2-(4-甲氧苯基)-3-羟基-2,3-二氢-1,5-苯并硫氮杂卓酮的制备方法
文献发布时间:2023-06-19 09:44:49
技术领域
本发明涉及一种化合物的制备方法,尤其涉及一种地尔硫卓手性中间体的制备方法,属于化学制药技术领域。
背景技术
地尔硫卓(diltiazem) 是苯并硫氮卓类钙拮抗剂,属于选择性钙通道阻滞剂,化学名为顺-(+)-5-[(2-二甲基氨基)乙基]-2-(4-甲氧基苯基)-3-乙酰氧基-2,3-二氢-1,5-苯并硫氮杂卓-4-(5H)-酮盐酸盐。地尔硫卓由日本田边公司七十年代初研发并上市。目前合成地尔硫卓的路线主要是先合成D-cis-2-(4-甲氧苯基)-3-羟基-2,3-二氢-1,5-苯并硫氮杂卓-4(5H)-酮(即2-(4-甲氧苯基)-3-羟基-2,3-二氢-1,5-苯并硫氮杂卓酮),再通过N-烷基化、O-乙酰化两步反应得到产品。
作为地尔硫卓合成的关键手性中间体2-(4-甲氧苯基)-3-羟基-2,3-二氢-1,5-苯并硫氮杂卓酮的合成现有合成方法报道主要有:
(一) 以大茴香醛和氯乙酸甲酯(氯代乙酸光学结构的酯)缩合,再和邻氨基苯硫酚取代环合:
WO2014030106;Tetrahedron:Asymmetry,Volume 20Issue 24,Pages 2854-2860;中国药物化学杂志,Volume 19,Issue 6,Pages 412-415;Journal of Organic Chemistry,Volume 57,Issue 3,Pages 851-856;
以上路线中间需要拆分,总体收率不高。
(二)不以大茴香醛为起始原料,如Synthesis,Issue 15,Pages 2405-2409,以2-氯-3-(4-甲氧基苯基)-3-氧代-丙酸甲酯为起始原料;WO2004058734和US20040127704以4-碘苯甲醚为起始原料;WO 9413828以3-(4-甲氧基苯基)-丙烯酸乙酯为起始原料等。这些路线大多路线较复杂,且反应需要使用昂贵的试剂,并且成本都比高。
发明内容
为了解决上述技术问题,本发明的目的在于提供一种地尔硫卓合成的手性中间体的制备方法。
为了实现上述目的,本发明提供了一种地尔硫卓合成的手性中间体(2-(4-甲氧苯基)-3-羟基-2,3-二氢-1,5-苯并硫氮杂卓酮)的制备方法,该制备方法包括:
在氮气保护下,加入邻氨基苯硫酚、第一溶剂、有机碱,搅拌溶解后,控制温度为-30℃至30℃,滴加丙炔酸甲酯,搅匀,-30℃至30℃,反应0.3-5小时,至邻氨基苯硫酚完全反应 ,蒸去低沸点物质,升温至90℃-180℃,反应0.5-6小时,冷却,水洗,浓缩冷却,析出晶体,抽滤,烘干,得到中间产物1;
向第二溶剂、催化剂一中加入中间产物1,控制反应温度为-30℃至60℃,滴加过氧化叔丁醇,反应完全,过滤,回收催化剂,洗涤,浓缩,得到中间产物2;
氮气保护下,将中间产物2和第三溶剂搅拌均匀,控制反应温度为-30至30℃,滴加4-甲氧基苯基负离子溶液,反应完全,控制反应温度为0℃-30℃,加入苯甲酸、偶氮二甲酸二乙酯和三苯基膦,控温反应完全,滴加稀酸溶液,分层,分液,萃取,后处理得到中间产物3;
将中间产物3与溶剂混合,加入无机弱碱,40℃-100℃下反应1-6小时,冷却,抽滤,浓缩,得到2-(4-甲氧苯基)-3-羟基-2,3-二氢-1,5-苯并硫氮杂卓酮。
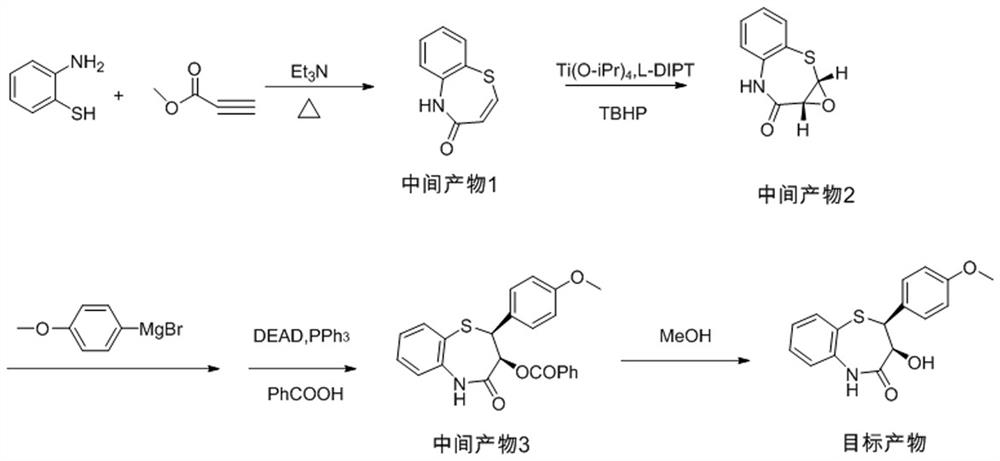
本发明的地尔硫卓合成的手性中间体(2-(4-甲氧苯基)-3-羟基-2,3-二氢-1,5-苯并硫氮杂卓酮)的制备方法以邻氨基苯硫酚和丙炔酸甲酯为原料先取代、再环合得中间产物1,中间产物1经过烯烃的不对称环氧化得到中间产物2,中间产物2再和苯负离子反应,再构型翻转、水解得到地尔硫卓手性中间体。合成线路如图1所示。
本发明的地尔硫卓手性中间体(2-(4-甲氧苯基)-3-羟基-2,3-二氢-1,5-苯并硫氮杂卓酮)的制备方法,具体包括以下步骤:
第一步:以邻氨基苯硫酚和丙炔酸甲酯为原料,在较低温度取代,再在较高温度下环合得到中间产物1;
第二步:中间产物1经过不对称环氧化得到中间产物2;
第三步:中间产物2再和苯负离子反应,邻位再构型翻转;
第四步:水解得到产品。
其中第一步取代环合收率75-80%,第二步不对称氧化收率75-80%,第三步:碳负离子取代、构型翻转反应收率70-85%。第四步的收率为95%-98%。该制备方法中环合步骤得到的Z构型烯烃为空间张力最小,所以选择性高,不对称环合氧化,立体选择性和收率都较高,然后为碳负离子SN2反应,构型单一,再邻位构型翻转,所以避开了手性拆分,同时路线合理,总收率较高。
在本发明中,制备中间产物1时,是在氮气保护下,反应瓶中加入邻氨基苯硫酚、第一溶剂、催化量的有机碱,控温,滴加丙炔酸甲酯,搅匀,保温反应至邻氨基苯硫酚检测反应完全,升温,蒸去低沸点物质和未反应的丙炔酸甲酯,再升温,控温反应完全,冷却,水洗,浓缩冷却,析出晶体,抽滤,烘干,得中间产物1。如图2所示。
在本发明的一具体实施方式中,第一溶剂为沸点为150℃以下芳烃或取代芳烃;优选第一溶剂为甲苯、氯苯、二甲苯中的一种或几种的组合。
在本发明的一具体实施方式中,有机碱为极性非质子试剂;优选有机碱为三乙胺、N,N-二甲基甲酰胺、N,N-二甲基苯胺中的一种或几种的组合。
在本发明的一具体实施方式中,邻氨基苯硫酚、有机碱、第一溶剂、丙炔酸甲酯的当量比为1.0eq:0.01eq-0.2eq:2-30eq:1eq-1.3eq。
在本发明中,制备中间产物2时,在反应器中,加入溶剂,催化剂、中间产物1,控温,控制内温然后滴加入过氧化叔丁醇,反应完全,过滤,用水洗涤,浓缩,得到中间产物2。如图3所示。
在本发明的一具体实施方式中,第二溶剂为C5以下卤代烃或四氢呋喃;优选C5以下卤代烃为二氯甲烷、三氯甲烷、1,2-二氯乙烷中的一种或几种的组合。
在本发明的一具体实施方式中,所述催化剂一为四异丙醇钛与L-(+)-酒石酸二异丙酯的混合物,四异丙醇钛与L-(+)-酒石酸二异丙酯的质量比根据实际需要调整(可以为1:0.1~9)。
在本发明的一具体实施方式中,第二溶剂、催化剂一、中间产物1、过氧化叔丁醇的当量比为2-30eq:0.01eq-0.2eq:1.0eq:1eq-1.5eq。
在本发明中,制备中间产物3时,反应瓶中,加入中间产物2和第三溶剂,控温,4-甲氧基苯基负离子溶液,反应完全,加入苯甲酸、偶氮二甲酸二乙酯、三苯基膦,反应完全,缓慢滴加稀盐酸,分液,萃取相,有机相旋干,得到中间产物3。如图4所示。
在本发明的一具体实施方式中,4-甲氧基苯基负离子溶液为(4-甲氧基苯基)溴化镁的四氢呋喃溶液或4-甲氧基苯基锂离子溶液。
其中,(4-甲氧基苯基)溴化镁的四氢呋喃溶液按照以下步骤制备得到的:
氮气保护下,加入溶剂、镁屑、碘,升温到30℃-80℃,加入少量4-甲氧基溴苯,引发后,30℃-80℃滴加4-甲氧基溴苯,回流0.5-2小时,得到(4-甲氧基苯基)溴化镁的四氢呋喃溶液。
在本发明的一具体实施方式中,制备(4-甲氧基苯基)溴化镁的四氢呋喃溶液时,溶剂、镁屑、碘、4-甲氧基溴苯当量比2eq-30eq:1.0eq-1.3eq:0.01eq-0.05eq:1eq。
在本发明的一具体实施方式中,第三溶剂为四氢呋喃和/或沸点120℃以下的醚类;优选沸点120℃以下的醚类为乙醚、异丙醚、甲基叔丁基醚中的一种或几种的组合。
在本发明的一具体实施方式中,中间产物2、第三溶剂、4-甲氧基苯基负离子溶液、苯甲酸、偶氮二甲酸二乙酯、三苯基膦、稀酸的当量比为1.0eq:2eq-30eq:1.0-1.2eq:1eq-1.2eq:1eq-5eq:1eq-5eq:2.0-10.0eq。
在本发明中,制备最终产物2-(4-甲氧苯基)-3-羟基-2,3-二氢-1,5-苯并硫氮杂卓酮时,是在反应瓶中,加入中间产物3和溶剂,加入无机弱碱,反应完全,后处理,得到地尔硫卓手性中间体(2-(4-甲氧苯基)-3-羟基-2,3-二氢-1,5-苯并硫氮杂卓酮)。如图5所示。
在本发明的一具体实施方式中,无机弱碱为碳酸钠、碳酸钾、碳酸氢钠等中的一种或几种的组合。
在本发明的一具体实施方式中,中间产物3、溶剂、无机弱碱的当量比为1.0eq:2eq-30eq:0.5eq-2.5eq。
本发明的2-(4-甲氧苯基)-3-羟基-2,3-二氢-1,5-苯并硫氮杂卓酮的选择性合成方法,旨在路线新颖环保、经济、提高原料使用效率,更加适应生产需要。而且本发明的制备方法的产品从收率高,产品纯度高。
本发明的2-(4-甲氧苯基)-3-羟基-2,3-二氢-1,5-苯并硫氮杂卓酮的制备方法,中间产物1成环过程中,Z结构烯烃张力小,所以收率高,后续环氧化为不对称环氧化反应,得到中间产物2光学纯度高,产率高,中间产物2和4-甲氧基苯基负离子为SN2反应,再邻位经过构型翻转,得到产物。整个应过程,无需拆分,且路线总收率较高、经济。
附图说明
图1为本发明的地尔硫卓手性中间体的合成线路图。
图2为本发明的中间产物1的合成线路图。
图3为本发明的中间产物2的合成线路图。
图4为本发明的中间产物3的合成线路图。
图5为本发明的地尔硫卓手性中间体(2-(4-甲氧苯基)-3-羟基-2,3-二氢-1,5-苯并硫氮杂卓酮)的合成线路图。
图6为本发明实施例5的地尔硫卓手性中间体(2-(4-甲氧苯基)-3-羟基-2,3-二氢-1,5-苯并硫氮杂卓酮)的核磁谱图。
具体实施方式
实施例1
中间产物1:
1000ml反应瓶,氮气保护,加入邻氨基苯硫酚(62.6g,0.5mol,1.0eq)、甲苯500ml、三乙胺(10.1g,0.1mol,0.2eq),降温到-18℃,滴加入丙炔酸甲酯(46.2g,0.55mol,1.1eq),控制温度-18℃,搅匀,保温半小时,至邻氨基苯硫酚检测反应完全,升温,蒸去低沸点物质,再升温至甲苯回流,反应2小时,冷却,水洗,浓缩冷却,析出晶体,抽滤,烘干,中间产物1,69.9g,收率78.9%,纯度99.2%。
实施例2
中间产物2:
1000ml反应器中,加入二氯甲烷500ml,催化剂一(10g,四异丙醇钛和L-(+)-酒石酸二异丙酯按质量比1:1组成的混合物),加入中间体1(53g,0.3mol,1.0eq),冷却到0℃,控制内温0℃然后滴加入过氧化叔丁醇(28.4g,0.315mol,1.05eq)反应半小时,过滤,用水洗涤,浓缩,得到中间产物2(44.1g,0.228mol),收率76%,纯度98.5%。
实施例3
1000ml的反应瓶中,氮气保护下,加入四氢呋喃300ml,加入镁屑12g,碘0.1g,加入少量4-甲氧基溴苯,引发后,回流滴加4-甲氧基溴苯,共计加入4-甲氧基溴苯(39.3g,0.21mol),回流1小时,得到(4-甲氧基苯基)溴化镁的四氢呋喃溶液。
实施例4
中间产物3
1000ml反应瓶中,把中间体2(38.6g,0.2mol)和四氢呋喃200ml加入,降温到-10℃,滴加上述实施例3的溶液(含(4-甲氧基苯基)溴化镁:0.21mol,1.05eq),反应半小时,反应完全,加入苯甲酸(25.6g,0.21mol,1.05eq),再加入偶氮二甲酸二乙酯(DEAD)(70g,0.4mol,2.0eq),三苯基膦(105g,0.4mol,2.0eq),低温搅拌反应,过滤,缓慢滴加稀盐酸,分层,二氯甲烷萃取有机相,有机相旋干,得到中间产物3(63.2g,0.156mol),收率78%,纯度98%。
实施例5
500ml反应瓶中,加入中间产物3(63.2g,0.156mol,1.0eq),加入甲醇300ml,加入碳酸钠(9.1g,0.086mol,0.55eq),搅拌升温至内温50℃,反应2小时,冷却,抽滤,减压蒸去甲醇,得到产品地尔硫卓手性中间体(2-(4-甲氧苯基)-3-羟基-2,3-二氢-1,5-苯并硫氮杂卓酮)(45.2g,0.15mol)收率96%,纯度99%。地尔硫卓手性中间体(2-(4-甲氧苯基)-3-羟基-2,3-二氢-1,5-苯并硫氮杂卓酮)的核磁谱图如图6所示:1HNMR(CDCl
对比例1
中间产物1:
1000ml反应瓶,氮气保护,加入邻氨基苯硫酚(62.6g,0.5mol,1.0eq)、甲苯500ml、三乙胺(10.1g,0.1mol,0.2eq),降温到15℃,滴加入丙炔酸甲酯(46.2g,0.55mol,1.1eq),控制温度15℃,搅匀,保温半小时,至邻氨基苯硫酚检测反应完全,升温,蒸去低沸点物质,再升温至甲苯回流,反应2小时,冷却,水洗,浓缩冷却,析出晶体,抽滤,烘干,中间产物1,收率75.8%,纯度98.5%。
对比例2
中间产物2:
1000ml反应器中,加入二氯甲烷500ml,催化剂一(10g,四异丙醇钛和L-(+)-酒石酸二异丙酯按质量比1:1组成的混合物),加入实施例1的中间体1(53g,0.3mol,1.0eq),冷却到10℃,控制内温10℃然后滴加入过氧化叔丁醇(28.4g,0.315mol,1.05eq)反应半小时,过滤,用水洗涤,浓缩,得到中间产物2,收率70%,纯度98%。
对比例3
中间产物3
1000ml反应瓶中,把实施例2的中间体2(38.6g,0.2mol)和四氢呋喃200ml加入,降温到5℃,滴加上述实施例3的溶液(含(4-甲氧基苯基)溴化镁:0.21mol,1.05eq),反应半小时,反应完全,加入苯甲酸(25.6g,0.21mol,1.05eq),再加入偶氮二甲酸二乙酯(DEAD)(70g,0.4mol,2.0eq),三苯基膦(105g,0.4mol,2.0eq),低温搅拌反应,过滤,缓慢滴加稀盐酸,分层,二氯甲烷萃取有机相,有机相旋干,得到中间产物3,收率75%,纯度96%。
对比例4
500ml反应瓶中,加入实施例4的中间产物3(63.2g,0.156mol,1.0eq),加入甲醇300ml,加入碳酸钠(9.1g,0.086mol,0.55eq),搅拌升温至内温60℃,反应2小时,冷却,抽滤,减压蒸去甲醇,得到产品地尔硫卓手性中间体(2-(4-甲氧苯基)-3-羟基-2,3-二氢-1,5-苯并硫氮杂卓酮),收率95.5%,纯度99%。
上述实施例只为说明本发明的技术构思及特点,其目的在于让熟悉此项技术的人士能够了解本发明的内容并据以实施,并不能以此限制本发明的保护范围。凡根据本发明精神实质所作的等效变化或修饰,都应涵盖在本发明的保护范围之内。
- 2-(4-甲氧苯基)-3-羟基-2,3-二氢-1,5-苯并硫氮杂卓酮的制备方法
- (2S-顺)-(+)-2,3-二氢-3-羟基-2-(4-甲氧苯基)-1,5-苯并硫氮杂卓-4(5H)-酮在制备治疗肝炎的药物中的用途