是2时,判断所述单个测试样本为静默携带者;
当p_silent值在S2中计算的P_silent值分布范围内但所述单个测试样本的SMN1基因的拷贝数不是2,判断所述单个测试样本为疑似静默携带者;
其他情况时均判断所述单个测试样本为非静默携带者。
进一步的,本发明提出所述S1中寻找所述对照区间的步骤包括:
S101、用MLPA平台验证所述所有样本的SMN1基因和SMN2基因的实际拷贝数,使用生信分析流程进行处理后得到Bam文件;
S102、预先筛选出两拷贝基因的Bed区间,统计所述所有样本在全外显子组的Bed区间内的覆盖度;
S103、把所述所有样本的覆盖度校正到100X,得到样本校正后覆盖度;
S104、根据所述所有样本校正后覆盖度计算相关性和方差,查找相关性好且方差值低的Bed区间作为对照区间。
进一步的,本发明提出所述对照区间为相关性好且方差值低的前5个Bed区间。
进一步的,本发明提出所述S2的步骤包括:
S201、统计所述所有样本在SMN1基因和SMN2基因7号外显子和8号外显子的总覆盖度并校正,得到SMN1基因和SMN2基因7号外显子和8号外显子的校正后总覆盖度;
S202、统计所述所有样本在5个所述对照区间的总覆盖度并校正,得到对照区间的校正覆盖度均值;
S203、统计所述所有样本的3个点突变的覆盖度并校正,得到3个点突变的校正后覆盖度;所述3个点突变的覆盖度包括7号外显子上的c.840C>T位点的覆盖度、8号外显子上的c.*239G>A位点的覆盖度和7号内含子上的g.27134T>G位点的覆盖度;计算SMN1基因的校正后覆盖度在7号外显子、8号外显子的ratio值;计算SMN2基因的的校正后覆盖度在7号外显子、8号外显子的ratio值;
S204、根据所述SMN1基因和SMN2基因7号外显子和8号外显子的校正后总覆盖度、对照区间的校正覆盖度均值、所述ratio值,计算SMN1基因的7号外显子和8号外显子的拷贝数p_e7_s1值和p_e8_s1值;计算SMN2基因的7号外显子和8号外显子的拷贝数p_e7_s2值和p_e8_s2值;根据p_e7_s1值和p_e8_s1值计算p1值;根据p_e7_s2值和p_e8_s2值计算p2值。
进一步的,本发明提出所述校正均采用对应的批次内中位数覆盖度进行校正。
进一步的,本发明提出SMN1基因在7号外显子上的ratio值和p_e7_s1值的计算公式为:
ratio_e7_s1=rc_e7_s1/(rc_e7_s1+rc_e7_s2);
cn_e7_s1=rc_e7_s1_total/rc_control;
cn_e7_s2=rc_e7_s2_total/rc_control;
p_e7_s1=ratio_e7_s1*(cn_e7_s1+cn_e7_s2)*2;
SMN1基因在8号外显子上的ratio值和p_e8_s1值的计算公式为:
ratio_e8_s1=rc_e8_s1/(rc_e8_s1+rc_e8_s2);
cn_e8_s1=rc_e8_s1_total/rc_control;
cn_e8_s2=rc_e8_s2_total/rc_control;
p_e8_s1=ratio_e8_s1*(cn_e8_s1+cn_e8_s2)*2;
SMN1基因的p1值的计算公式为:
p1=(p_e7_s1+p_e8_s1)/2
SMN2基因在7号外显子上的ratio值和p_e7_s2值的计算公式为:
ratio_e7_s2=rc_e7_s2/(rc_e7_s1+rc_e7_s2);
p_e7_s2=ratio_e7_s2*(cn_e7_s1+cn_e7_s2)*2;
SMN2基因在8号外显子上的ratio值和p_e8_s2值的计算公式为:
ratio_e8_s2=rc_e8_s2/(rc_e8_s1+rc_e8_s2);
p_e8_s2=ratio_e8_s2*(cn_e8_s1+cn_e8_s2)*2;
SMN2基因的p2值的计算公式为:
p2=(p_e7_s1+p_e8_s1)/2
p_silent值的计算公式为:
p_silent=[g.27134T>G位点的校正后覆盖度]
P_silent值的计算公式为:
P_silent=[min{p_silent_sample1,p_silent_sample1,...,p_silent_sampleN},5000]
公式中的变量名称含义如下:
rc_e7_s1:用批次内中位数校正后的SMN1的7号外显子上的c.840C>T位点的校正覆盖度,
rc_e8_s1:用批次内中位数校正后的SMN1的8号外显子上的c.*239G>A位点的校正覆盖度,
rc_e7_s2:用批次内中位数校正后的SMN2的7号外显子上的c.840C>T位点的校正覆盖度,
rc_e8_s2:用批次内中位数校正后的SMN2的8号外显子上的c.*239G>A位点的校正覆盖度,
rc_control:用批次内中位数校准后的control region上的覆盖度,
rc_e7_s1_total:SMN1在exon7上的校正后总覆盖度,
rc_e8_s1_total:SMN1在exon8上的校正后总覆盖度,
rc_e7_s2_total:SMN2在exon7上的校正后总覆盖度,
rc_e8_s2_total:SMN2在exon8上的校正后总覆盖度,
cn_e7_s1:SMN1在exon7上的拷贝数系数,
cn_e8_s1:SMN1在exon8上的拷贝数系数,
cn_e7_s2:SMN2在exon7上的拷贝数系数,
cn_e8_s2:SMN2在exon8上的拷贝数系数,
ratio_e7_s1:SMN1在exon7上的ratio值,
ratio_e8_s1:SMN1在exon8上的ratio值,
ratio_e7_s2:SMN2在exon7上的ratio值,
ratio_e8_s2:SMN2在exon8上的ratio值,
p_e7_s1:SMN1在exon7上的拷贝数概率值,
p_e8_s1:SMN1在exon8上的拷贝数概率值,
p_e7_s2:SMN2在exon7上的拷贝数概率值,
p_e8_s2:SMN2在exon8上的拷贝数概率值,
p1:单样本的SMN1基因相应拷贝数时的概率值,
p2:单样本的SMN2基因相应拷贝数时的概率值,
P1:所述所有样本根据相应拷贝数统计的p1值的分布范围,
P2:所述所有样本根据相应拷贝数统计的p2值的分布范围,
p_silent:单个样本的g.27134T>G位点的校正后覆盖度,
P_silent:静默携带者阈值分布范围,是样本中排除离群值后所有p_silent的最小值到5000,(5000是最大限定值,理论上该值为该位点上的最大矫正覆盖度)。
进一步的,本发明提出计算所述S3中单个测试样本的SMN1基因的7号外显子和8号外显子的拷贝数p1、SMN2基因的7号外显子和8号外显子的拷贝数p2的步骤包括:
S301、分别统计单个测试样本的SMN1基因和SMN2基因上的7号外显子、8号外显子的总覆盖度、5个对照区间的覆盖度和3个点突变的覆盖度,并分别进行校正得到单个测试样本的SMN1基因和SMN2基因上的7号外显子和8号外显子的校正后总覆盖度、5个对照区间的校正覆盖度均值和3个点突变的校正后覆盖度;所述3个点突变的覆盖度包括7号外显子上的c.840C>T位点的覆盖度、8号外显子上的c.*239G>A位点的覆盖度、7号内含子上的g.27134T>G位点的覆盖度;
S302、根据所述单个测试样本的SMN1基因和SMN2基因上的7号外显子和8号外显子的校正后总覆盖度、5个对照区间的校正覆盖度均值和3个点突变的校正后覆盖度,计算SMN1基因和SMN2基因的校正后覆盖度在7号外显子和8号外显子的ratio值;
S303、计算单个测试样本SMN1基因和SMN2基因上的7号外显子和8号外显子的拷贝数p1值和p2值。
进一步的,本发明提出对单个测试样本的SMN1基因和SMN2基因7号外显子和8号外显子的总覆盖度的校正采用S201中所述的SMN1基因和SMN2基因上的7号外显子和8号外显子的校正后总覆盖度的中位数进行;
对单个测试样本的5个对照区间的覆盖度的校正采用S202中所述的对照区间的校正覆盖度均值的中位数进行;
对单个测试样本的3个点突变的覆盖度的校正采用S203中所述的3个点突变的校正后覆盖度的中位数进行。
有益效果
本发明提供了一种在WES数据中检测单样本SMN基因拷贝数的方法,所以有效解决了目前现有技术中不能精确检测单样本SMN基因拷贝数的问题,进而达到了如下技术效果:
本发明通过预先使用已知SMN基因实际拷贝数的阴性样本和已知SMN基因实际拷贝数的阳性样本构建SMN1基因和SMN2基因拷贝数分值数据集来检测单个样本的基因拷贝数,通过在全外显子Bed区间内寻找与SMN基因拷贝数高相关性的对照区间;利用该区域的reads覆盖度校正不同样本间的批次效应,有效提升了检测方法的准确性,同时还可以检测出发生了g.27134T>G点突变的SMN1 2+0静默携带者。
应当理解,前述构思以及在下面更加详细地描述的额外构思的所有组合只要在这样的构思不相互矛盾的情况下都可以被视为本公开的发明主题的一部分。
结合附图从下面的描述中可以更加全面地理解本发明教导的前述和其他方面、实施例和特征。本发明的其他附加方面例如示例性实施方式的特征和/或有益效果将在下面的描述中显见,或通过根据本发明教导的具体实施方式的实践中得知。
附图说明
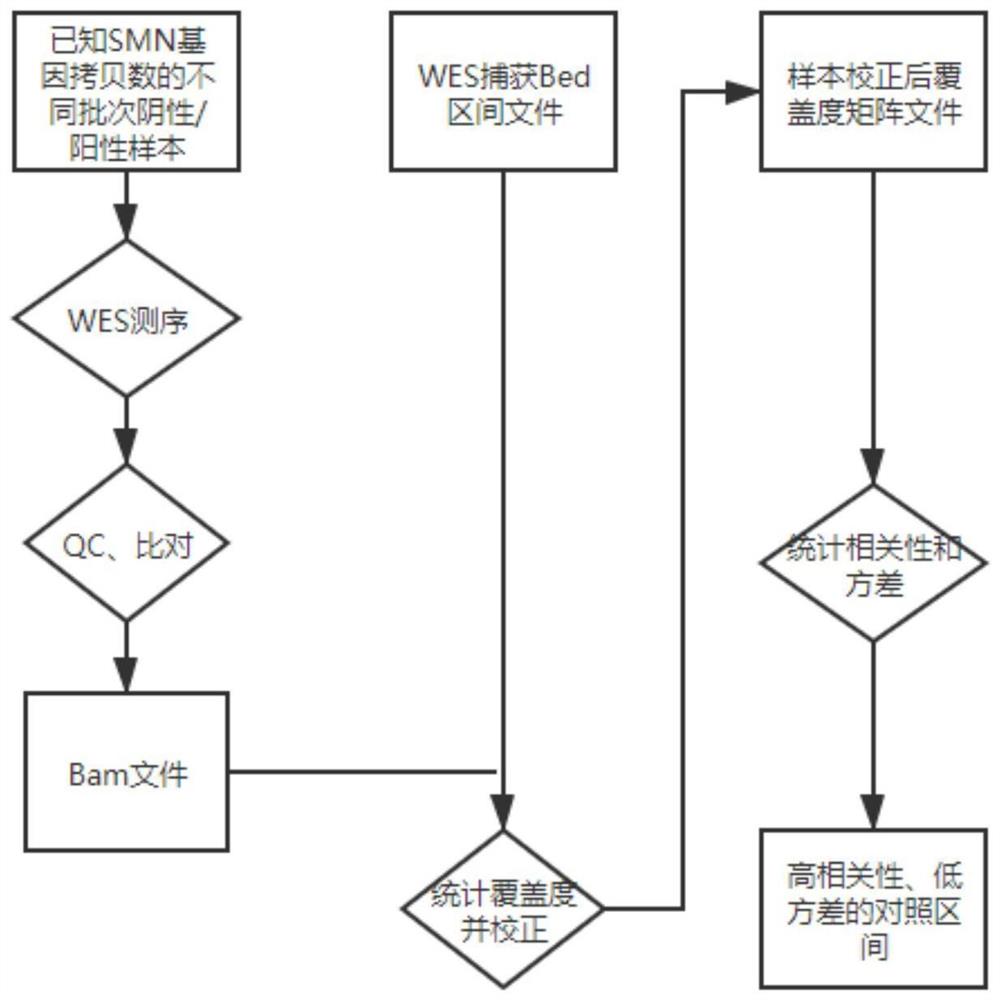
图1为本发明中在全外显子Bed区间内寻找与SMN基因拷贝数高相关性的对照区间的流程图;
图2为本发明中计算所有样本的SMN1基因和SMN2基因拷贝数P值分布范围流程图;
图3为本发明中判断单个测试样本的SMN1基因和SMN2基因拷贝数p值和静默携带者状态流程图。
具体实施方式
在本公开中参照附图来描述本发明的各方面,附图中示出了许多说明的实施例。本公开的实施例不必定义在包括本发明的所有方面。应当理解,上面介绍的多种构思和实施例,以及下面更加详细地描述的那些构思和实施方式可以以很多方式中任意一种来实施,这是因为本发明所公开的构思和实施例并不限于任何实施方式。另外,本发明公开的一些方面可以单独使用,或者与本发明公开的其他方面的任何适当组合来使用。
为解决现有技术中不能精确检测单样本SMN基因拷贝数且不能同时检测出特殊的SMN1 2+0携带者状态的问题,本发明预先使用已知SMN基因实际拷贝数的阴性样本和已知SMN基因实际拷贝数的阳性样本构建SMN1基因和SMN2基因拷贝数分值数据集来检测单个样本的基因拷贝数,通过在全外显子Bed区间内寻找与SMN基因拷贝数高相关性的对照区间;利用该区域的reads覆盖度校正不同样本间的批次效应,有效提升了检测方法的准确性,同时还可以检测出发生了g.27134T>G点突变的SMN1 2+0静默携带者。实现了精确检测单样本SMN基因拷贝数且检测出发生了g.27134T>G点突变的SMN1 2+0静默携带者的目的。
具体实施时,本申请提供了一种在WES数据中检测单样本SMN基因拷贝数的方法,
S1、如图1所示,收集500个不同批次WES数据的已知SMN基因实际拷贝数的阴性样本和已知SMN基因实际拷贝数的阳性样本,在全外显子Bed区间内寻找与SMN基因拷贝数高相关性的对照区间;
S2、利用所述对照区间的resds覆盖度校正不同批次的所述阴性样本和阳性样本间的批次效应,定义所述不同批次WES数据的已知SMN基因实际拷贝数的阴性样本和已知SMN基因实际拷贝数的阳性样本为所有样本,计算所述所有样本的SMN1基因的相应拷贝数时的P1值分布范围和SMN2基因的相应拷贝数时的P2值分布范围。
统计所述所有样本中已经验证为是静默携带者的样本的7号内含子的g.27134T>G位点的校正后覆盖度P_silent值分布范围,后期可以根据该覆盖度P_silent值分布范围和单个样本的SMN1基因的拷贝数为2的证据,判断单样本是否为静默携带者;
S3、计算单个测试样本的SMN1基因的7号外显子和8号外显子的p1值、SMN2基因的7号外显子和8号外显子的p2值,根据S2计算所得的P1值和P2值的分布范围判断本步骤中p1值和p2值所对应的SMN1基因和SMN2基因的拷贝数;
统计单个测试样本的7号内含子上的g.27134T>G位点的覆盖度p_silent值;根据所述p_silent值和所述单个测试样本的SMN1基因的拷贝数,判断该单个测试样本状态:;
当p_silent值在S2中计算的P_silent值分布范围内且所述单个测试样本的SMN1基因的拷贝数是2时,判断所述单个测试样本为静默携带者;
当p_silent值在S2中计算的P_silent值分布范围内但所述单个测试样本的SMN1基因的拷贝数不是2,判断所述单个测试样本为疑似静默携带者;
其他情况时均判断所述单个测试样本为非静默携带者。
具体实施时,本发明提出所述S1中寻找所述对照区间的步骤包括:如图1所示,
S101、用MLPA平台验证所述所有样本的SMN1基因和SMN2基因的实际拷贝数,使用生信分析流程进行处理后得到Bam文件;
S102、预先筛选出两拷贝基因的Bed区间,统计所述所有样本在全外显子组的Bed区间内的覆盖度;
S103、把所述所有样本的覆盖度校正到100X,得到样本校正后覆盖度;
S104、根据所述所有样本校正后覆盖度计算相关性和方差,查找相关性好且方差值低的前5个Bed区间作为对照区间,在后续计算平均值或中位数时以达到降低校正偏差的作用。
具体实施时,本发明提出所述S2的步骤包括:
S201、统计所述所有样本在SMN1基因和SMN2基因7号外显子和8号外显子的总覆盖度并采用对应的批次内中位数覆盖度进行校正,得到SMN1基因和SMN2基因7号外显子和8号外显子的校正后总覆盖度;
S202、统计所述所有样本在5个所述对照区间的总覆盖度并采用对应的批次内中位数覆盖度进行校正,得到对照区间的校正覆盖度均值;
S203、统计所述所有样本的3个点突变的覆盖度并采用对应的批次内中位数覆盖度进行校正,得到3个点突变的校正后覆盖度;所述3个点突变的覆盖度包括7号外显子上的c.840C>T位点的覆盖度、8号外显子上的c.*239G>A位点的覆盖度和7号内含子上的g.27134T>G位点的覆盖度;根据公式计算SMN1基因的校正后覆盖度在7号外显子、8号外显子的ratio值;计算SMN2基因的的校正后覆盖度在7号外显子、8号外显子的ratio值;
S204、根据所述SMN1基因和SMN2基因7号外显子和8号外显子的校正后总覆盖度、对照区间的校正覆盖度均值、所述ratio值,计算SMN1基因的7号外显子和8号外显子的拷贝数p_e7_s1值和p_e8_s1值;计算SMN2基因的7号外显子和8号外显子的拷贝数p_e7_s2值和p_e8_s2值;根据p_e7_s1值和p_e8_s1值计算p1值;根据p_e7_s2值和p_e8_s2值计算p2值。
具体实施时,SMN1基因在7号外显子上的ratio值和p_e7_s1值的计算公式为:
ratio_e7_s1=rc_e7_s1/(rc_e7_s1+rc_e7_s2);
cn_e7_s1=rc_e7_s1_total/rc_control;
cn_e7_s2=rc_e7_s2_total/rc_control;
p_e7_s1=ratio_e7_s1*(cn_e7_s1+cn_e7_s2)*2;
SMN1基因在8号外显子上的ratio值和p_e8_s1值的计算公式为:
ratio_e8_s1=rc_e8_s1/(rc_e8_s1+rc_e8_s2);
cn_e8_s1=rc_e8_s1_total/rc_control;
cn_e8_s2=rc_e8_s2_total/rc_control;
p_e8_s1=ratio_e8_s1*(cn_e8_s1+cn_e8_s2)*2;
SMN1基因的p1值的计算公式为:
p1=(p_e7_s1+p_e8_s1)/2
SMN2基因在7号外显子上的ratio值和p_e7_s2值的计算公式为:
ratio_e7_s2=rc_e7_s2/(rc_e7_s1+rc_e7_s2);
p_e7_s2=ratio_e7_s2*(cn_e7_s1+cn_e7_s2)*2;
SMN2基因在8号外显子上的ratio值和p_e8_s2值的计算公式为:
ratio_e8_s2=rc_e8_s2/(rc_e8_s1+rc_e8_s2);
p_e8_s2=ratio_e8_s2*(cn_e8_s1+cn_e8_s2)*2;
SMN2基因的p2值的计算公式为:
p2=(p_e7_s1+p_e8_s1)/2
本实施例中所述500个所有样本的SMN1基因的相应拷贝数时的P1值分布范围和SMN2基因的相应拷贝数时的P2值分布范围如下表1:
表1所有样本的SMN1基因的相应拷贝数时的P1和SMN2基因的相应拷贝数时的P2值
500个所有样本中有6个静默携带者的P_silen值的范围是[0.39,0.65]。
具体实施时,本发明提出计算所述S3中单个测试样本的SMN1基因的7号外显子和8号外显子的拷贝数p1、SMN2基因的7号外显子和8号外显子的拷贝数p2的步骤包括:
S301、统计单个测试样本的SMN1基因和SMN2基因上的7号外显子、8号外显子的总覆盖度并通过采用S201中所述的SMN1基因和SMN2基因上的7号外显子和8号外显子的校正后总覆盖度的中位数进行校正后得到单个测试样本的SMN1基因和SMN2基因上的7号外显子和8号外显子的校正后总覆盖度;
统计5个对照区间的覆盖度和3个点突变的覆盖度,并通过采用S202中所述的对照区间的校正覆盖度均值的中位数进行校正后得到5个对照区间的校正覆盖度均值;
统计3个点突变的校正后覆盖度;所述3个点突变的覆盖度包括7号外显子上的c.840C>T位点的覆盖度、8号外显子上的c.*239G>A位点的覆盖度、7号内含子上的g.27134T>G位点的覆盖度,并采用采用S203中所述的3个点突变的校正后覆盖度的中位数进行校正得到3个点突变的校正后覆盖度;
S302、计算SMN1基因和SMN2基因的校正后覆盖度在7号外显子和8号外显子的ratio值;
S303、计算单个测试样本SMN1基因的7号外显子和8号外显子的拷贝数p1值和SMN2基因上的7号外显子和8号外显子的拷贝数p2值。
依上述步骤分别计算500个已知SMN基因实际拷贝数的测试样本的SMN1基因的7号外显子和8号外显子的拷贝数p1值和SMN2基因上的7号外显子和8号外显子的拷贝数p2值如表2所示,并得出SMN1基因和SMN2基因的拷贝数及SMN1 2+0静默携带者的状态。
表2 15个单个测试样本的SMN1基因和SMN2基因的拷贝数及SMN1 2+0静默携带者的状态
通过上表可知,通过本方法计算出的上述15个测试样本的SMN基因实际拷贝数与已知SMN基因实际拷贝数相同,SMN1 2+0的状态与通过RFLP的检测结果一致。验证本发明提供的方法可以精确检测单样本SMN基因拷贝数同时判断样本SMN1 2+0静默携带者的状态。
显然所描述的实施例是本发明的一部分实施例,而不是全部的实施例。基于所描述的本发明的实施例,本领域普通技术人员在无需创造性劳动的前提下所获得的所有其他实施例,都属于本发明保护的范围。除非另作定义,此处使用的技术术语或者科学术语应当为本发明所属领域内具有一般技能的人士所理解的通常意义。
本发明专利申请说明书以及权利要求书中使用的“第一”、“第二”以及类似的词语并不表示任何顺序、数量或者重要性,而只是用来区分不同的组成部分。同样,除非上下文清楚地指明其它情况,否则单数形式的“一个”“一”或者“该”等类似词语也不表示数量限制,而是表示存在至少一个。
- 一种在WES数据中检测单样本SMN基因拷贝数的方法
- 以SMNP作为对照检测SMN基因拷贝数的方法