一种乐伐替尼衍生物及其制备方法和用途
文献发布时间:2024-04-18 19:58:26
技术领域
本发明属于医药技术领域,具体地说,涉及一种乐伐替尼衍生物及其制备方法和用途。
背景技术
乐伐替尼是由日本卫材公司研发的多靶点酪氨酸激酶抑制剂,可适用于甲状腺癌、肾癌、神经胶质瘤、肝癌、卵巢癌等实体瘤的临床治疗(药学学报,2010(2):16)。乐伐替尼的主要作用为抑制一种酪氨酸激酶,这种酪氨酸激酶主要影响并参与肿瘤细胞的增殖及致癌信号通路,同时乐伐替尼还能够选择性的抑制血管内皮生长因子(VEGF)受体的激酶活性。乐伐替尼作为多靶点酪氨酸激酶抑制剂,包括维持正常细胞功能在内,乐伐替尼还能起到抑制病理性血管、肿瘤的生成,延缓癌症进展的作用(中国药物化学杂志,2015(4):332)。乐伐替尼2015年2月获FDA批准上市。2015年7月乐伐替尼又获得治疗晚期和/或转移性肾细胞癌的突破性药物资格。
乐伐替尼作为一种多靶点的蛋白酶抑制剂,服用时对人体正常细胞也会产生危害,因此会产生副作用。乐伐替尼常见的副作用主要包括高血压、心功能不全、动脉血栓栓塞等。因此,本发明的目的是提供一种具有与乐伐替尼相近的酪氨酸激酶抑制活性且副作用较小的乐伐替尼衍生物。
发明内容
本发明的目的是提供一种乐伐替尼衍生物,该化合物具有通式(I)所示的结构:
其中,R1、R2分别独立地选自对位、邻位或者间位取代的H或者卤素中的任意一种;X为O或S;R3为H或氨基酸。
该化合物或其药学可接受的盐具有酪氨酸激酶抑制活性。
优选地,上述卤素选自F、Cl、Br中的任意一种。
优选地,R3选自苯丙氨酸、蛋氨酸、缬氨酸、丙氨酸、赖氨酸、异亮氨酸、甘氨酸、和脯氨酸中的任意一种。
优选地,本发明的化合物包括
本发明的另一目的是提供一种该化合物或其药学上可接受的盐的制备方法,该制备方法包括以下步骤:以4-氨基苯酚为原料,通过二碳酸二叔丁酯对氨基进行保护,再与6-氯烟酰胺进行反应,后续脱除酰化后再与苯胺反应制得所述的化合物。
优选地,该制备方法还包括以下步骤:将上述反应制得的化合物在缩合剂的作用下与氨基酸反应,得到所述化合物的氨基酸衍生物,反应式如下所示:
本发明的又一目的是提供一种药物组合物,该药物组合物含有上述化合物或其药学上可接受的盐。
本发明还有一个目的是提供上述化合物或其药学可接受的盐在制备治疗肿瘤药物中的应用。
优选地,上述肿瘤选自:胰脏癌、胃癌、大肠癌、乳癌、前列腺癌、肺癌、肾癌、脑肿瘤、血液癌、卵巢癌、肝癌或甲状腺癌中的任意一种。
本发明的乐伐替尼衍生物化合物具有与乐伐替尼相似的抗肿瘤细胞增殖活性,并且对正常细胞的毒性更小。具有优异的抗癌作用和良好的安全性,可用于制备治疗肿瘤的药物。
附图说明
图1:本发明化合物与对照组乐伐替尼对小鼠前列腺癌细胞RM-1增殖的影响。
具体实施方式
以下将结合部分实施例进一步地说明本发明的技术方案,下述实施例不构成对本发明的任何限制。
本发明的试剂材料均为市售产品,均为分析纯。
实施例1化合物1的合成
第一步:向500mL四氢呋喃中加入4-羟基苯胺10.9g(10mol),二羰基二叔丁酯26.1g(12mol)以及三乙胺20.2g(20mol),室温搅拌4个小时后,将反应液用500mL水洗涤,加入200mL石油醚室温打浆一小时,加入无水硫酸钠干燥,真空减压旋干溶剂,得到化合物(A)19.6g,收率为94%。
1H NMR(400MHz,DMSO-d6)9.71(s,1H),9.32(s,1H),7.40-7.33(m,2H),6.28-6.23(m,2H),1.52(s,9H)。
第二步:向200mL二甲基亚砜中加入6-氯烟酰胺15.7g(10mol),化合物(A)25.1g(12mol)以及碳酸铯65.1g(20mol),90℃下搅拌过夜,反应液降至室温后,倒入1000mL水中,搅拌1个小时,过滤,干燥得到化合物(B)30.3g,收率为92%。
1H NMR(400MHz,DMSO-d6)9.85(s,1H),8.24-7.96(m,2H),9.85(s,2H),7.28-6.98(m,5H),1.52(s,9H)。
第三步:向50mL甲醇中加入化合物(B)32.9g(10mol),加入2N的盐酸50mL,室温下搅拌6h后向反应液中加入200mL乙醚,持续搅拌1小时,抽滤,得到固体,干燥得到化合物(C)26.5g,收率为86%。
1H NMR(400MHz,DMSO-d6)9.02(s,1H),7.86-7.62(m,3H),7.40-7.33(m,2H),7.21(s,2H),7.13-6.87(m,3H),5.88(s,2H)。
第四步:向300mL四氢呋喃中加入化合物(C)30.8g(10mol)、苯胺13.9g(15mol)、N,N'-羰基二咪唑24.3g(15mol)和三乙胺20.4g(20mol),60℃加热回流搅拌过夜。反应液用800mL水洗涤,萃取,无水硫酸钠干燥,减压蒸出溶剂,甲醇重结晶,得到化合物1固体29.2g,收率84%。
1H NMR(400MHz,DMSO-d6)8.76(s,1H),8.35(s,1H),8.21-7.89(m,2H),8.03(s,2H),7.40-7.07(m,6H),6.92(m,1H),6.88-6.84(m,3H)。
化合物1的合成路线如下所示:
实施例2化合物2的合成
0℃条件下,将N,N-二甲基甘氨酸1.8g(17.4mmol)、1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐4.0g(20.9mmol)、4-二甲氨基吡啶(1.6g,13.0mmol)加入到60mL的无水四氢呋喃中,反应30分钟后,将化合物1 3.0g(8.7mmol)加入反应体系,转移至室温,反应24小时。反应液过滤,向滤液中加入200mL1mol/L的稀盐酸溶液,搅拌,抽滤,滤液用饱和碳酸钠溶液调节pH至10,析出大量白色固体,抽滤,100mL水洗滤饼,滤饼烘干得粗品。将粗品溶于2mol/L的稀盐酸100mL中,过滤除去不溶物,滤液再用饱和碳酸钠调节pH至10,析出大量白色固体,抽滤,100mL水洗滤饼,滤饼烘干。将固体溶于80mL无水丙酮中,加入浓盐酸2mL,室温搅拌过夜,析出产品盐酸盐,抽滤,滤饼用50mL无水丙酮洗涤,滤饼于55℃烘箱中干燥。得到化合物2固体3.42g,收率94%。LC-MS:420.1668[M+H]+。
实施例3化合物3的合成
以化合物1和2-吗啉基乙酸为原料,参照实施例2的方法获得化合物3,LC-MS:476.1926[M+H]+,合成路线如下所示。
实施例4化合物的体外酪氨酸激酶的抑制活性筛选
将一系列梯度浓度的受试化合物,在室温条件下与特定浓度的酪氨酸激酶溶液共同孵育5min,之后加入适量的酶反应底物、ATP,启动酶反应过程,30min后,向酶反应体系中加入适量的反应终止液和检测液,孵育1h后,在多功能酶标仪上,测定特定化合物浓度下的酶活力,并计算不同浓度的化合物对酶活力的抑制活性,之后根据四参数方程,拟合不同浓度的化合物对酶活力的抑制活性,计算出IC50值,结果见表1。
表1化合物对体外酪氨酸激酶的抑制活性
由表1可知,本发明化合物的体外酪氨酸激酶的抑制活性明显低于乐伐替尼。提示本发明的化合物在该实验条件下的体外酪氨酸激酶抑制活性不如乐伐替尼。
实施例5化合物的细胞毒性测试
本发明采用CCK8法测定乐伐替尼和实施例化合物1-3对人的肝癌细胞HepG-2的作用。
实验步骤:
(1)细胞复苏:将人肝癌细胞HepG-2的冻存管从液氮中取出,置于39℃水浴条件下,使之快速融化,然后转移到15mL离心管中,加入10mL含10%FBS的培养液,以1000r/min的转速离心5min,弃除培养基,重新加入含10%FBS和双抗的培养液,转移到培养瓶中培养。
(2)细胞接种:取处于对数生长期的细胞,去除培养瓶中的培养液,用PBS将细胞润洗一遍,膜酶消化后离心,用含10%胎牛血清的培养基重悬,计数并调整到合适浓度(细胞密度:5*10
(3)化合物处理:将乐伐替尼和本发明的三个化合物,即化合物1、化合物2、化合物3分别配置成5组加药组,浓度分别为0.01μmol/L,0.05μmol/L,0.2μmol/L,1μmol/L,5μmol/L,分别取200μL加药组溶液到步骤(1)的96孔板相应孔中,置于二氧化碳细胞培养箱中培养72小时。
(4)细胞活力检测:去除培养基,加入CCK8,每孔10μL,置于二氧化碳培养箱培养3小时,酶标仪450nm处读取吸光值。
(5)计算增殖细胞抑制率:
计算方法:抑制率(%)=[(读数Vehicle-读数compound)/(读数Vehicle-读数compound)]*100%。
实验结果:
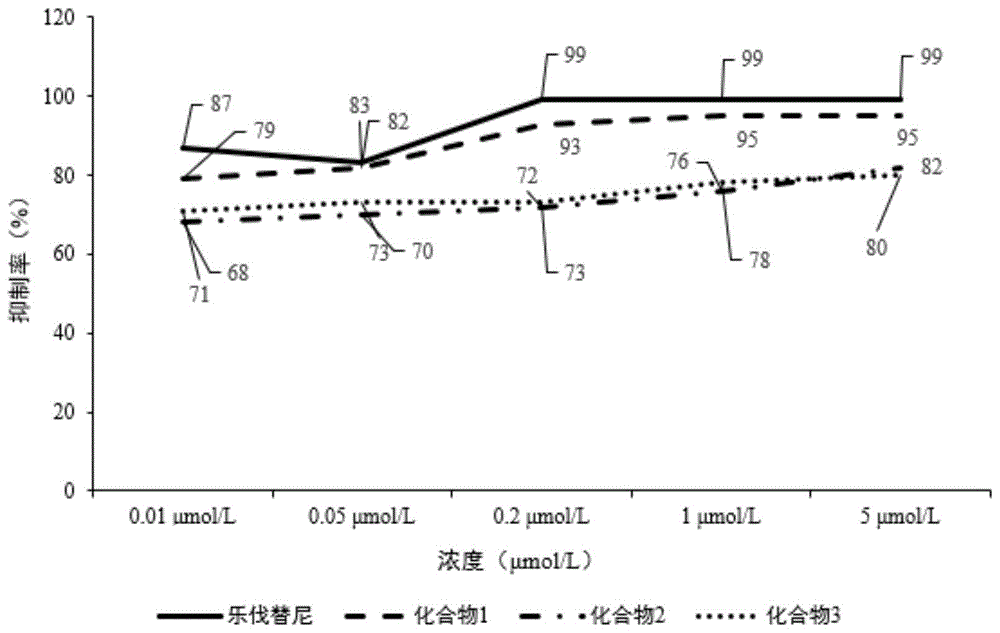
实验结果如表2和图1所示,其中表2的数据分别在图1中标明,更直观地显示实验结果。对照药物乐伐替尼及本发明的化合物1、2和3的细胞增殖抑制率均高达50%以上,具有显著的抗细胞增殖的作用,因此具有显著的体外抗肿瘤活性。其中本发明的化合物1,化合物2,化合物3的效果虽然不如乐伐替尼,但也具有显著的体外抗肿瘤活性,具有进一步的研究价值。
表2化合物对细胞增殖的抑制率
综上所述,乐伐替尼衍生物虽然体外酪氨酸激酶的抑制活性和细胞增殖抑制率都稍差于乐伐替尼,但由于乐伐替尼药物副作用大,而衍生物仍具有较好的生物活性,可视为新化学结构的多靶点抑制剂和抗肿瘤药物。
以上实例只为说明本发明的技术构思及特点,其目的在于让熟悉此项技术的人是能够了解本发明的内容并据以实施,并不能以此限制本发明的保护范围。凡根据本发明精神实质所做的等效变换或修饰,都应涵盖在本发明的保护范围之内。
- 一种吡唑并嘧啶衍生物及其制备方法和在药物制备中的用途
- 一种吡喃衍生物的用途及其制备方法
- 一种四氢异喹啉类衍生物的制备方法及用途
- 一种6-取代平板霉素衍生物、制备方法、药物制剂及用途
- 一种噻唑酰胺衍生物及其制备方法和用途
- 一种乐伐替尼衍生物及制备方法和用途
- 一种乐伐替尼衍生物及制备方法和用途