一种级联放大CRISPR-Dx检测体系
文献发布时间:2024-05-31 01:29:11
技术领域
本发明涉及核酸检测技术领域,特别涉及一种级联放大CRISPR-Dx检测体系。
背景技术
核酸检测技术(CRISPR-based Diagnostics,CRISPR-Dx)是基于CRISPR-Cas12和CRISPR-Cas13家族的反式切割特性开发的检测技术。
长期以来,聚合酶链式反应(PCR)及其衍生技术如常规PCR、荧光定量PCR、数字微滴PCR等一直是核酸检测金标准。但PCR反应需要变性、退火、延伸三个步骤,对设备和操作人员要求较高、需要高温条件、容易出现非特异性扩增、反应时间较长(1~2小时),使其应用局限于医疗检验机构。重组酶聚合酶等温扩增技术(RPA)检测反应较快(20分钟),可以在常温下反应,但其特异性较差,易出现非特异性扩增,易受环境核酸污染的影响而出现假阳性结果。CRISPR-Dx可以在室温条件下快速反应,且拥有良好的特异性,但单独使用时灵敏度不足,在核酸浓度较低时易出现漏诊。
由于CRISPR-Dx的灵敏度仍有待优化,有研究团队将其与RPA相结合,达到了显著提高了灵敏度的目的。可惜的是RPA易出现非特异性扩增,导致结合后的检测技术精确性降低。
现有技术倾向于采用级联放大的方法,通过将与模板结构类似的脱氧核糖核酸(DNA)底物加入反应体系,达到无需扩增而可以以极高的灵敏度和特异性极低浓度DNA模板的效果,如:1、通过加入DNA/RNA底物达到级联放大效果,但检测限为pM级,灵敏度不足,且反应时间需要100分钟,需要cas12、cas13两种酶、DNA/RNA底物成本较高;2、将有信号放大功能的Csm6酶与Cas13酶相串联,可以在20分钟内实现对低至50aM核酸的检测,但将酶串联的成本极高;3、通过加入含有探针的RNA底物达到级联放大效果,检测限低至5aM,但反应时间需要4小时,且底物结构较复杂,RNA探针合成成本较高。
发明内容
本发明目的在于公开了一种级联放大CRISPR-Dx检测体系,以解决现有技术中所存在的一个或多个技术问题,提供至少一种有益的选择或创造条件。
为实现上述目的,本发明通过以下技术方案实现:
本发明第一方面在于提供一种级联放大CRISPR-Dx检测体系。
本发明第一方面提供的检测体系包括底物片段、通用CrRNA和Lbcas12a,所述底物片段的核苷酸序列如SEQ ID No:1或SEQ ID No:2所示,所述通用CrRNA的核苷酸序列如SEQID No:3所示。所述底物片段依次包括第一互补区、环形区和第二互补区,所述第一互补区和所述第二互补区互补配对。所述Lbcas12a与所述通用CrRNA结合后形成通用CrRNA-Lbcas12a复合物。通用CrRNA-Lbcas12a复合物对底物片段所述环形区的单链进行切割,使所述底物片段结构发生改变。结构改变后的底物可以作为模板被通用CrRNA识别,从而进一步激活更多通用CrRNA-Lbcas12a复合物,实现级联放大反应。
所述底物片段的核苷酸序列为:
5’-GGGGGCAACGGATATGACTGGGACAACTTTTTTTTTTTTTTTTATTTCCCAGTCATATCCG-3’(SEQ ID No:1)或,
5’-GGGGGCAACGGATATTACTGGGACAACTTTTTTTTTTTTTTTTATTTCCCAGTAATATCCGTTGCCCCC-3’(SEQ ID No:2)。
所述通用CrRNA的核苷酸序列为:
5’-UAAUUUCUACUAAGUGUAGAUGUUGUCCCAGUCAUAUCCGUUGCTATTATT-3’(SEQ ID No:3)。
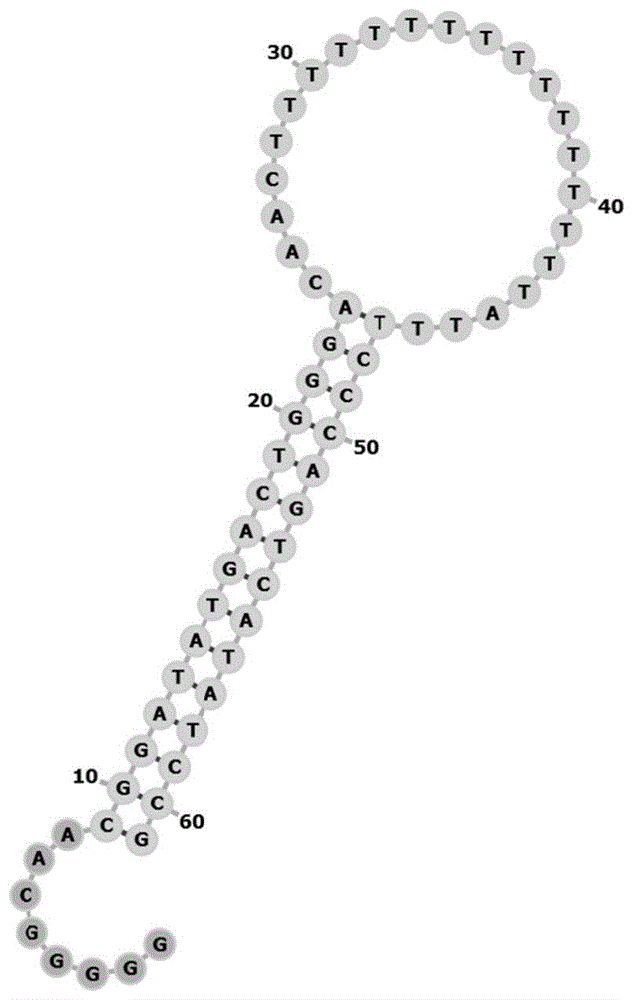
在本发明第一方面的一些实施方式中,所述底物片段经退火处理后折叠形成具有茎状部分和环状部分的茎环结构,所述第一互补区和所述第二互补区互补配对形成双链的茎状部分,所述环形区的两端分别与所述第一互补区和所述第二互补区连接形成单链的环状部分。
在本发明第一方面的一些实施方式中,所述退火处理的反应体系包括退火缓冲液、所述底物片段和无酶水。所述DNA退火缓冲液可选择市售产品,例如碧云天的D0251。
在本发明第一方面的一些实施方式中,所述退火处理的反应程序为:98℃,持续1分钟;以每分钟下降1℃的速率降至25℃;25℃保存。所述退火处理过程通过升温打开氢键,匀速降温使得所述第一互补区能够与所述第二互补区相互识别配对,从而形成所述茎环结构。
在本发明第一方面的一些实施方式中,所述检测体系还包括缓冲液、氯化镁溶液、RNA酶抑制剂、检测CrRNA和探针。所述CRISPR-Dx检测体系具有多种拓展的可能性,可以根据不同的使用场景和使用需求包括但不限于制备为荧光检测试剂盒或胶体金试纸条等。荧光检测试剂盒通过含有待测样品的实验组与不添加待测样品的空白组进行对照实验,由荧光值比值进行判读,从而得出待测样品中是否含有模板片段。
在本发明第一方面的一些实施方式中,所述探针的5’端连接有荧光基团、3’端连接有荧光猝灭基团。
在本发明第一方面的一些实施方式中,所述探针的核苷酸序列为5’-TTATTATT-3’。
在本发明第一方面的一些实施方式中,所述检测CrRNA的核苷酸序列如SEQ IDNo:4所示。如SEQ ID No:4所示的检测CrRNA用于检测ERBB2基因。进一步可以根据其他基因的保守序列设计相应的检测CrRNA也能够利用本检测体系实现级联放大的检查反应。
在本发明第一方面的一些实施方式中,所述缓冲液为10×HOLMES缓冲液。
在本发明第一方面的一些实施方式中,所述检测体系的体积为20μL,包括10×HOLMES缓冲液2μL,浓度为25mM的氯化镁溶液1.5μL,40U/μL的RNA酶抑制剂0.25μL,浓度为1μM的所述通用CrRNA 0.25μL,浓度为1μM的所述检测CrRNA0.25μL,浓度为10pmol/μL的Lbcas12a 0.5μL,浓度为10mM的所述探针1μL,稀释200倍含有所述底物片段的底物溶液1μL,无酶水12.25μL,待测样品溶液1μL。
本发明的优点在于:
(1)通过在CRISPR-Dx反应体系中加入所述底物片段和所述通用CrRNA,所述底物片段的环形区能产生空间位阻,使底物片段无法直接被被通用CrRNA-Lbcas12a复合物识别。当靶gDNA存在时通用CrRNA-Lbcas12a复合物会顺式切割靶gDNA,而后反式切割环形区的单链区,使底物片段发生变构,环形区变为线性。失去空间位阻的底物片段可以被通用CrRNA-Lbcas12a复合物识别,从而激活更多cas蛋白,反式切割更多探针,产生更高荧光。
(2)灵敏度高则不需要使用RPA扩增,避免了RPA的非特异性扩增,提高了特异性。
(3)本发明加入的底物仅包含DNA,理论上稳定性要高于RNA底物。
(4)由于底物仅包含DNA,所以反应只需要加入cas12酶而无需cas13酶,使得成本更低。
(5)由于本发明以实验组荧光值与对空白荧光值的比值作为判读标准,而此比值在10~30分钟的范围内很稳定,因此反应时间20分钟即可满足诊断要求,解决了现有技术中CRISPR-Dx反应时间很长的问题。
附图说明
图1是实施例1所述茎环结构的示意图;
图2是实施例2中优化荧光法CRISPR-Dx检测体系检测时间的折线图;
图3是实施例2中优化荧光法CRISPR-Dx检测体系DNA底物组合稀释倍数的柱状图;
图4是实施例2中优化荧光法CRISPR-Dx检测体系MgCl
图5是实施例2中优化荧光法CRISPR-Dx检测体系Lbcas12a用量的柱状图;
图6是实施例2中优化荧光法CRISPR-Dx检测体系探针用量的柱状图;
图7是实施例2中荧光法CRISPR-Dx检测体系的特异性验证曲线图;
图8是实施例2中荧光法CRISPR-Dx检测体系的特异性验证柱状图;
图9是实施例2中添加不同浓度gDNA的荧光值曲线图;
图10是实施例2中添加不同浓度gDNA的倍数柱状图;
图11是实施例2中不同gDNA浓度的倍数线性回归图;
图12是实施例2中传统CRISPR-Dx检测体系与所述荧光法CRISPR-Dx检测体系检测ASFV阳性猪血临床样本的对比折线图;
图13是实施例2中所述荧光法CRISPR-Dx检测体系检测ASFV阳性(5例)和阴性(5例)猪血临床样本的倍数柱状图;
图14是实施例2中所述荧光法CRISPR-Dx检测体系检测ASFV阳性和阴性猪血临床样本的平均倍数柱状图;
图15是实施例3中传统CRISPR-Dx检测体系(不加入底物)试纸条法检测高浓度ASFV gDNA的结果图;
图16是实施例3中添加不同浓度gDNA的试纸条法结果图;
图17是实施例3中添加ASFV阳性和阴性猪血临床样本的试纸条法结果图;
图18为实施例4中荧光法CRISPR-Dx检测体系使用不同浓度gDNA检测ERBB2基因的荧光值曲线图;
图19为实施例4中荧光法CRISPR-Dx检测体系使用不同浓度gDNA检测ERBB2基因的倍数柱状图;
图20为实施例5中试纸条法CRISPR-Dx检测体系使用不同浓度gDNA检测ERBB2基因的结果图。
具体实施方式
以下实施例中未作具体说明的分子生物学试验方法,均参照《分子克隆实验指南》(第三版)或者按照方法和产品说明书进行;所述方法生物材料,如无特殊说明,均可从商业途径获得。
实施例1:构建辅助检测非洲猪瘟病毒(ASFV)的DNA底物。
本实施例以ASFV为检测对象,根据其P72基因的T8位点设计DNA底物,获得两组共2条底物片段,其核苷酸序列如表1所示。
表1、DNA底物中各底物片段的核酸序列
所述底物片段需要升温打开双键,再退火处理后形成如图1所示的茎环结构。所述退火体系如表2所示。
表2、DNA底物的退火体系
所述退火体系的反应程序为:98℃,持续1分钟;以每分钟下降1℃的速率降至25℃;25℃保存。
如果反应体系中存在T8模板片段,将会激活cas12酶的反式切割活性,从而对所述茎环结构的单链区域进行切割,使其结构发生改变。
实施例2:利用DNA底物构建荧光法CRISPR-Dx检测体系。
将实施例1所述R13d作为底物片段,构建荧光法CRISPR-Dx检测体系,以获得特异性和灵敏度均令人满意的试剂盒。试剂盒的主要试剂构成包括所述底物片段、无酶水、缓冲液、氯化镁溶液、RNA酶抑制剂、Lbcas12a、通用CrRNA和荧光探针。
荧光CRISPR-Dx检测系统的反应时间和各成分的剂量需要优化。由于希望加入gDNA(实验组)与加入水(空白组)的荧光值差异尽量大,需要选择荧光值倍数较高的反应体系(倍数=加入DNA底物与gDNA(实验组)的荧光值/加入底物与水(空白组)的荧光值)。如图2所示,首先测试了三个反应时间(10、20和30分钟),发现荧光信号倍数在10分钟时达到平稳。但10分钟时绝对荧光值较低,且倍数精确性略低(标准差较大),故选用20分钟为反应时间。底物片段溶液稀释倍数的优化如图3所示,200倍稀释的DNA底物实验组与空白组荧光信号比值最高,从而被选择用于下游实验;如图4所示,对氯化镁溶液(MgCl
表3、荧光法CRISPR-Dx检测体系
所述通用CrRNA的核苷酸序列如下:
5’-UAAUUUCUACUAAGUGUAGAUGUUGUCCCAGUCAUAUCCGUUGCTAT TATT-3’(SEQ IDNo.3)。所述探针的序列为:5’-TTATTATT-3’,探针的5’端连接有FAM,3’端连接有BHQ1。
由于荧光法CRISPR-Dx检测体系需要以实验组荧光值与对空白荧光值的比值作为判读依据,因此使用时以无酶水替代待测样品溶液设置对空白,将对空白的荧光值作为背景减去。
使用均值+3倍标准差的方法确定了实验组荧光值与对空白荧光值的阈值,即阳性与阴性判读标准。如图7、8所示,加入含有模板靶DNA的比值与加入其它相同浓度干扰DNA的比值有显著差异,且阈值=均值+3×标准差=1.018+3×0.128=1.402≈1.5,故将阈值定为1.5倍,大于等于此值被定义为阳性。
确定阈值后可以根据荧光值进行结果判读。需要注意的是,0时刻的荧光值应作为背景减去。
结果判读:
实验组荧光值与对空白荧光值的比值≥1.5,应判读为阳性。
实验组荧光值与对空白荧光值的比值<1.5,应判读为阴性。
如图9、10所示,本方法的检测限为166fM。模板gDNA浓度在83~332fM时,实验组与空白组的荧光比值与模板gDNA浓度存在线性关系(图11),可以根据方程推测出理论上该方法的检测限为129.68fM。
而不加入DNA底物时即使T8序列模板浓度高达2491fM也将被诊断为阴性。由此可知本实施例所述荧光法CRISPR-Dx检测体系加入将检测限降低了15倍多。
对临床样本进行了测试。共有10个样本,其中5个是猪血ASFV检测呈阳性,另外5个检测呈阴性。均通过qPCR方法进行了验证。使用DNA提取试剂盒从中提取gDNA并进行检测。如图12所示,仅添加具有靶DNA的阳性样本不会增加检测曲线,表明结果为阴性;添加底物进行检测后,实验组与空白组的荧光值之比为1139.67/665.67=1.71>1.5,表明结果为阳性。这表明本实施例的检测体系对于直接检测病毒核酸具有更低的检测限。图13和图14显示了阳性猪血与阴性猪血荧光值比值的显著差异。以1.5倍为阈值,阳性样本比值均大于阈值,阴性样本比值均小于阈值,因此可以看到此检测体系的特异度良好。
实施例3:利用DNA底物构建胶体金试纸条CRISPR-Dx检测体系。
同样,实施例1提供的底物片段也可应用于胶体金试纸条。
侧流层析胶体金试纸条法则不需要设置对空白。由于反应体系的载体存在区别,因此DNA底物溶液的稀释倍数需要降低,经优化试验确认50倍稀释的底物片段浓度较佳。胶体金试纸条法具体反应体系、反应温度条件如表4所示。而后可进行结果判读。
表4、胶体金试纸条CRISPR-Dx检测体系
结果判断方法:
控制线(C线)显色、检测线(T线)显色,应判读为阳性。
控制线(C线)显色、检测线(T线)不显色,应判读为阴性。
C线、T线均不显色,应判读为检测无效。
如图16所示,本方法的检测限为17fM。而不加入底物时即使模板DNA浓度高达2491fM(如图15所示),也被诊断为阴性,加入底物的方法将检测限降低了100多倍。临床样本的检测(如图17所示)也显示出与荧光法一致的准确结果。
实施例4:利用荧光法CRISPR-Dx检测体系检测ERBB2基因。
按实施例2提供的方法构建靶向MKN-45细胞系的ERBB2基因的CRISPR-Dx检测体系,具体如表5所示。
表5、靶向ERBB2基因的CRISPR-Dx检测体系
所述检测CrRNA的核苷酸序列如下:
5’-UAAUUUCUACUAAGUGUAGAUCCUCAACACUUUGAUGGCCA-3’(SEQ ID No:4)。所述底物片段溶液中含有的底物片段为实施例1中记载的R13d。所述荧光探针同实施例2。
使用传统CRISPR-Dx检测体系作为对照组。检测结果如图20所示。可见加入人类模板gDNA浓度越高,荧光曲线越高,倍数越大,且加入317、361、529ng/μL gDNA后倍数均大于1.5倍(如图18、19所示,加入317ng/μL gDNA,20分钟时实验组荧光值/空白组荧光值=621/241=2.58>1.5;加入361ng/μL gDNA,20分钟时实验组荧光值/空白组荧光值=760/241=3.15>1.5;加入529ng/μL gDNA,20分钟时实验组荧光值/空白组荧光值=1072/241=4.45>1.5),检测结果为阳性;而传统CRISPR-Dx方法即使浓度高达529ng/μL也不起线,检测结果为阴性,因此本发明检测限(LOD)低于传统CRISPR-Dx方法。
实施例5:利用胶体金试纸条CRISPR-Dx检测体系检测ERBB2基因。
按实施例3提供的方法构建靶向ERBB2基因的胶体金试纸条,其检测体系如表6所示。
表6、胶体金试纸条CRISPR-Dx检测体系
如图20所示,本方法的能够检出浓度为529ng/μL的gDNA样本。
对于本领域技术人员而言,显然本发明不限于上述示范性实施例的细节,而且在不背离本发明的精神或基本特征的情况下,能够以其他的具体形式实现本发明。因此,无论从哪一点来看,均应将实施例看作是示范性的,而且是非限制性的,本发明的范围由所附权利要求而不是上述说明限定,因此旨在将落在权利要求的等同要件的含义和范围内的所有变化囊括在本发明内。
- 一种用于转基因CaMV35S启动子的定量检测的可视化级联放大功能核酸传感器
- 一种沙门氏菌定量检测的可视化级联放大功能核酸传感器
- 一种用于免疫检测低丰度生物标志物信号级联放大的荧光树状大分子的制备方法及其应用
- 一种基于级联放大快速定量检测阪崎肠杆菌的方法