一种高弹性、高载药量的栓塞微球及其制备方法
文献发布时间:2023-06-19 13:46:35
技术领域
本发明涉及医用高分子材料领域,尤其涉及一种高弹性、高载药量的栓塞微球及其制备方法。
背景技术
肝癌是全球最常见的10种肿瘤之一,而中国是原发性肝癌发病率最高的国家,占全球患者的50%以上。针对原发性肝癌的治疗方法而言,涵盖手术切除、肝移植、微波消融、经肝动脉化疗栓塞(TACE)、放射治疗等治疗方案。临床实践和研究表明,作为经肝动脉栓塞治疗技术,是不能手术切除的中晚期肝癌患者首选和最有效的治疗方案,可以带来更长的生存期,具有显著的临床效果。
目前TACE作用机理主要是通过栓塞剂进行超选择栓塞肿瘤的供血动脉,造成肿瘤供血动脉闭塞以及在二级血管深处注入细胞毒性化疗药物,以达到药物治疗的“首过效应”,并诱导肿瘤病灶缺血坏死。因此,理想的TACE治疗应该是使化疗药物在肿瘤病灶内达到最大和持续浓度,使外周血药浓度低,以及精准栓塞肿瘤病灶的供血动脉。载药微球栓塞技术作为TACE新型的介入治疗方案,由于微球粒径大小分布均一,有利于靶向选择相应的血管,同时微球具备载药及缓慢释放化疗药物功能,达到良好的介入手术效果,因此,制备可载药、可缓控释放化疗药物、生物相容性好的高分子栓塞微球是目前的研究热点。
目前国内外上市的微球栓塞剂有聚乙烯醇栓塞微球、壳聚糖微球、海藻酸钠微球、白蛋白微球等。但是每一种微球产品都具有不同的弹性、压缩性、顺应性等生物化学特性,这些性质对微球产品的功效产生很大的影响,也决定其临床使用范围。具体缺陷及不足如下:(1)例如海藻酸钠微球由于其采用离子交联聚合,导致其弹性较差,不可任意压缩修复,微球在经导管输送后,容易发生微球挤碎,导致栓塞发生漂移风险,同时使血管栓塞不紧密;(2)例如壳聚糖微球具有一定的降解能力,但是其力学性能较差,不能承受血流的冲击,限制其临床应用;(3)而目前市场上已有聚乙烯醇栓塞微球,不具有生物降解性,作为可载药的聚乙烯醇栓塞微球,需要对聚乙烯醇进行改性衍生化,引入较多的毒理学物质,导致其工艺复杂,其次聚乙烯醇栓塞微球的载药量仍比较低,而且其在血管内释放化疗药物较快,使得药物在靶肿瘤组织中滞留时间短,影响治疗效果。同时目前微球粒径范围(100-300um、300-500um)分布宽,粒径不均一,微球尺寸大小无法得到精确调控,不能完全栓塞目标血管,未能实现超选择治疗。这些因素极大地降低了载药微球栓塞剂的临床疗效。
因此,为了克服上述技术的不足,本领域的技术人员致力于开发一种具有极好的可变弹性、载药量高、药物缓控释放时间长、粒径分布均一的新型栓塞微球及其制备方法,使微球与血管完美契合,栓塞更彻底,同时缓慢释放化疗药物,使肿瘤区的药物浓度长时间维持在较高水平,降低体循环中药物浓度,提高治疗效果。
发明内容
有鉴于现有技术的上述缺陷,本发明所要解决的技术问题是如何开发一种具有极好的可变弹性、载药量高、药物缓控释放时间长、粒径分布均一的栓塞微球。
为实现上述目的,本发明提供了一种高弹性、高载药量的栓塞微球,由聚乙烯醇大分子为母体、成环接枝剂、含双键小分子桥连剂、离子型交联剂四者发生无规共聚制备得到,栓塞微球的结构为以小分子交联体伸缩振动、聚乙烯醇大分子为骨架的网络结构,栓塞微球可变弹性高、载药量高、药物缓控释放时间长、粒径分布均一。
一种高弹性、高载药量的栓塞微球的制备方法,包括以下步骤:
步骤1、合成改性的聚乙烯醇衍生物,通过成环接枝剂对聚乙烯醇大分子链上羟基发生化学反应,形成含有稳定六元环和含有双键的功能性侧基的大分子水凝胶结构,得到改性的聚乙烯醇衍生物;
步骤2、合成栓塞微球,反应体系分为分散相和连续相,分散相中由改性的聚乙烯醇衍生物、含双键小分子桥连剂、离子型交联剂、第一引发剂组成;连续相中以油溶性溶剂、表面活性剂、第二引发剂组成,将分散相通过分散装置分散在连续相中,通过流体剪切力,制备得到栓塞微球。
进一步地,步骤1还包括:
步骤1.1、于烧瓶中加入80-120g聚乙烯醇大分子并加500-1000ml水升温至92-98℃溶解,使聚乙烯醇大分子完全溶解;
步骤1.2、冷却至室温后,向其中加入0.5-5g的成环接枝剂,搅拌速度100-300r/min,再向反应体系中加入一定量催化剂,滴加结束后继续搅拌4-24h,待反应体系出现粘稠状,证明形成以配位键或缩醛结构的水凝胶,停止搅拌,采用在线pH检测,并调节反应体系至中性,最后收集产物,经透析、脱盐处理,得到改性的聚乙烯醇衍生物。
进一步地,步骤2还包括:
步骤2.1、将离子型交联剂、第一引发剂、含双键小分子桥连剂、改性的聚乙烯醇衍生物按照5~80:0.5~5:0.1~2:80~100比例投料,搅拌均匀,形成分散相均相液;
步骤2.2、将表面活性剂、第二引发剂、油溶性溶剂按照5~50:800-1000:0.5~2比例投料,搅拌均匀,形成连续相均相液;
步骤2.3、将分散相均相液与连续相均相液按照1:3~30比例分散至反应体系中,搅拌速度100-600r/min,分散相的分散装置为60-1000um点胶针头、50-1000um筛网进行分散液滴,以实现液滴精准控制,使分散相均一化分散;
步骤2.4、待分散相分散均一,在惰性气体保护下,升温使发生自由基聚合反应,然后在40-70℃条件下保温1~5h,反应结束后,洗涤、过滤、干燥,再经过溶胀、灭菌,制备出高弹性、高载药量的栓塞微球。
进一步地,聚乙烯醇大分子聚合度范围在1000-2000,分子量为5-15万,具有良好的生物相容性。
进一步地,成环接枝剂包括含烯硼酸结构衍生物或烯醛结构衍生物,烯硼酸结构衍生物为戊-4-烯-1-基硼酸、乙烯基苯硼酸、4-烯丙基氨甲酰基苯硼酸中的一种,烯醛结构衍生物为丙烯醛、丙烯醛缩二乙醇中的一种。
进一步地,含双键小分子桥连剂为含有单个或两个以上双键结构的衍生物,优选为丙烯酰胺、N,N-亚甲基双丙烯酰胺中的一种。
进一步地,离子型交联剂为含有负电荷的磺酸根衍生物,优选为含有离子型丙烯酰胺单体。
进一步地,含有离子型丙烯酰胺单体为2-丙烯酰胺-2-甲基丙磺酸钠或2-丙烯酰胺基-2-甲基丙磺酸中的一种。
进一步地,栓塞微球粒径为75-125μm、150-250μm、350-450μm、550-650μm、750-850μm或900-1200μm。
本发明技术效果如下:
(1)本发明通过引入含双键小分子桥连剂,使小分子交联的链状结构与聚乙烯醇大分子网状结构相结合,由于链状结构具有一定的伸缩振动,大大提高整体聚乙烯醇栓塞微球的弹性性能。
(2)本发明通过改性的聚乙烯醇大分子侧链基具有稳定的六元环结构,同时其含有双键的功能性侧基与含双键小分子桥连剂、交联剂进行双键聚合,大大提高离子型丙烯酰胺单体的转化率、含量与空间密度,使载药量大大提升。同时由于空间密度提升,其阴离子基团的电场分布均匀,因此药物能够缓慢释放,同时达到梯度释放。
(3)本发明同时引入含有双键的功能性侧基聚乙烯醇大分子、含双键小分子桥连剂、含有双键的离子型交联剂单体,三者发生无规共聚反应,无规共聚比常规聚乙烯醇微球的嵌段共聚相比,其力学性能、耐热性能、透明度都显著提升。
(4)本发明制备栓塞微球粒径均一,通过控制小分子桥连剂比例改变分散相的表面张力,使其在同样流体剪切力作用下,依靠自身表面张力,粒度均匀分布在流动相中,微观上制备微球粒径分布均一。
(5)本发明栓塞微球制备工艺上引入特定的分散装置,将分散相通过局域限制液滴尺寸的装置,有序加入连续相中,宏观上制备微球粒径分布均一;
(6)本发明栓塞微球与血管契合,实现超选择栓塞治疗,同时微球结构中富含阴离子集团,可与带正电荷的化疗药物(如阿霉素、伊立替康等)相互吸引形成离子键而加载较高的化疗药物,当载药微球进入人体后,体液中的其他离子同药物分子竞争,能够进行长时间缓慢释放化疗药物,使肿瘤区的药物浓度维持在较高水平,提高介入治疗效果。
以下将结合附图对本发明的构思、具体结构及产生的技术效果作进一步说明,以充分地了解本发明的目的、特征和效果。
附图说明
图1是本发明的一个较佳实施例的75-125um栓塞微球的20X显微镜照片;
图2是本发明的一个较佳实施例的900-1200um栓塞微球的4X显微镜照片;
图3是本发明的一个较佳实施例的900-1200um栓塞微球的压缩形变受力图;
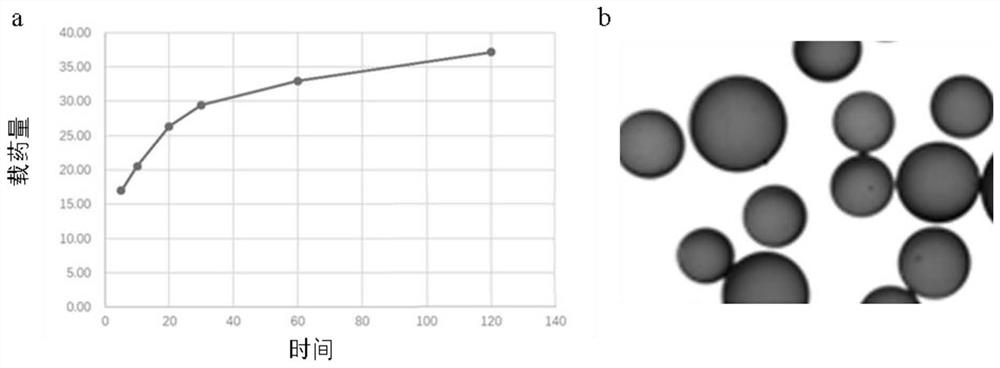
图4是本发明的一个较佳实施例的栓塞微球载药性能测试结果,其中a是载药量结果,b是载药微球载药后的显微镜照片;
图5是本发明的一个较佳实施例的栓塞微球药物释放性能测试结果。
具体实施方式
以下参考说明书附图介绍本发明的多个优选实施例,使其技术内容更加清楚和便于理解。本发明可以通过许多不同形式的实施例来得以体现,本发明的保护范围并非仅限于文中提到的实施例。
实施例1
聚乙烯醇衍生物合成
于反应烧瓶中加入600ml纯化水,再加入100g聚乙烯醇大分子,控制搅拌速度100r/min,搅拌均匀后升温至92-98℃溶解,保温2h,冷却至室温后,形成聚乙烯醇水凝胶。聚乙烯醇大分子聚合度范围在1000-2000,分子量为5-15万,具有良好的生物相容性。
向溶解好的600ml聚乙烯醇水凝胶其中加入1g的乙烯基苯硼酸,搅拌30min以混合均匀,再用1M盐酸调节反应体系pH值至3-4,室温搅拌24h。用截留分子量5000的透析袋在水溶液中透析,去除未反应的小分子量化合物和杂质,减压除去水,待反应体系出现粘稠状,证明形成以配位键生产的水凝胶,停止反应,收集产物,得到改性的聚乙烯醇衍生物;
栓塞微球的合成
称取10g 2-丙烯酰胺-2-甲基丙磺酸钠、0.8g过硫酸钾、1.2g丙烯酰胺于烧杯中,加入磁子,放置于磁力搅拌器上,转速300r/min,搅拌10min,再将烧杯中溶液倒入含有上述制备的100g聚乙烯醇衍生物烧杯中,继续搅拌30min,混合均匀,形成分散相均相液。
在惰性气体保护下,于烧瓶中加入1500ml液体石蜡中,再加入15g吐温80溶液,启动搅拌,转速300r,10min搅拌均匀,形成连续相均相液。
再将上述分散相均相液通过注射器连接的60um点胶针头,50-1000um筛网进行分散液滴,缓慢注入反应体系中,时间约1h,再用移液枪吸取1.5ml四甲基乙二胺,加入体系,开始升高温度至60℃,开始计时,保温2.5h,反应结束后,经过丙酮的洗涤、过滤、干燥,制备75-125um微球粗品,再经过溶胀、灭菌,制备出高弹性、高载药量、粒径均一的75-125um栓塞微球,通过20X显微镜观察微球如图1。
实施例2
聚乙烯醇衍生物合成
于反应烧瓶中加入600ml纯化水,再加入100g聚乙烯醇大分子,控制搅拌速度100r/min,搅拌均匀后升温至92-98℃溶解,保温2h,冷却至室温后,形成聚乙烯醇水凝胶。
向溶解好的500ml聚乙烯醇水凝胶中加入4g的丙烯醛,搅拌60min且速度150r/min,再向反应体系中缓慢滴加60ml浓盐酸,滴加结束后继续搅拌8h,反应结束后,向体系缓慢滴加2mol/L氢氧化钠,采用在线pH监测,调节反应体系至中性7.0,并继续搅拌30min,最后收集产物,经透析、脱盐处理,得到改性的聚乙烯醇衍生物;
栓塞微球的合成
称取10g 2-丙烯酰胺-2-甲基丙磺酸钠、0.8g过硫酸钾、1.2g丙烯酰胺于烧杯中,加入磁子,放置于磁力搅拌器上,转速300r/min,搅拌10min,再将烧杯中溶液倒入含有上述制备的100g聚乙烯醇衍生物烧杯中,继续搅拌30min,混合均匀,形成分散相均相液。
在惰性气体保护下,于烧瓶中加入1500ml液体石蜡中,再加入15g吐温80溶液,启动搅拌,转速300r,10min搅拌均匀,形成连续相均相液。再将上述分散相均相液通过注射器连接的60um点胶针头,缓慢注入反应体系中,时间约1h,再用移液枪吸取1.5ml四甲基乙二胺,加入体系,开始升高温度至60℃,开始计时,保温2.5h,反应结束后,经过丙酮的洗涤、过滤、干燥,制备75-125um微球粗品,再经过溶胀、灭菌,制备出高弹性、高载药量、粒径均一的75-125um栓塞微球。
实施例3
聚乙烯醇衍生物合成
于反应烧瓶中加入600ml纯化水,再加入100g聚乙烯醇大分子,控制搅拌速度100r/min,搅拌均匀后升温至92-98℃溶解,保温2h,冷却至室温后,形成聚乙烯醇水凝胶。
向溶解好的600ml聚乙烯醇水凝胶其中加入1g的乙烯基苯硼酸,搅拌30min以混合均匀,再用1M盐酸调节反应体系pH值至3-4,室温搅拌24h。用截留分子量5000的透析袋在水溶液中透析,去除未反应的小分子量化合物和杂质,减压除去水,待反应体系出现粘稠状,证明形成以配位键生产的水凝胶,停止反应,收集产物,得到改性的聚乙烯醇衍生物;
栓塞微球的合成
称取15g 2-丙烯酰胺-2-甲基丙磺酸钠、0.6g过硫酸钾、0.5gN,N-亚甲基双丙烯酰胺于烧杯中,加入磁子,放置于磁力搅拌器上,转速300r/min,搅拌10min,再将烧杯中溶液倒入含有上述制备的100g聚乙烯醇衍生物烧杯中,继续搅拌30min,混合均匀,形成分散相均相液。
于烧瓶中加入1500ml乙酸丁酯中,再加入20g纤维素物质溶液,启动搅拌,转速150r,10min搅拌均匀,形成连续相均相液。在惰性气体保护下,再将上述分散相均相液通过注射器连接的800um点胶针头,缓慢注入反应体系中,时间约20min,再用移液枪吸取1.0ml四甲基乙二胺,加入体系,开始升高温度至62℃,开始计时,保温3.5h,反应结束后,经过丙酮的洗涤、过滤、干燥,制备900-1200um微球粗品,再经过溶胀、灭菌,制备出高弹性、高载药量、粒径均一的900-1200um栓塞微球,通过显微镜4X观察微球如图2。
实施例4
聚乙烯醇衍生物合成
于反应烧瓶中加入600ml纯化水,再加入100g聚乙烯醇大分子,控制搅拌速度100r/min,搅拌均匀后升温至92-98℃溶解,保温2h,冷却至室温后,形成聚乙烯醇水凝胶。
向溶解好的500ml聚乙烯醇水凝胶中加入4g的丙烯醛,搅拌60min且速度150r/min,再向反应体系中缓慢滴加60ml浓盐酸,滴加结束后继续搅拌8h,反应结束后,向体系缓慢滴加2mol/L氢氧化钠,采用在线pH监测,调节反应体系至中性7.0,并继续搅拌30min,最后收集产物,经透析、脱盐处理,得到改性的聚乙烯醇衍生物;
栓塞微球的合成
称取15g 2-丙烯酰胺-2-甲基丙磺酸钠、0.6g过硫酸钾、0.5gN,N-亚甲基双丙烯酰胺于烧杯中,加入磁子,放置于磁力搅拌器上,转速300r/min,搅拌10min,再将烧杯中溶液倒入含有上述制备的100g聚乙烯醇衍生物烧杯中,继续搅拌30min,混合均匀,形成分散相均相液。
于烧瓶中加入1500ml乙酸丁酯中,再加入20g纤维素物质溶液,启动搅拌,转速150r,10min搅拌均匀,形成连续相均相液。在惰性气体保护下,再将上述分散相均相液通过注射器连接的800um点胶针头,缓慢注入反应体系中,时间约20min,再用移液枪吸取1.0ml四甲基乙二胺,加入体系,开始升高温度至62℃,开始计时,保温3.5h,反应结束后,经过丙酮的洗涤、过滤、干燥,制备900-1200um微球粗品,再经过溶胀、灭菌,制备出高弹性、高载药量、粒径均一的900-1200um栓塞微球。
实施例1-4制备得到的栓塞微球性能测试
1、微球弹性性能测试
选用物性测定仪来测试900-1200um微球的强度和弹性。测定过程中,设置力量感应元、合适探头、选择压缩速度0.2mm/s、压缩形变80%,保持时间10s,返回速度0.2mm/s。保证数据的可重复性,可用性。如图3结果显示,微球压缩形变80%未破碎,且微球受力台阶平稳显示其保持力良好,证明其优异的弹性性能。
2、微球载药性能测试
称取0.25g 100-150um湿的栓塞微球,放入20mL西林瓶中,加入4ml 2.5mg/ml的上阿霉素溶液,振荡摇匀。分别第5、10、20、30、60、120min定时取样40uL,加入5mL生理盐水中稀释,在485nm处测定吸光度。将所测的吸光度代入标准曲线方程,计算微球载药量。结果如图4a所示,微球最大载药量达38mg/g;且载药微球仍保持较好的球形,如图4b所示。随着更优条件的制备,微球载药量可进一步提升。
3、载药微球药物释放性能测试
使用已载阿霉20mg/g微球(载药量阿霉素/克微球),置于250mL三角瓶中,加入100mLpH7.4的PBS磷酸盐缓冲液,在37℃水浴中搅拌释放。分别在30、60、90、120、180、240min定时取1mL释放液,加入3mL生理盐水稀释,于485nm测定吸光度,将测得的吸光度代入阿霉素标准曲线方程式,计算药物释放量。结果如图5所示,载药微球缓慢释放药物,且释放线性较好,因此制备微球能够达到缓控释放,在临床应用上,能够在病灶处进行长时间缓慢释放化疗药物,且药物的突释浓度不高,大大降低对周围组织的炎症反应,具有重要意义。
以上详细描述了本发明的较佳具体实施例。应当理解,本领域的普通技术无需创造性劳动就可以根据本发明的构思作出诸多修改和变化。因此,凡本技术领域中技术人员依本发明的构思在现有技术的基础上通过逻辑分析、推理或者有限的实验可以得到的技术方案,皆应在由权利要求书所确定的保护范围内。