治疗与S1P1受体相关的病况的方法
文献发布时间:2023-06-19 10:27:30
技术领域
本发明提供适用于治疗鞘氨醇1-磷酸盐亚型1(S1P
鞘氨醇-1-磷酸盐(S1P)受体1-5构成具有七个跨膜结构域的G蛋白偶联受体家族。称为S1P
鉴于对可用于治疗S1P
已作为重要的新化合物出现,参见PCT专利申请案第PCT/US2009/004265号,所述申请案特此以全文引用的方式并入。化合物1或其药学上可接受的盐、溶剂合物或水合物是意图用于治疗鞘氨醇1-磷酸盐亚型1(S1P
许多S1P
需要有效治疗需要用化合物1或其药学上可接受的盐、溶剂合物或水合物治疗的个体。本公开满足此需要且还提供相关优点。
对贯穿本申请案的任何参考文献的引用都不应被解释为承认所述参考文献是本申请案的现有技术。
发明内容
本发明提供一种治疗患有鞘氨醇1-磷酸盐亚型1(S1P
还提供了一种向有需要的患者投与选自(R)-2-(7-(4-环戊基-3-(三氟甲基)苄氧基)-1,2,3,4-四氢环戊并[b]吲哚-3-基)乙酸(化合物1)或其药学上可接受的盐、水合物或溶剂合物的鞘氨醇1-磷酸盐亚型1(S1P
还提供了一种向有需要的患者投与选自(R)-2-(7-(4-环戊基-3-(三氟甲基)苄氧基)-1,2,3,4-四氢环戊并[b]吲哚-3-基)乙酸(化合物1)或其药学上可接受的盐、水合物或溶剂合物的鞘氨醇1-磷酸盐亚型1(S1P
还提供了一种向有需要的患者投与选自以下的鞘氨醇1-磷酸盐亚型1(S1P
还提供了一种向有需要的患者投与选自(R)-2-(7-(4-环戊基-3-(三氟甲基)苄氧基)-1,2,3,4-四氢环戊并[b]吲哚-3-基)乙酸(化合物1)或其药学上可接受的盐和/或同位素变体的鞘氨醇1-磷酸盐亚型1(S1P
将在专利公开进行时更详细地阐述本文所公开的本发明的这些和其它方面。
附图说明
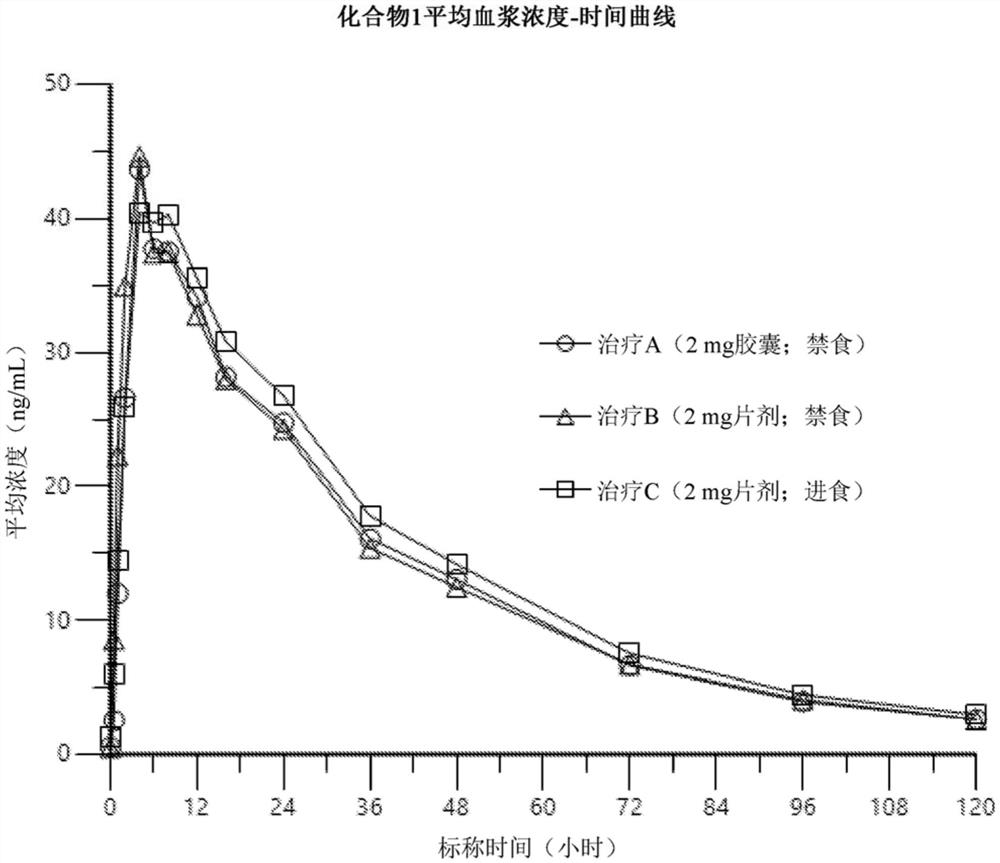
图1显示了在进食与禁食条件下投与的化合物1的片剂调配物的平均血浆浓度-时间曲线。
具体实施方式
如本说明书中所使用,以下字词和短语一般意图具有如下文所阐述的含义,除使用其的上下文另外指示的方面外。
化合物1:如本文所使用,“化合物1”意指(R)-2-(7-(4-环戊基-3-(三氟甲基)苄氧基)-1,2,3,4-四氢环戊并[b]吲哚-3-基)乙酸,包括其结晶形式。作为非限制性实例,化合物1可以如描述于WO 2010/011316(以全文引用的方式并入本文中)中的无水非溶剂化结晶形式存在。作为另一个非限制性实例,化合物1的L-精氨酸盐可以如描述于WO 2010/011316和WO2011/094008(其中的每一者以全文引用的方式并入本文中)中的无水非溶剂化结晶形式存在。作为另一个非限制性实例,化合物1的钙盐可以如描述于WO 2010/011316(以全文引用的方式并入本文中)中的结晶形式存在。
投与:如本文所使用,“投与”意指提供化合物或其它疗法、救治或治疗,使得个体内化化合物。
共同投与:如本文所使用,“共同投与”(“co-administer”/“co-administration”)和其变体意指将至少两种药物相继地,同时地或因此在时间上彼此接近地(例如,在同一天或同一周或30天的时间内,或足够接近以至于可以在血浆中同时检测出至少两种药物中的每一种)投与患者。当共同投与时,两种或更多种活性剂可以作为相同组合物的一部分共同调配或作为单独调配物投与。此在本文中也可称作“同时”投与或其变体。
开处方:如本文所使用,“开处方”意指命令、批准或推荐使用药物或其它疗法、救治或治疗。在一些实施例中,医护从业者可以口头建议、推荐或批准对个体使用化合物、给药方案或其它治疗。在此情况下,医护从业者可或可不提供对化合物、给药方案或治疗的处方。此外,医护从业者可或可不提供所推荐的化合物或治疗。例如,医护从业者可以建议个体在何处获得化合物而非提供化合物。在一些实施例中,医护从业者可以向个体提供化合物、给药方案或治疗的处方。例如,医护从业者可以向个体提供书面或口头处方。处方可以写在纸上或电子媒体上,如计算机文件,例如,在手持计算机设备上。例如,医护从业者可以将一张纸或电子媒体转化成具有化合物、给药方案或治疗的处方。此外,处方可以(口头)阐述、传真(书面)或经由互联网电子提交到药房或配药处。在一些实施例中,可以给予个体化合物或治疗的样品。如本文所使用,给出化合物的样品构成化合物的隐含处方。世界上的不同健康护理系统使用用于开处和/或投与化合物或治疗的不同方法,且本公开涵盖这些方法。
处方可以包括例如个体的姓名和/或身份信息,如出生日期。此外,例如,处方可以包括:药物名称、药物强度、剂量、投与频率、投与途径、待分配的数量或量、再配药数量、医师姓名、医师签名等。此外,例如,处方可以包括DEA号和/或州号。
医护从业者可以包括例如医师、护士、护士从业者或其它相关医护专业人员,其可以开处或投与用于治疗鞘氨醇1-磷酸盐亚型1(S1P
预防(PREVENT/PREVENTING/PREVENTION):如本文所使用,术语“预防(prevent/preventing/prevention)”如预防鞘氨醇1-磷酸盐亚型1(S1P
治疗(treat/treating/treatment):如本文所使用,术语“治疗(treat/treating/treatment)”意指向已显示疾病或病况的至少一种症状或先前已显示疾病或病况的至少一种症状的个体投与疗法。例如,“治疗”可以包括缓解、缓和或改善疾病或病况症状;防止其它症状;改善症状的潜在代谢病因,抑制疾病或病况,例如使疾病或病况的发展停滞,缓解疾病或病况,致使疾病或病况消退,缓解由疾病或病况所导致的病况,或使疾病或病况的症状停止。例如,关于病症的术语“治疗”意指减轻与特定病症相关的一种或多种症状的严重程度。因此,治疗病症并不一定意指减轻与病症相关的所有症状的严重程度且不一定意指完全减轻与病症相关的一种或多种症状的严重程度。
耐受:如本文所使用,如果对个体投与一定剂量的化合物不会导致不可接受的不良事件或不可接受的不良事件的组合,那么称个体“耐受”所述剂量的化合物。所属领域的技术人员将理解,耐受是一种主观的量度,且一个个体可耐受的东西可能对另一个个体不耐受。例如,一个个体可能不能够耐受头痛,而第二个个体可能发现头痛可耐受但无法耐受呕吐,但对于第三个个体而言,单独的头痛或单独的呕吐是可耐受的,但所述个体不能够耐受头痛与呕吐的组合,即使每一个的严重程度比单独经历时小。
不良事件:如本文所使用,“不良事件”是与用化合物1或其药学上可接受的盐、溶剂合物或水合物治疗相关的不良医学事件发生。在一个实施例中,不良事件是选自:白细胞减少症、便秘、腹泻、恶心、腹痛、嗜中性白细胞减少症、呕吐、背痛和月经异常。在一个实施例中,不良事件是心传导阻滞,例如,一级房室心传导阻滞。在一个实施例中,不良事件是急性心率降低。在一个实施例中,不良事件是异常肺功能测试发现,如低于80%的FEV1,FVC。在一个实施例中,不良事件是异常肝功能测试,如升高的ALT&AST>2X ULN。在一个实施例中,不良事件是黄斑水肿。
需要治疗和有需要的:如本文所使用,“需要治疗”和“有需要的”在涉及治疗时可互换使用,以意指由护理人员(例如,就人类而言,医师、护士、护士从业人员等;就动物而言,兽医,包括非人类哺乳动物)作出的个体或动物需要或将受益于治疗的判断。此判断是基于护理人员的专业知识领域内的多种因素而作出的,但包括以下知识:个体或动物由于可由本发明的化合物治疗的疾病、病况或病症而生病或将生病。因此,本发明的化合物可以以防护性或预防性方式使用;或本发明的化合物可以用于缓解、抑制或改善疾病、病况或病症。
个体:如本文所使用,“个体”意指任何动物,包括哺乳动物,优选小鼠、大鼠、其它啮齿动物、兔子、狗、猫、猪、牛、绵羊、马或灵长类动物,且最优选人类。在一些实施例中,人类个体称为“患者”。
急性心率降低:如本文所使用,“急性心率降低”意指心率从正常的窦性心律降低到,例如,每分钟搏动10次或更多次(bpm),如小于约5bpm,例如小于约4bpm或小于约3bpm或小于2bpm,即在投与药物后数小时(例如1-3小时)内达到最大值,且之后心率恢复到给药前的值。
正常窦性心律:如本文所使用,“正常窦性心律”意指个体在未进行治疗时的窦性心律。对正常窦性心律的评估在医师的能力范围内。正常窦性心律将通常产生60-100bpm范围内的心率。
剂量:如本文所使用,“剂量”意指在一个特定时间给予个体的用于治疗或预防疾病或病症的化合物1或其药学上可接受的盐、溶剂合物或水合物的量。
标准剂量:如本文所使用,“标准剂量”意指给予个体的用于治疗或预防疾病或病症的化合物1或其药学上可接受的盐、溶剂合物或水合物的剂量。在一些实施例中,投与标准剂量实现外围血液淋巴细胞计数的目标降低,例如基线降低至少35%,如至少40%、如至少45%、如至少50%、如至少55%、如至少60%、如至少65%、如至少70%。在一些实施例中,投与标准剂量实现基线降低约35%至约70%,如约40%至约65%,如约50%至约65%。在一些实施例中,投与标准剂量实现目标外围血液淋巴细胞计数,例如每微升小于1000个淋巴细胞,如每微升400-800个淋巴细胞。目标剂量可根据待治疗的疾病的性质和严重程度而变化。
治疗有效量:如本文所使用,药剂、化合物、药物、组合物或组合的“治疗有效量”是无毒且在向受试者或患者(例如人类受试者或患者)投与后有效产生一些所期望的治疗效果的量。受试者的精确治疗有效量可视例如受试者的身材和健康状况、病况的性质和程度、所选择用于投与的疗法或疗法的组合和所属领域中的技术人员已知的其它变量而定。给定情形的有效量通过常规实验测定且在临床医师的判断内。在一些实施例中,治疗有效量是标准剂量。
禁食个体:如本文所使用,“禁食个体”意指未进食任何食物的个体,即在投与化合物1或其药学上可接受的盐、水合物或溶剂合物之前,已禁食至少6-8小时,如约8小时,且所述个体不吃任何食物且在投与化合物1或其药学上可接受的盐、水合物或溶剂合物之后继续禁食至少1小时。在某些实施例中,个体还可在禁食期间避免摄取某些非食物物质。例如,在某些实施例中,个体在禁食期间不摄取任何补充剂和/或药物。在某些实施例中,个体在禁食期间不摄取任何高卡路里液体。在某些实施例中,个体在禁食期间不摄取除水以外的任何液体。在某些实施例中,个体可摄取少量低卡路里饮料,如茶、咖啡或稀果汁。
梅奥诊所评分(MCS):如本文所使用,“梅奥诊所评分”或“MCS”意指经设计以测量溃疡性结肠炎疾病活性且由至多4个分项评分组成:大便频率、直肠出血,柔性直肠乙状结肠镜检查的结果和医师的总体评估,每个分量的范围是0至3(0=正常,1=轻度,2=中度,3=重度)。因此,总评分在0至12范围内,其中较高评分指示较严重疾病。6分梅奥评分是基于每天使用电子病历收集的大便频率和直肠出血PRO得出的,且不包括内窥镜检查的结果和医师的总体评估。3分梅奥评分是基于大便频率、直肠出血和内窥镜检查的结果,且总评分范围是0至9。2分梅奥评分是基于直肠出血和内窥镜检查的结果,且总评分范围是0至6。医师的总体评估认可了MCS的其它三个标准检查结果,个体每天的腹部不适和一般健康状况的记录和其它观察结果,如身体检查结果和个体的表现。
轻度至中度活动性溃疡性结肠炎:如本文所使用,“轻度至中度活动性溃疡性结肠炎”意指特征在于4分量MCS为4至10的溃疡性结肠炎。
中度至重度活动性溃疡性结肠炎:如本文所使用,“中度至重度活动性溃疡性结肠炎”意指特征在于3分量MCS为4至9,包括内窥镜评分≥2且直肠出血评分≥1的溃疡性结肠炎。3分量MCS使用完整MCS的4分量中的3分量(内窥镜检查结果、直肠出血和大便频率)。
临床缓解:如本文所使用,关于溃疡性结肠炎的“临床缓解”意指3分量梅奥诊所评分如下:内窥镜评分(使用柔性直肠乙状结肠镜检查)为0或1,直肠出血评分为0,且大便频率评分为0或1,相对于基线分项评分降低≥1分。
临床反应:如本文所使用,关于溃疡性结肠炎的“临床反应”意指3分量梅奥诊所评分降低了≥2分,且相对于基线降低≥30%,同时直肠出血评分降低≥1或绝对直肠出血评分为0或1。
内窥镜改善:如本文所使用,关于溃疡性结肠炎的“内窥镜改善”是指特征在于梅奥内窥镜分项评分(使用柔性直肠乙状结肠镜检查)≤1分的溃疡性结肠炎。
内窥镜缓解:如本文所使用,关于溃疡性结肠炎的“内窥镜缓解”意指特征在于梅奥诊所评分=0的柔性直肠乙状结肠镜检查结果的溃疡性结肠炎。
直肠出血的改善:如本文所使用,关于溃疡性结肠炎的“直肠出血的改善”意指相对于基线变化<0。
组织学愈合/组织学改善:如本文所使用,关于溃疡性结肠炎的“组织学愈合(histologic healing/histological healing)”、“组织学改善(histologicimprovement/histological improvement)”意指在吉布斯指数(Geboes Index)上的评分<3.1。
组织学缓解:如本文所使用,关于溃疡性结肠炎的“组织学缓解(histologicremission/histological remission)”意指在吉布斯指数上的评分<2.0。
粘膜愈合:如本文所使用,“粘膜愈合”是内窥镜改善和组织学缓解。
大便频率的改善:如本文所使用,关于溃疡性结肠炎的“大便频率的改善”意指相对于基线变化<0。
5-氨基水杨酸盐:如本文所使用,“5-氨基水杨酸盐”意指一类药物,其包括例如
免疫抑制剂:如本文所使用,“免疫抑制剂”意指一类药物,其包括例如
糖皮质类固醇:如本文所使用,“糖皮质类固醇”意指一类药物,其包括例如
TNFα拮抗剂:如本文所使用,“TNFα拮抗剂”或“肿瘤坏死因子-α拮抗剂”意指一类药物,其包括例如
整联蛋白受体拮抗剂:如本文所使用,“整联蛋白受体拮抗剂”意指一类药物,其包括例如
药物组合物:如本文所使用,“药物组合物”意指包含至少一种活性成分(如化合物1)的组合物;包括但不限于化合物1的盐、溶剂合物和水合物,由此所述组合物适于研究哺乳动物(例如但不限于人类)的特定有效结果。所属领域的一般技术人员将理解并了解适合于确定活性成分是否具有基于所属领域的技术人员需要的所期望的有效结果的技术。
激动剂:如本文所使用,“激动剂”意指与G蛋白偶联受体(如S1P
拮抗剂:如本文所使用,“拮抗剂”意指与激动剂(如内源性配体)在同一位点竞争性结合到受体的部分,但其不激活由受体活性形式引发的细胞内反应,且从而可以抑制激动剂或部分激动剂的细胞内反应。在不存在激动剂或部分激动剂的情况下,“拮抗剂”不会减少基线细胞内应答。
反向激动剂:如本文所使用,“反向激动剂”意指与受体的内源性形式或与受体的组成型活化形式结合且抑制由正常碱基水平活性以下的受体的活性形式引发的基线细胞内应答,所述正常碱基水平活性在不存在激动剂或部分激动剂的情况下观察到,或降低GTP与膜的结合的部分。在一些实施例中,在反向激动剂的存在下基线细胞内应答被抑制了至少30%。在一些实施例中,在反向激动剂的存在下基线细胞内应答被抑制了至少50%。在一些实施例中,与不存在反向激动剂的情况下的基线应答相比,在反向激动剂的存在下,基线细胞内反应被抑制了至少75%。
水合物:如本文所使用,“水合物”意指本发明的化合物或其盐,其进一步包括通过非共价分子间力结合的化学计量或非化学计量的量的水。
安全人群:如本文所使用,“安全人群”意指接受研究药物的所有随机受试者。
溶剂合物:如本文所使用,术语“溶剂合物”意指本发明的化合物或其盐,其进一步包括通过非共价分子间力结合的化学计量或非化学计量的量的溶剂。优选的溶剂是挥发性、无毒和/或可接受的以微量向人类投与。
根据本发明的化合物可任选地作为药学上可接受的盐而存在,包括由药学上可接受的无毒酸(包括无机酸和有机酸)制备的药学上可接受的酸加成盐。代表性的酸包括但不限于乙酸、苯磺酸、苯甲酸、樟脑磺酸、柠檬酸、乙烯磺酸、二氯乙酸、甲酸、富马酸、葡萄糖酸、谷氨酸、马尿酸、氢溴酸、盐酸、羟乙基磺酸、乳酸、马来酸、苹果酸、扁桃酸、甲磺酸、粘液酸、硝酸、草酸、双羟萘酸、泛酸、磷酸、琥珀酸、硫酸、酒石酸、草酸、对甲苯磺酸等,如Berge等人,《药物科学期刊(Journal of Pharmaceutical Sciences)》,66:1-19(1977)列出的那些药学上可接受的盐,其以全文引用的方式并入本文中。
酸加成盐可作为化合物合成的直接产物获得。在替代方案中,可将游离碱溶解在含有适当酸的合适溶剂中,且通过蒸发溶剂或以其它方式分离盐和溶剂来分离盐。本发明的化合物可使用所属领域的技术人员已知的方法与标准低分子量溶剂形成溶剂合物。
应当理解,当提及化合物1时使用短语“药学上可接受的盐、溶剂合物和水合物”或短语“药学上可接受的盐、溶剂合物和水合物”时,其包括化合物1的药学上可接受的溶剂合物和/或水合物、化合物1的药学上可接受的盐以及化合物1的药学上可接受的盐的药学上可接受的溶剂合物和/或水合物。还应理解,当提及作为盐的化合物1时使用短语“药学上可接受的溶剂合物和水合物”或短语“药学上可接受的溶剂合物和水合物”时,其包括此类盐的药学上可接受的溶剂合物和/或水合物。
所属领域的技术人员将显而易见的是,本文所描述的剂型可包含化合物1或其药学上可接受的盐或溶剂合物或水合物作为活性成分。此外,化合物1的各种水合物和溶剂合物和其盐将可用作制备药物组合物的中间体。除本文提到的那些之外,制备和鉴别合适的水合物和溶剂合物的典型方法是所属领域的技术人员众所周知的;参见例如,K.J.Guillory,《多晶型、水合物、溶剂合物和无定形固体的产生在:药物固体中的多态性(Generation of Polymorphs,Hydrates,Solvates,and Amorphous Solids,in:Polymorphism in Pharmaceutical Solids)》中的202-209页,Harry G.Brittain编,第95卷,马塞尔德克公司(Marcel Dekker,Inc.),纽约,1999。因此,本公开的一个方面涉及开处和/或投与化合物1的水合物和溶剂合物和/或其药学上可接受的盐的方法,其可以通过所属领域已知的方法来分离和表征,如热重分析(TGA)、TGA-质谱法、TGA-红外光谱法、粉剂X射线衍射(XRPD)、卡尔飞世尔(Karl Fisher)滴定、高分辨率X射线衍射等。有几个商业实体提供常规鉴别溶剂合物和水合物的快速和有效的服务。提供这些服务的示例公司包括威明顿医药技术公司(Wilmington PharmaTech)(特拉华州威明顿市),Avantium技术公司(Avantium Technologies)(阿姆斯特丹)和Aptuit公司(康涅狄格州格林威治)。
本公开包括存在于本发明化合物、盐、溶剂合物和水合物中的原子的所有同位素。同位素包括那些具有相同原子数但不同质量数的原子。本发明的一个方面包括本发明化合物、盐、溶剂合物和水合物中的一个或多个原子的每一组合,其用具有相同原子数但不同质量数的原子替换。一个此实例是将在本发明化合物、盐、溶剂合物和水合物之一中发现的天然最丰富的同位素(如
本发明的化合物可以转化为“前药”。术语“前药”意指用所属领域已知的特定化学基团修饰的化合物,且当投与个体时,其经历生物转化以得到母体化合物。因此,前药可以视为本发明的化合物,其含有一种或多种以短暂方式使用的专门的无毒保护基团,以改变或消除化合物的特性。在一个普遍的方面,“前药”方法用于促进口服吸收。在T.Higuchi和V.Stella,《作为新颖递送系统的前药(Pro-drugs as Novel Delivery Systems)》《美国化学会会议论文集(A.C.S.Symposium Series)》第14卷;和在《药物设计中的生物可逆载体(Bioreversible Carriers in Drug Design)》,Edward B.Roche编,美国药学协会和帕加马出版社(American Pharmaceutical Association and Pergamon Press),1987中进行了深入的论述,两者都以全文引用的方式并入本文中。
AUC:如本文所使用,“AUC”是指给药事件后活性药物成分或代谢产物的血浆浓度随时间变化的曲线下面积或积分。
AUC
AUC
Cmax:如本文所使用,Cmax(或C
t
t
如本文所使用,“具有狭义治疗指数的物质”是指属于美国食品和药物管局理或其任何后续机构颁布的狭义治疗指数的任何定义内的物质,例如,在血液中具有小于2倍的半数致死剂量(LD50)和半数有效剂量(ED50)值差异或具有小于2倍的最小毒性浓度和最小有效浓度差异的物质;且为了安全有效地使用所述物质,需要仔细调定和患者监测。
如本文所使用,当物质可以通过酶对物质的作用而化学转化时,所述物质是酶活性的“底物”。“酶活性”广泛地指酶的比活性(即,酶每mg或摩尔酶转化底物的速率)以及此类转化的代谢作用。因此,当物质的存在可以降低酶的比活性或比活性的代谢作用时,所述物质是酶活性的“抑制剂”,而不涉及此类降低的确切机理。例如,物质可以是通过竞争性、非竞争性、变构或其它类型的酶抑制、通过减少酶的表达或其它直接或间接机理的酶活性的抑制剂。类似地,当物质的存在可以增加酶的比活性或比活性的代谢作用时,所述物质是酶活性的“诱导剂”,而不涉及此类增加的确切机理。例如,物质可以通过增加反应速率、通过增加酶的表达、通过变构激活或其它直接或间接机理来成为酶活性的诱导剂。对酶活性的这些影响中的任一者可以在单一样品、供体或患者中在给定浓度的活性剂下进行,而不考虑临床意义。物质有可能是酶活性的底物、抑制剂或诱导剂。例如,物质可以是一种机理的酶活性抑制剂和另一种机理的酶活性诱导剂。物质关于酶活性的功能(底物、抑制剂或诱导剂)可以取决于环境条件。CYP3A4的抑制剂,诱导剂和底物的列表可以例如在以下中找到:
http://www.genemedrx.com/Cytochrome_P450_Metabolism_Table.php和其它网站。
当在本文所公开的方法中使用整数时,可以在整数之前插入术语“约”。
在整个说明书中,除非上下文另有要求,否则词语“包含(comprise)”或其变化形式(如“包含(comprises/comprising)”将理解为暗示包括一个陈述的步骤或元件或整数或步骤或元件或整数组但不排除任何其它步骤或元件或整数或元件或整数组。
在整个说明书中,除非另外具体说明或上下文另外要求,否则提及单个步骤、物质组合物、步骤组或物质组合物组应理解为涵盖那些步骤、物质组合物、步骤组或物质组合物组中的一者和多者(即,一者或多者)。
除非另外具体说明,否则本文中所描述的每一实施例将被加以必要修正以适用每一和每个其它实施例。
所属领域的技术人员将了解本文所描述的本发明除具体描述的那些内容以外容许进行变化和修改。应了解,本发明包括所有此类变化和修改。除非另外具体说明,否则本发明还包括本说明书中单独或共同提及或指出的所有步骤、特征、组合物和化合物,以及任何和所有组合或所述步骤或特征中的任何两个或更多个。
本发明不受本文中所描述的具体实施例的范围限制,所述具体实施例意图仅出于示例的目的。功能上等效的产品、组合物和方法清楚地在如本文所描述的本发明的范围内。
应了解,为了清楚起见,在单独实施例的上下文中所描述的本发明的某些特征还可以提供于单一实施例中的组合中。相反,为了简洁起见,在单一实施例的上下文中所描述的本发明的各种特征也可以单独地或以任何合适的子组合形式提供。例如,叙述开处和/或投与化合物1或其药学上可接受的盐、溶剂合物或水合物的方法可以分为两种方法;叙述处方化合物1或其药学上可接受的盐、溶剂合物或水合物的一种方法,以及叙述投与化合物1或其药学上可接受的盐、溶剂合物或水合物的另一种方法,此外,例如,叙述开处化合物1或其药学上可接受的盐、溶剂合物或水合物的方法,且叙述投与化合物1或其药学上可接受的盐、溶剂合物或水合物的本发明的单独方法可以组合成叙述开处和/或投与化合物1或其药学上可接受的盐、溶剂合物或水合物的单一方法。
本文提供共同投与化合物1与另一种药物的方法。本文还提供当与另一种药物共同投与时减少化合物1的剂量的方法。本文还提供当与化合物1共同投与时减少另一种药物的剂量的方法。本文还提供当向个体投与另一种药物时中断投与化合物1的方法。本文还提供当向个体投与化合物1时中断投与另一种药物的方法。本文还提供当向个体投与另一种药物时继续投与化合物1的方法。本文还提供当向个体投与化合物1时继续投与另一种药物的方法。本文还提供监测共同投与化合物1和另一种药物的个体的方法。本文还提供当与另一种药物共同投与时调定化合物1的剂量的方法。本文还提供当与化合物1共同投与时调定另一种药物的剂量的方法。本文还提供需要前述方法的组合的方法。
提供一种治疗患有鞘氨醇1-磷酸盐亚型1(S1P
还提供了一种向有需要的患者投与选自(R)-2-(7-(4-环戊基-3-(三氟甲基)苄氧基)-1,2,3,4-四氢环戊并[b]吲哚-3-基)乙酸(化合物1)或其药学上可接受的盐、水合物或溶剂合物的鞘氨醇1-磷酸盐亚型1(S1P
还提供了一种向有需要的患者投与选自(R)-2-(7-(4-环戊基-3-(三氟甲基)苄氧基)-1,2,3,4-四氢环戊并[b]吲哚-3-基)乙酸(化合物1)或其药学上可接受的盐、水合物或溶剂合物的鞘氨醇1-磷酸盐亚型1(S1P
在一些实施例中,所述方法进一步包含告知患者或医护人员向也正在被投与CYP2C8抑制剂、CYP2C9抑制剂、UDP-葡糖醛酸基转移酶(UGT)酶UGT1A1抑制剂或UDP-葡糖醛酸基转移酶(UGT)酶UGT1A6抑制剂的患者投与所述S1P
在一些实施例中,所述方法进一步包含告知患者或医护人员向也正在被投与CYP2C8抑制剂、CYP2C9抑制剂、UDP-葡糖醛酸基转移酶(UGT)酶UGT1A1抑制剂或UDP-葡糖醛酸基转移酶(UGT)酶UGT1A6抑制剂的患者投与S1P
在一些实施例中,也向患者投与CYP2C8抑制剂。在一些实施例中,也向患者投与CYP2C9抑制剂。在一些实施例中,也向患者投与UDP-葡糖醛酸基转移酶(UGT)酶UGT1A1抑制剂。在一些实施例中,也向患者投与UDP-葡糖醛酸基转移酶(UGT)酶UGT1A6抑制剂。
在一些实施例中,CYP2C8抑制剂是吉非罗齐(gemfibrozil)、利托那韦(ritonavir)、氯吡格雷(clopidogrel)、洛匹那韦(lopinavir)、地拉罗司(deferasirox)、拉帕替尼(lapatinib)、甲氧苄啶(trimethoprim)、噻唑烷二酮(thiazolidinediones)、孟鲁司特(montelukast)、槲皮素(quercetin)、坎地沙坦酯(candesartan cilexetil)(坎地沙坦的碳酸环己酯前药)、扎鲁司特(zafirlukast)、克霉唑(clotrimazole)、非洛地平(felodipine)、糠酸莫米松(mometasone furoate)、沙美特罗(salmeterol)、雷洛昔芬(raloxifene)、非诺贝特(fenofibrate)、利托那韦、左甲状腺素(levothyroxine)、他莫昔芬(tamoxifen)、氯雷他定(loratadine)、奥昔布宁(oxybutynin)、甲羟孕酮(medroxyprogesterone)、辛伐他汀(simvastatin)、酮康唑(ketoeonazole)、乙炔雌二醇(ethinyl estradiol)、螺内酯(spironolactone)、洛伐他汀(lovastatin)、硝苯地平(nifedipine)或厄贝沙坦(irbesartan)。在一些实施例中,CYP2C8抑制剂是吉非罗齐。
在一些实施例中,CYP2C9抑制剂是胺碘酮(amiodarone)、双硫仑(disulfram)、去氧氟尿苷(doxifluridine)、依非韦伦(efavirenz)、氟康唑(fluconazole)、伊马替尼(imatinib)、来氟米特(lefluonomide)、甲硝唑(metronidazole)、咪康唑(miconazole)、苯妥英(phenytoin)、磺胺甲恶唑(sulfamethoxazole)、磺胺苯吡唑(sulfapenazone)、西地那非(sildenafil)、扎鲁司特、伐地昔布(valdecoxib)、双氯芬酸(diclofenac)、伏立康唑(voriconazole)、他莫昔芬、氯沙坦(losartan)、华法林(warfarin)、依托度酸(etodolac)、甲芬那酸(mefenamic acid)、美洛昔康(meloxicam)、舒洛芬(suprofen)、厄贝沙坦(irbesartan)、布洛芬(ibuprofen)、氟伐他汀(fluvastatin)、舍曲林(sertraline)、氟伏沙明(fluvoxamine)、泮托拉唑(pantoprazole)、罗格列酮(rosiglitazone)、兰索拉唑(lansoprazole)、利托那韦、尼卡地平(nicardipine)、阿瑞吡坦(aprepitant)、地拉韦啶(delavirdine)、地氯雷他定(desloratadine)、格列苯脲(glyburide)、酮康唑、吉非罗齐、醋硝香豆素(acenocoumarol)、阿伐麦布(avasimibe)、罗伐他汀(rosuvastatin)、洛伐他汀(lovastatin)、辛伐他汀、阿托伐他汀(atorvastatin)、西立伐他汀(cerivastatin)、奎宁(quinine)、氯氮平(clozapine)或安定(diazepam)。在一些实施例中,CYP2C9抑制剂是氟康唑。在一些实施例中,CYP2C9抑制剂是吉非罗齐。
在一些实施例中,CYP2C9抑制剂是噻氯匹定(ticlopidine)、奥美拉唑(omeprazole)、帕罗西汀(paroxetine)、缬沙坦(valsartan)、硼替佐米(bortezomib)、丙戊酸(valproic acid)、奈韦拉平(nevirapine)、氮卓斯汀(azelastine)、氯诺昔康(氯诺昔康)、非诺贝特(fenofibrate)、异烟肼(isoniazid)、苯基丁氮酮(phenylbutazone)、丙磺舒(probenecid)、磺胺苯吡唑(sulfaphenazole)、替尼泊甙(teniposide)、依曲韦林(etravirine)、磺胺嘧啶(sulfadiazine)、苯磺唑酮(sulfinpyrazone)、磺胺异恶唑(sulfisoxazole)、甲氧苄氨嘧啶(trimethoprim)、来氟米特(leflunomide)、尼洛替尼(nilotinib)、乙胺嘧啶(pyrimethamine)、索拉非尼(sorafenib)、卡培他滨(capecitabine)、氟尿嘧啶(fluorouracil)、西他塞坦(sitaxentan)、反苯环丙胺(tranylcypromine)、氨基比林(aminophenazone)、氯吡格雷(clopidogrel)、维拉帕米(verapamil)、依托昔布(etoricoxib)、丙泊酚(propofol)、酮洛芬(ketoprofen)、塞曲司特(seratrodast)、磺胺恶唑(sulfamoxole)、氨氯地平(amlodipine)、氨酚喹(amodiaquine)、阿那曲唑(anastrozole)、阿托伐醌(atovaquone)、氯霉素(chloramphenicol)、环孢灵(cyclosporine)、西米替丁(cimetidine)、克霉唑(clotrimazole)、可卡因(cocaine)、秋水仙碱(colchicine)、胆钙化醇(cholecalciferol)、环嗪(cyclizine)、右旋芬氟拉明(dexfenfluramine)、右旋丙氧吩(dextropropoxyphene)、双香豆素(dicoumarol)、地尔硫卓(diltiazem)、双硫醒(disulfiram)、肾上腺素(epinephrine)、依普罗沙坦(eprosartan)、乙醇、非洛地平(felodipine)、氟卡尼(flecainide)、组胺(histamine)、茚地那韦(indinavir)、咯匹那韦(lopinavir)、氯雷他定、醋酸甲羟孕酮(medroxyprogesterone acetate)、甲醋唑胺(methazolamide)、吗氯贝胺(moclobemide)、莫达非尼(modafinil)、奈非那韦(nelfinavir)、硝苯地平(nifedipine)、尼鲁米特(nilutamide)、尼伐地平(nilvadipine)、奥氮平(olanzapine)、苯丁胺(phentermine)、吡格列酮(pioglitazone)、普仑司特(pranlukast)、普伐他汀(pravastatin)、普鲁米近(promethazine)、普罗帕酮(propafenone)、奎尼定(quinidine)、芸香苷(rutin)、沙奎那韦(saquinavir)、司来吉兰(selegiline)、磺胺二甲啶(sulfadimethoxine)、磺胺甲二唑(sulfamethizole)、磺胺(sulfanilamide)、磺胺吡啶(sulfapyridine)、替加色罗(tegaserod)、甲巯咪唑(methimazole)、甲硫达嗪(thioridazine)、噻康唑(tioconazole)、托卡朋(tolcapone)、三唑苯二氮(triazolam)、曲格列酮(troglitazone)、比卡鲁胺(bicalutamide)、雷贝拉唑(rabeprazole)、莫达非尼(armodafinil)、己烯雌酚(diethylstilbestrol)、阿戈美拉汀(agomelatine)、那可汀(noscapine)、氯维地平(clevidipine)、顺氯氨铂(cisplatin)、人血清蛋白、硫康唑(sulconazole)、维莫德吉(vismodegib)、瑞格非尼(regorafenib)、吉非替尼(gefitinib)、帕瑞昔布(parecoxib)、鲁玛卡托(lumacaftor)、阿比特龙(abiraterone)、替卡格雷(ticagrelor)、赛立替尼(ceritinib)、氟尿苷(floxuridine)、克立硼罗(crisaborole)、米哚妥林(midostaurin)、贝利司他(belinostat)、利非斯特(lifitegrast)、大黄酸(rhein)、双醋瑞因(diacerein)、二十二碳六烯酸、托匹司他(topiroxostat)、珠卡赛辛(zucapsaicin)、司替戊醇(stiripentol)、洛贝格列酮(lobeglitazone)、二苯噻庚英(dosulepin)、苯溴马隆(benzbromarone)、马尼地平(manidipine)、恩西地平(enasidenib)、坎地沙坦、鲁卡帕利(rucaparib)、艾沙康唑(isavuconazole)、黑升麻(cimicifuga racemose)、庚苯吡酮(nabilone)、磺胺乙酰异恶唑(acetyl sulfisoxazole)或姜黄素(curcumin)。
在一些实施例中,UDP-葡糖醛酸基转移酶(UGT)酶UGT1A1抑制剂是腺嘌呤、丙泊酚、消炎痛(indomethacin)、尼洛替尼、帕唑帕尼(pazopamb)、瑞格非尼、氟硝西泮、厄洛替尼、索拉非尼、恩西地平、哌仑他韦(pibrentasvir)、格拉卡匹伟(glecaprevir)、芦卡帕尼(rucaparib)、埃格列净(ertugliflozin)、或福坦替尼(fostamatinib)。
在一些实施例中,UDP-葡糖醛酸基转移酶(UGT)酶UGT1A6抑制剂是曲格列酮。
在一些实施例中,UGT底物是双氯芬酸。
还提供了一种向有需要的患者投与鞘氨醇1-磷酸盐亚型1(S1P
还提供了一种向有需要的患者投与鞘氨醇1-磷酸盐亚型1(S1P
在一些实施例中,所述方法进一步包含监测患者与膜转运蛋白的底物相关的毒性和临床反应的体征和症状。
在一些实施例中,所述方法进一步包含基于患者耐受与膜转运蛋白的底物相关的一种或多种暴露相关的不良反应的能力,减少投与患者的膜转运蛋白的底物的量。
在一些实施例中,所述方法进一步包含告知患者或医护人员共同投与S1P
在一些实施例中,所述方法进一步包含告知患者或医护人员共同投与S1P
在一些实施例中,监测毒性和临床反应的体征和症状包含监测膜转运蛋白的底物的血清浓度。
在一些实施例中,监测毒性和临床反应的体征和症状包含确定患者是否经历与膜转运蛋白的底物的血清浓度相关的一种或多种暴露相关的不良反应。
在一些实施例中,监测毒性和临床反应的体征和症状包含监测膜转运蛋白的底物的功效。
在一些实施例中,膜转运蛋白是P-糖蛋白(Pgp)。在一些实施例中,膜转运蛋白是BCRP(乳腺癌抗性蛋白)。在一些实施例中,膜转运蛋白是OATP1B1。
在一些实施例中,膜转运蛋白是P-糖蛋白。在一些实施例中,膜转运蛋白的底物选自地高辛(digoxin)、洛哌丁胺(loperamide)、小蘖碱(berberine)、伊立替康(irinotecan)、阿霉素(doxorubicin)、长春碱(vinblastine)、紫杉醇(paclitaxel)和非索非那定(fexofenadine)。
在一些实施例中,膜转运蛋白是BCRP。在一些实施例中,膜转运蛋白的底物选自米托蒽醌(mitoxantrone)、甲氨蝶呤(methotrexate)、拓扑替康(topotecan)、伊马替尼(imatinib)、伊立替康、他汀类药物(statins)、硫酸盐缀合物和卟啉(porphyrin)。
在一些实施例中,膜转运蛋白是OATP1B1。在一些实施例中,膜转运蛋白的底物选自溴磺酚酞(bromosulphophthalein)、雌酮-3-硫酸盐、雌二醇-17β-葡糖苷酸、他汀类药物、瑞格列奈(repaglinide)、缬沙坦、奥美沙坦(olmesartan)、胆红素葡糖苷酸、胆红素和胆汁酸。在一些实施例中,膜转运蛋白的底物是利福平(rifampin)。
在一些实施例中,膜转运蛋白是OATP1B3。在一些实施例中,膜转运蛋白的底物是利福平。
在一些实施例中,剂型在禁食条件下投与。在一些实施例中,剂型在进食条件下投与。
在一些实施例中,所述方法是非性别特异性的。
在一些实施例中,也向个体投与独立地选自口服皮质类固醇和氨基水杨酸盐的一种或多种药剂。
在一些实施例中,不向个体投与独立地选自那他珠单抗(natalizumab)、依法珠单抗(efalizumab)和利妥昔单抗(rituximab)的一种或多种药剂。在一些实施例中,如果向个体投与独立地选自那他珠单抗、依法珠单抗和利妥昔单抗的一种或多种药剂,那么不开始化合物1的投与。在一些实施例中,如果向个体投与独立地选自那他珠单抗、依法珠单抗和利妥昔单抗的一种或多种药剂,那么中断化合物1的投与。在一些实施例中,如果向个体投与独立地选自那他珠单抗、依法珠单抗和利妥昔单抗的一种或多种药剂,那么减少化合物1的剂量。在一些实施例中,还未向个体投与生物制剂。在一些实施例中,还未向个体投与两种或更多种生物制剂。在一些实施例中,还未向个体投与三种或更多种生物制剂。在一些实施例中,不向个体投与生物制剂。在一些实施例中,不向个体投与两种或更多种生物制剂。在一些实施例中,不向个体投与三种或更多种生物制剂。
在一些实施例中,在无调定的情况下投与标准剂量;且个体不经历严重相关不良事件。
在一些实施例中,如果个体不患有活动性感染,那么治疗有效量等效于约0.5mg至约5.0mg的化合物1。在一些实施例中,如果个体患有活动性感染,那么不向个体1投与化合物1。在一些实施例中,活动性感染是严重的活动性感染。在一些实施例中,所述方法进一步包含监测个体的活动性感染。在一些实施例中,所述方法进一步包含如果个体出现活动性感染,那么中断投与。
在一些实施例中,所述方法进一步包含在投与化合物1或其药学上可接受的盐、水合物或溶剂合物期间监测不良事件,且任选地中断或终止化合物1或其药学上可接受的盐、水合物或溶剂合物的投与。
在一些实施例中,治疗进一步包含监测投与期间的心率、监测投与期间的肺功能或监测投与期间的肝功能。
在一些实施例中,治疗进一步包含监测投与期间的心率。
在一些实施例中,治疗进一步包含监测投与期间的肺功能。
在一些实施例中,治疗进一步包含监测投与期间的肝功能。
在一些实施例中,所述方法降低由鞘氨醇1-磷酸盐亚型1(S1P
在一些实施例中,不良事件是严重不良事件。
在一些实施例中,严重不良事件是选自白细胞减少症、便秘、腹泻、恶心、腹痛、嗜中性白细胞减少症、呕吐、背痛和月经异常。
在一些实施例中,所述方法不导致严重不良事件。
在一些实施例中,投与标准剂量,且基本上不诱发个体的急性心率降低或心传导阻滞。
在一些实施例中,投与化合物1而未引起心率降低超过6bpm。
在一些实施例中,投与化合物1而没有在其它S1P受体调节剂的情况下所见的心率的首剂效应。在一些实施例中,投与化合物1而没有在其它S1P受体调节剂的情况下所见的AV传导的首剂效应。
在一些实施例中,先前向个体投与至少一种选自以下的药剂:TNF拮抗剂、整联蛋白拮抗剂和免疫抑制剂。
在一些实施例中,个体对至少一种药剂反应不足、丧失反应或不耐受。
在一些实施例中,个体在过去的3个月中表现出对选自口服5-氨基水杨酸盐、皮质类固醇、免疫抑制剂、TNFα拮抗剂和整联蛋白拮抗剂中的至少一种药剂反应不足、丧失反应或不耐受。在一些实施例中,个体在过去的6个月中表现出对选自口服5-氨基水杨酸盐、皮质类固醇、免疫抑制剂、TNFα拮抗剂和整联蛋白拮抗剂中的至少一种药剂反应不足、丧失反应或不耐受。在一些实施例中,个体在过去的9个月中表现出对选自口服5-氨基水杨酸盐、皮质类固醇、免疫抑制剂、TNFα拮抗剂和整联蛋白拮抗剂中的至少一种药剂反应不足、丧失反应或不耐受。在一些实施例中,个体在过去的1年中表现出对选自口服5-氨基水杨酸盐、皮质类固醇、免疫抑制剂、TNFα拮抗剂和整联蛋白拮抗剂中的至少一种药剂反应不足、丧失反应或不耐受。在一些实施例中,个体在过去的2年中表现出对选自口服5-氨基水杨酸盐、皮质类固醇、免疫抑制剂、TNFα拮抗剂和整联蛋白拮抗剂中的至少一种药剂反应不足、丧失反应或不耐受。在一些实施例中,个体在过去的3年中表现出对选自口服5-氨基水杨酸盐、皮质类固醇、免疫抑制剂、TNFα拮抗剂和整联蛋白拮抗剂中的至少一种药剂反应不足、丧失反应或不耐受。在一些实施例中,个体在过去的4年中表现出对选自口服5-氨基水杨酸盐、皮质类固醇、免疫抑制剂、TNFα拮抗剂和整联蛋白拮抗剂中的至少一种药剂反应不足、丧失反应或不耐受。在一些实施例中,个体在过去的5年中表现出对选自口服5-氨基水杨酸盐、皮质类固醇、免疫抑制剂、TNFα拮抗剂和整联蛋白拮抗剂中的至少一种药剂反应不足、丧失反应或不耐受。
在一些实施例中,在无调定的情况下投与标准剂量。
在一些实施例中,个体在投与标准剂量之前禁食。
在一些实施例中,治疗包含诱导和/或维持临床反应;改善粘膜的内窥镜外观;和/或诱导和/或维持临床缓解。
在一些实施例中,治疗包含诱导和/或维持组织学改善。
在一些实施例中,治疗包含诱导和/或维持组织学缓解。
在一些实施例中,治疗包含诱导和/或维持粘膜愈合。
在一些实施例中,在投与之前,个体的3分量梅奥诊所评分为至少6。
在一些实施例中,所述方法导致个体的3分量梅奥诊所评分改善。在一些实施例中,所述方法导致个体的2分量梅奥诊所评分改善。在一些实施例中,所述方法导致个体的总梅奥诊所评分改善。
在治疗发炎性肠病的方法的一些实施例中,例如溃疡性结肠炎,如中度至重度活动性溃疡性结肠炎,治疗导致内窥镜改善,例如改善粘膜的内窥镜外观。
在治疗发炎性肠病的方法的一些实施例中,例如溃疡性结肠炎,如中度至重度活动性溃疡性结肠炎,治疗诱导临床缓解。在治疗发炎性肠病的方法的一些实施例中,例如溃疡性结肠炎,如中度至重度活动性溃疡性结肠炎,治疗维持临床缓解。在治疗发炎性肠病的方法的一些实施例中,例如溃疡性结肠炎,如中度至重度活动性溃疡性结肠炎,治疗诱导且维持临床缓解。
在治疗发炎性肠病的方法的一些实施例中,例如溃疡性结肠炎,如中度至重度活动性溃疡性结肠炎,治疗诱导临床反应。在治疗发炎性肠病的方法的一些实施例中,例如溃疡性结肠炎,如中度至重度活动性溃疡性结肠炎,治疗维持临床反应。在治疗发炎性肠病的方法的一些实施例中,例如溃疡性结肠炎,如中度至重度活动性溃疡性结肠炎,治疗诱导且维持临床反应。
在一些实施例中,治疗使个体中的淋巴细胞计数减少至少40%。在一些实施例中,治疗使个体中的淋巴细胞计数减少至少45%、50%、55%、60%或65%。
在治疗发炎性肠病的方法的一些实施例中,例如溃疡性结肠炎,如中度至重度活动性溃疡性结肠炎,治疗导致不含皮质类固醇的缓解。
在治疗发炎性肠病的方法的一些实施例中,例如溃疡性结肠炎,如中度至重度活动性溃疡性结肠炎,治疗导致内窥镜缓解。
在治疗发炎性肠病的方法的一些实施例中,例如溃疡性结肠炎,如中度至重度活动性溃疡性结肠炎,治疗导致直肠出血改善。
在治疗发炎性肠病的方法的一些实施例中,例如溃疡性结肠炎,如中度至重度活动性溃疡性结肠炎,治疗导致组织学愈合。
在治疗发炎性肠病的方法的一些实施例中,例如溃疡性结肠炎,如中度至重度活动性溃疡性结肠炎,治疗导致大便频率改善。
在治疗发炎性肠病的方法的一些实施例中,例如溃疡性结肠炎,如中度至重度活动性溃疡性结肠炎,治疗进一步包含监测粪便钙卫蛋白的水平。
在治疗发炎性肠病的方法的一些实施例中,例如溃疡性结肠炎,如中度至重度活动性溃疡性结肠炎,治疗进一步包含监测C-反应蛋白(CRP)的水平。
在一些实施例中,治疗是减轻溃疡性结肠炎的体征和/或症状。在一些实施例中,治疗是减轻溃疡性结肠炎的体征。在一些实施例中,治疗是减轻溃疡性结肠炎的症状。在一些实施例中,治疗是减轻克罗恩氏病的体征和/或症状。在一些实施例中,治疗是减轻克罗恩氏病的体征。在一些实施例中,治疗是减轻克罗恩氏病的症状。
在一些实施例中,治疗是诱导和/或维持临床缓解。在一些实施例中,治疗是诱导和维持临床缓解。在一些实施例中,治疗是诱导和/或维持临床缓解和/或临床反应。在一些实施例中,治疗是诱导和维持临床缓解和临床反应。在一些实施例中,治疗是诱导临床缓解和/或临床反应。在一些实施例中,治疗是维持临床缓解和/或临床反应。在一些实施例中,治疗是诱导临床缓解和临床反应。在一些实施例中,治疗是维持临床缓解和临床反应。在一些实施例中,治疗是诱导和/或维持临床缓解和/或粘膜愈合。在一些实施例中,治疗是诱导和维持临床缓解和粘膜愈合。在一些实施例中,治疗是诱导和维持粘膜愈合。在一些实施例中,治疗是诱导和维持临床缓解。在一些实施例中,治疗是诱导临床缓解。在一些实施例中,治疗是诱导粘膜愈合。在一些实施例中,治疗是维持临床缓解。在一些实施例中,治疗是维持粘膜愈合。在一些实施例中,治疗是在诱导应答者中实现和/或维持临床缓解。在一些实施例中,治疗是在诱导应答者中实现和维持临床缓解。在一些实施例中,治疗是在诱导应答者中实现临床缓解。在一些实施例中,治疗是在诱导应答者中维持临床缓解。在一些实施例中,治疗是诱导和/或维持临床反应。在一些实施例中,治疗是诱导和维持临床反应。在一些实施例中,治疗是诱导临床反应。在一些实施例中,治疗是维持临床反应。在一些实施例中,治疗是诱导内窥镜改善。在一些实施例中,治疗是维持内窥镜改善。在一些实施例中,治疗是实现内窥镜改善。在一些实施例中,治疗改善内窥镜缓解。在一些实施例中,治疗是维持内窥镜缓解。在一些实施例中,治疗是诱导组织学愈合。在一些实施例中,治疗是维持组织学愈合。在一些实施例中,治疗是改善大便频率。在一些实施例中,治疗是维持大便频率的改善。在一些实施例中,治疗是改善粘膜的内窥镜外观。在一些实施例中,治疗是维持粘膜的内窥镜改善。在一些实施例中,治疗是在诱导期间改善粘膜的内窥镜外观。在一些实施例中,治疗消除了对皮质类固醇使用的需要。在一些实施例中,治疗允许减少皮质类固醇的使用。在一些实施例中,治疗允许使用较低剂量的皮质类固醇。在一些实施例中,治疗是实现不含皮质类固醇的缓解。在一些实施例中,治疗是维持不含皮质类固醇的缓解。在一些实施例中,治疗是改善直肠出血。在一些实施例中,治疗是维持直肠出血的改善。在一些实施例中,治疗是提高内窥镜分项评分。在一些实施例中,治疗是维持内窥镜分项评分的提高。
在一些实施例中,已使用2分量梅奥诊所评分来诊断溃疡性结肠炎。例如,在一些实施例中,已使用范围为0至9的直肠出血和内窥镜检查结果的评分来诊断溃疡性结肠炎。在一些实施例中,已使用3分量梅奥诊所评分来诊断溃疡性结肠炎。例如,在一些实施例中,已使用范围为0至9的大便频率、直肠出血和内窥镜检查结果的评分来诊断溃疡性结肠炎。在一些实施例中,已使用总梅奥评分来诊断溃疡性结肠炎。例如,在一些实施例中,已使用范围为0至12的大便频率、直肠出血、内窥镜检查结果和医师总体评估的评分来诊断溃疡性结肠炎。
在一些实施例中,使用2分量梅奥诊所评分测量溃疡性结肠炎的改善。在一些实施例中,使用3分量梅奥诊所评分测量溃疡性结肠炎的改善。在一些实施例中,使用总梅奥评分测量溃疡性结肠炎的改善。在一些实施例中,通过临床缓解来测量溃疡性结肠炎的改善。在一些实施例中,通过淋巴细胞减少来测量溃疡性结肠炎的改善。在一些实施例中,通过内窥镜改善来测量溃疡性结肠炎的改善。在一些实施例中,通过6分梅奥评分来测量溃疡性结肠炎的改善。例如,在一些实施例中,通过大便频率和直肠出血来测量溃疡性结肠炎的改善。在一些实施例中,溃疡性结肠炎的改善在统计学上是显著的。
在一些实施例中,不推荐在患有活动性、重度感染的个体中使用化合物1。在一些实施例中,不推荐在患有活动性感染的个体中使用化合物1。在一些实施例中,不推荐在患有重度感染的个体中使用化合物1。在一些实施例中,不推荐在患有活动性、重度感染的个体中使用化合物1,直到感染受到控制。在一些实施例中,不推荐在患有活动性感染的个体中使用化合物1,直到感染受到控制。在一些实施例中,不推荐在患有重度感染的个体中使用化合物1,直到感染受到控制。在一些实施例中,在活动性感染期间未开始投与化合物1。在一些实施例中,监测个体感染。在一些实施例中,如果个体出现感染,那么停止化合物1的投与。在一些实施例中,如果感染变得严重,那么停止化合物1的投与。在一些实施例中,如果个体出现感染,那么中断化合物1的投与。在一些实施例中,不向患有感染的个体投与化合物1。在一些实施例中,在活动性感染期间不投与化合物1。在一些实施例中,在活动性感染期间未开始投与化合物1;监测个体在投与期间是否出现感染;且如果感染变得严重,那么停止投与。在一些实施例中,感染是轻度的。在一些实施例中,感染是中度的。在一些实施例中,感染是重度的。在一些实施例中,感染是严重的。在一些实施例中,感染是严重的不良事件。在一些实施例中,感染是呼吸道感染。
在一些实施例中,投与化合物1而未引起严重不良事件。在一些实施例中,投与化合物1而未引起与心率有关的严重不良事件。在一些实施例中,投与化合物1而未引起与心率变化有关的严重不良事件。在一些实施例中,投与化合物1而未引起与心率升高有关的严重不良事件。在一些实施例中,投与化合物1而未引起与心动过缓有关的严重不良事件。在一些实施例中,投与化合物1而未引起与AV阻断有关的严重不良事件。在一些实施例中,投与化合物1而未引起与AV传导有关的严重不良事件。在一些实施例中,投与化合物1而未引起心动过缓。在一些实施例中,投与化合物1而未引起AV阻滞。在一些实施例中,在治疗的第一天投与化合物1而未引起超过轻度的心率降低(例如>10bpm)。在一些实施例中,投与化合物1而没有在其它S1P受体调节剂的情况下所见的首剂效应。在一些实施例中,投与化合物1而没有在其它S1P受体调节剂的情况下所见的首剂心血管效应。在一些实施例中,投与化合物1而没有心率的症状变化。在一些实施例中,投与化合物1而没有心律的症状变化。在一些实施例中,化合物1无需调定即可投与以避免在其它S1P受体调节剂的情况下所见的首剂效应。
在一些实施例中,投与化合物1而不增加肝功能测试(LFT)。在一些实施例中,投与化合物1而不引起LFT升高。在一些实施例中,投与化合物1而不使ALT增加。在一些实施例中,投与化合物1而不使AST增加。在一些实施例中,投与化合物1而不使ALT增加>3X ULN。在一些实施例中,投与化合物1而不使ALT增加>2.5X ULN。在一些实施例中,投与化合物1而不使ALT增加>2X ULN。在一些实施例中,投与化合物1而不使ALT增加>1.5X ULN。在一些实施例中,投与化合物1而不使AST增加>3X ULN。在一些实施例中,投与化合物1而不使AST增加>2.5X ULN。在一些实施例中,投与化合物1而不使AST增加>2X ULN。在一些实施例中,投与化合物1而不使AST增加>1.5X ULN。在一些实施例中,投与化合物1而不使胆红素增加。在一些实施例中,投与化合物1而不使胆红素增加>3X ULN。在一些实施例中,投与化合物1而不使胆红素增加>2.5X ULN。在一些实施例中,投与化合物1而不使胆红素增加>2X ULN。在一些实施例中,投与化合物1而不使胆红素增加>1.5X ULN。在一些实施例中,投与化合物1而不使γ-谷氨酰转移酶(GGT)增加。在一些实施例中,投与化合物1而不使GGT增加>3X ULN。在一些实施例中,投与化合物1而不使GGT增加>2.5X ULN。在一些实施例中,投与化合物1而不使GGT增加>2X ULN。在一些实施例中,投与化合物1而不使GGT增加>1.5X ULN。
在一些实施例中,投与化合物1而未引起肺功能测试异常。在一些实施例中,投与化合物1而未引起黄斑水肿。
在一些实施例中,个体对用于治疗发炎性肠病的另一种药剂反应不足、丧失反应、不耐受,或表现出依赖性。在一些实施例中,个体对用于治疗发炎性肠病的另一种药剂反应不足。在一些实施例中,个体对用于治疗发炎性肠病的另一种药剂丧失反应。在一些实施例中,个体对用于治疗发炎性肠病的另一种药剂不耐受。在一些实施例中,个体需要连续的类固醇疗法。在一些实施例中,另一种药剂是选自以下的至少一种药剂:肿瘤坏死因子(TNF)拮抗剂、皮质类固醇、整联蛋白拮抗剂和免疫抑制剂以及氨基水杨酸盐。
在一些实施例中,个体对常规疗法反应不足、丧失反应或不耐受。在一些实施例中,个体对常规疗法反应不足。在一些实施例中,个体对常规疗法丧失反应。在一些实施例中,个体对常规疗法不耐受。在一些实施例中,常规疗法是选自:至少一种选自以下的药剂:肿瘤坏死因子(TNF)拮抗剂、皮质类固醇、整联蛋白拮抗剂和免疫抑制剂以及氨基水杨酸盐。
在一些实施例中,先前已对个体投与皮质类固醇和/或氨基水杨酸盐。在一些实施例中,先前已对个体投与肿瘤坏死因子(TNF)拮抗剂、整联蛋白拮抗剂和/或免疫抑制剂。
在一些实施例中,皮质类固醇是口服皮质类固醇。在一些实施例中,TNF拮抗剂是TNF-α阻断剂在一些实施例中,氨基水杨酸盐是5-氨基水杨酸盐。在一些实施例中,整联蛋白拮抗剂被称为整联蛋白受体拮抗剂。在一些实施例中,TNF拮抗剂被称为TNF阻断剂。在一些实施例中,免疫抑制剂被称为免疫调节剂。在一些实施例中,先前常规疗法被称为先前治疗。
在一些实施例中,未向个体投与治疗剂量的硫代嘌呤。在一些实施例中,未向个体投与治疗剂量的硫唑嘌呤。在一些实施例中,未向个体投与治疗剂量的6-巯基嘌呤。在一些实施例中,未向个体投与治疗剂量的硫鸟嘌呤(也称为硫代鸟嘌呤或6-硫鸟嘌呤)。
在一些实施例中,抑制剂是中度抑制剂。在一些实施例中,抑制剂是强抑制剂。在一些实施例中,诱导剂是中度诱导剂。在一些实施例中,诱导剂是强诱导剂。
在一些实施例中,当化合物1与CYP底物共同投与时要谨慎。在一些实施例中,当化合物1与UGT底物共同投与时要谨慎。在一些实施例中,当化合物1与OAT底物共同投与时要谨慎。在一些实施例中,当化合物1与强抑制剂共同投与时要谨慎。在一些实施例中,强抑制剂是强CYP抑制剂。在一些实施例中,强抑制剂是CYP2C8。在一些实施例中,当化合物1与中度抑制剂共同投与时要谨慎。在一些实施例中,中度抑制剂是中度CYP抑制剂。在一些实施例中,中度抑制剂是CYP2C9抑制剂。在一些实施例中,当化合物1与强诱导剂共同投与时要谨慎。在一些实施例中,强抑制剂是强CYP诱导剂。在一些实施例中,当化合物1与中度诱导剂共同投与时要谨慎。在一些实施例中,中度诱导剂是中度CYP抑制剂。在一些实施例中,中度诱导剂是CYP2C8诱导剂。在一些实施例中,中度诱导剂是CYP2C9诱导剂。
在一些实施例中,当与CYP底物一起使用时,化合物1的剂量受到限制。在一些实施例中,当使用UGT底物时,化合物1的剂量受到限制。在一些实施例中,当与OAT底物一起使用时,化合物1的剂量受到限制。在一些实施例中,当与强抑制剂一起使用时,化合物1的剂量受到限制。在一些实施例中,化合物1的剂量限于或限于约、不超过或不超过约0.25、0.3、0.4、0.5、0.6、0.7、0.75、0.8、0.9、1.0、1.1、1.2、1.3、1.4、1.5、1.6、1.7、1.75、1.8、1.9或2.0mg。在一些实施例中,当与强抑制剂一起使用时,化合物1以最低剂量投与。在一些实施例中,当与中度抑制剂一起使用时,化合物1的剂量受到限制。在一些实施例中,当与中度抑制剂一起使用时,化合物1以最低剂量投与。在一些实施例中,最低剂量是最低有效剂量。在一些实施例中,最低剂量是最低市售剂量。在一些实施例中,最低剂量是美国的最低市售剂量。在一些实施例中,化合物1的最低剂量为或为约0.25、0.3、0.4、0.5、0.6、0.7、0.75、0.8、0.9、1.0、1.1、1.2、1.3、1.4、1.5、1.6、1.7、1.75、1.8、1.9或2.0mg。
在一些实施例中,当与化合物1一起使用时,共同投与的化合物的剂量受到限制。在一些实施例中,共同投与的化合物是CYP底物。在一些实施例中,共同投与的化合物是UGT底物。在一些实施例中,共同投与的化合物的剂量是OAT底物。在一些实施例中,共同投与的化合物是强诱导剂。在一些实施例中,共同投与的化合物是强抑制剂。在一些实施例中,共同投与的化合物是中度诱导剂。在一些实施例中,共同投与的化合物是中度抑制剂。在一些实施例中,共同投与的化合物以最低剂量投与。在一些实施例中,最低剂量是最低有效剂量。在一些实施例中,最低剂量是最低市售剂量。在一些实施例中,最低剂量是美国的最低市售剂量。
在一些实施例中,化合物1不与CYP底物一起使用。在一些实施例中,化合物1不与UGT底物一起使用。在一些实施例中,化合物1不与OAT底物一起使用。在一些实施例中,化合物1不与强诱导剂一起使用。在一些实施例中,化合物1不与中度诱导剂一起使用。在一些实施例中,化合物1不与强抑制剂一起使用。在一些实施例中,化合物1不与中度抑制剂一起使用。
在一些实施例中,不推荐将化合物1与CYP底物同时使用。在一些实施例中,不推荐将化合物1与UGT底物同时使用。在一些实施例中,不推荐将化合物1与OAT底物同时使用。在一些实施例中,不推荐将化合物1与强诱导剂同时使用。在一些实施例中,不推荐将化合物1与中度诱导剂同时使用。在一些实施例中,不推荐将化合物1与强抑制剂同时使用。在一些实施例中,不推荐将化合物1与中度抑制剂同时使用。在一些实施例中,不推荐将化合物1与CYP2C8抑制剂同时使用。在一些实施例中,不推荐将化合物1与CYP2C8诱导剂同时使用。在一些实施例中,不推荐将化合物1与CYP2C9抑制剂同时使用。在一些实施例中,不推荐将化合物1与CYP2C8诱导剂同时使用。
一些实施例提供用(R)-2-(7-(4-环戊基-3-(三氟甲基)苄氧基)-1,2,3,4-四氢环戊并[b]吲哚-3-基)乙酸(化合物1)或其药学上可接受的盐、水合物或溶剂合物治疗个体的方法。
一些实施例提供安全投与(R)-2-(7-(4-环戊基-3-(三氟甲基)苄氧基)-1,2,3,4-四氢环戊并[b]吲哚-3-基)乙酸(化合物1)或其药学上可接受的盐、水合物或溶剂合物的方法。
一些实施例提供在个体接受CYP底物、OAT底物或UGT底物的同时投与每日剂量的化合物1的方法。在一些实施例中,化合物1的每日剂量不超过或不超过约0.25、0.3、0.4、0.5、0.6、0.7、0.75、0.8、0.9、1.0、1.1、1.2、1.3、1.4、1.5、1.6、1.7、1.75、1.8、1.9、2.0、2.1、2.2、2.25、2.3、2.4、2.5、2.6、2.7、2.75、2.8、2.9或3.0mg。在一些实施例中,化合物1的每日剂量小于或小于约0.25、0.3、0.4、0.5、0.6、0.7、0.75、0.8、0.9、1.0、1.1、1.2、1.3、1.4、1.5、1.6、1.7、1.75、1.8、1.9、2.0、2.1、2.2、2.25、2.3、2.4、2.5、2.6、2.7、2.75、2.8、2.9或3.0mg。
在一些实施例中,CYP底物是CYP2C8底物。
在一些实施例中,CYP底物是CYP2C9底物。
在一些实施例中,UGT底物是UGT1A1底物。
在一些实施例中,UGT底物是UGT1A4底物。
在一些实施例中,UGT底物是UGT1A6底物。
在一些实施例中,UGT底物是UGT1A7底物。
在一些实施例中,OAT底物是OATP1B1底物。
在一些实施例中,OAT底物是OATP1B3底物。
在一些实施例中,OAT底物是OAT1底物。
在一些实施例中,OAT底物是OAT3底物。
在一些实施例中,CYP2C8抑制剂、CYP2C9抑制剂、CYP2C8诱导剂或CYP2C9诱导剂是氟康唑。
在一些实施例中,CYP2C8抑制剂、CYP2C9抑制剂、CYP2C8诱导剂或CYP2C9诱导剂是吉非罗齐。
在一些实施例中,CYP2C8抑制剂、CYP2C9抑制剂、CYP2C8诱导剂或CYP2C9诱导剂是利福平。
在一些实施例中,OATP1B1的底物是利福平。
在一些实施例中,OATP1B3的底物是利福平。
在一些实施例中,共同投与的化合物是CYP2C8抑制剂。
在一些实施例中,共同投与的化合物是CYP2C8诱导剂。
在一些实施例中,共同投与的化合物是CYP2C9抑制剂。
在一些实施例中,共同投与的化合物是CYP2C9诱导剂。
在一些实施例中,共同投与的化合物是氟康唑。
在一些实施例中,共同投与的化合物是吉非罗齐。
在一些实施例中,共同投与的化合物是利福平。
在一些实施例中,小于将向也未被投与CYP底物、OAT底物、UGT底物、CYP2C8抑制剂、CYP2C9抑制剂、CYP2C8诱导剂、CYP2C9诱导剂、UGT1A1抑制剂或UGT1A6抑制剂的患者投与的量是比将向也未被投与CYP底物、OAT底物、UGT底物、CYP2C8抑制剂、CYP2C9抑制剂、CYP2C8诱导剂、CYP2C9诱导剂、UGT1A1抑制剂或UGT1A6抑制剂的患者投与的量小约、至少、或至少约1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、778、79、80、81、82、83、84、85、86、87、88、89或90%。
在一些实施例中,小于将向也未被投与CYP底物、OAT底物、UGT底物、CYP2C8抑制剂、CYP2C9抑制剂、CYP2C8诱导剂、CYP2C9诱导剂、UGT1A1抑制剂或UGT1A6抑制剂的患者投与的量是比将向也未被投与CYP底物、OAT底物、UGT底物、CYP2C8抑制剂、CYP2C9抑制剂、CYP2C8诱导剂、CYP2C9诱导剂、UGT1A1抑制剂或UGT1A6抑制剂的患者投与的量少约、至少、或至少约0.1、0.2、0.25、0.3、0.4、0.5、0.6、0.7、0.75、0.8、0.9、1.0、1.1、1.2、1.25、1.3、1.4、1.5、1.6、1.7、1.75、1.8、1.9或2.0mg。
一些实施例提供了向有需要的患者安全投与鞘氨醇1-磷酸盐亚型1(S1P
在一些实施例中,S1P
已显示对S1P
内皮S1P
对S1P
在临床试验中,FTY720引起不良事件(即,一过性无症状心动过缓),这可能是由于其对S1P
据报告FTY720至少在自身免疫性心肌炎的大鼠模型和急性病毒性心肌炎的小鼠模型中具有治疗功效(Kiyabayashi等人,《心血管药理学(J.Cardiovasc.)》,35:410-416,2000;Miyamoto等人,《美国大学心血管杂志(J.Am.Coll.Cardiol)》,37:1713-1718,2001);发炎性肠病(包括结肠炎)的小鼠模型(Mizushima等人,《发炎性肠病(Inflamm.BowelDis.)》,10:182-192,2004;Deguchi等人,《肿瘤学报告(Oncology Reports)》,16:699-703,2006;Fujii等人,《美国生理学、胃肠、肝脏生理学杂志(Am.J.Physiol.Gastrointest.Liver Physiol)》,291:G267-G274,2006;Daniel等人,《免疫学杂志(J.Immunol.)》,178:2458-2468,2007);进行性肾小球膜增生性肾小球肾炎的大鼠模型(Martini等人,《美国生理学杂志-肾脏生理学(Am.J.Physiol.Renal Physiol.)》,292:F1761-F1770,2007);在使用S1P
此外,据报告FTY720在大鼠和小鼠(人类多发性硬化症模型)的实验性自身免疫性脑脊髓炎(EAE)中具有治疗功效(Brinkmann等人,《生物化学杂志(J.Biol.Chem.)》,277:21453-21457,2002;Fujino等人,《药理学与实验治疗学杂志(J.Pharmacol Exp.Ther.)》,305:70-77,2003;Webb等人,《神经免疫学杂志(J.Neuroimmunol)》,153:108-121,2004;Rausch等人,《磁共振成像杂志(J.Magn.Reson,Imaging)》,20:16-24,2004;Kataoka等人,《细胞与分子免疫(Cellular&Molecular Immunology)》,2:439-448,2005;Brinkmann等人,《药理学和治疗学(Pharmacology&Therapeutics)》,115:84-105,2007;Baumruker等人,《研究药物专家评论(Expert Opin.Investig.Drugs)》,16:283-289,2007;Balatoni等人,《大脑研究公报(Brain Research Bulletin)》,74:307-316,2007)。此外,在临床试验中已发现FTY720对多发性硬化症具有治疗功效。在复发缓解型多发性硬化症的II期临床试验中,发现FTY720减少通过磁共振成像(MRI)检测到的病变的数目和患有多发性硬化症的个体的临床疾病活性(Kappos等人,《新英格兰医学杂志(N.Engl.J.Med.)》,355:1124-1140,2006;Martini等人,《研究药物专家评论(Expert Opin.Investig.Drugs)》,16:505-518,2007;Zhang等人,《小型-药物化学评论(Mini-Reviews in Medicinal Chemistry)》,7:845-850,2007;Brinkmann,《药理学和治疗学(Pharmacology&Therapeutics)》,115:84-105,2007)。已经报告了在患有缓解型-复发多发性硬化症的个体中使用FTY720进行的III期临床研究(Brinkmann,《药理学和治疗学(Pharmacology&Therapeutics)》,115:84-105,2007;Baumruker等人,《研究药物专家评论(Expert Opin.Investig.Drugs)》,16:283-289,2007;Dev等人,《药理学和治疗学(Pharmacology&Therapeutics)》,117:77-93,2008)。
据报告,FTY720具有抗病毒活性。在淋巴细胞性脉络丛脑膜炎病毒(LCMV)小鼠模型中已呈现了具体数据,其中用LCMV的Armstrong或克隆13毒株感染小鼠(Premenko-Lanier等人,《自然》,454,894,2008)。
据报告,FTY720削弱了被由土拉弗朗西斯菌(Francisella tularensis)感染的树突细胞向纵隔淋巴结的迁移,从而减少其细菌定殖。土拉弗朗西斯菌与兔热病(tularemia)、溃疡及淋巴结病感染、呼吸道感染和伤寒相关(E.Bar-Haim等人,《PLoS病原学(PLoS Pathogens》,4(11):e1000211,2008年11月21日出版;info:doi/10.1371/journal.ppat.1000211,2008)。
最近还报告,短期高剂量的FTY720在实验性自身免疫性葡萄膜视网膜炎中迅速减少了眼部浸润。在眼部炎症的早期给予FTY720可以快速预防视网膜损伤。据报告,FTY720不仅阻止靶器官的渗透,而且减少现有的渗透(Raveney等人,《眼科学档案(Arch.Ophthalmol)》,126(10),1390,2008)。
据报告,用FTY720进行的治疗通过减少附接到骨头表面的成熟破骨细胞的数目而减轻小鼠中的卵巢切除术诱导的骨质疏松。数据为S1P控制破骨细胞前体的迁移行为,动态调节骨矿物质稳态提供了证据(Ishii等人,《自然》,在线优先出版,2009年2月8日,doi:10.1038/nature07713)。
S1P
还报告了S1P
S1P
据报告,FTY720具有抑制病理性血管生成的治疗功效,如在肿瘤发展中可能发生的抑制作用。通过FTY720抑制血管形成被认为涉及S1P
据报告,给小鼠口服FTY720有效地阻断VEGF诱导的血管通透性,这是与血管形成、发炎和病理状况如败血病、缺氧和实体瘤生长相关的重要过程(T Sanchez等人,《生物化学杂志(J.Biol.Chem.)》,278(47),47281-47290,2003)。
环孢菌素A和FK506(钙调神经磷酸酶抑制剂)是用于防止移植器官排斥反应的药物。尽管它们有效延迟或抑制移植排斥反应,但已知经典的免疫抑制剂(如环孢菌素A和FK506)会引起多种不良副作用,包括肾毒性、神经毒性、β细胞毒性和胃肠道不适。在器官移植中存在对没有这些副作用的免疫抑制剂的未满足的需要,所述免疫抑制剂作为单一疗法或与经典免疫抑制剂组合有效用于抑制例如同种异体抗原反应性T细胞迁移至移植组织,从而延长移植物存活时间。
已显示,FTY720作为单一疗法和与经典免疫抑制剂包括环孢菌素A、FK506和RAD(mTOR抑制剂)的协同组合在移植排斥反应中具有治疗功效。已显示,不同于经典的免疫抑制剂环孢菌素A、FK506和RAD,FTY720具有延长移植物存活时间而不诱导全身免疫抑制的功效,且认为药物作用的此差异与观察到的组合的协同作用有关(Brinkmann等人,《移植进程(Transplant Proc.)》,33:530-531,2001;Brinkmann等人,《移植(Transplantation)》,72:764-769,2001)。
据报告,S1P
据报告,FTY720在不依赖前列腺素合成的神经性疼痛的保留神经损伤模型中减少疼痛性行为(O.Costu等人,《细胞与分子医学杂志(Journal of Cellular and MolecularMedicine)》12(3),995-1004,2008)。
据报告,FTY720削弱了小鼠接触超敏反应(CHS)的发生。在致敏阶段期间来自用FTY720处理的小鼠的免疫淋巴结细胞的过继转移实际上不能在接受者中诱导CHS反应(D.Nakashima等人,《皮肤病研究杂志(J.Investigative Dermatology)》(128(12),2833-2841,2008)。
据报告,预防性口服投与FTY720(1mg/kg,每周三次)完全阻止了C57BL/6小鼠中实验性自身免疫性重症肌无力(EAMG)的发展(T.Kohono等人,《生物学和医药学公报(Biological&Pharmaceutical Bulletin)》,28(4),736-739,2005)。
在一个实施例中,本发明涵盖作为S1P
使用化合物1的1期研究以0.1mg、0.35mg、1mg、3mg和5mg的单一剂量进行。化合物1作为L-精氨酸盐投与。0.1mg至3mg的较低剂量被受试者良好耐受,仅报告了较少的不良事件,其中最常见的是头痛和接触性皮炎。在>0.35mg的所有剂量中观察到心率的剂量依赖性降低,然而,在低于5mg剂量的剂量下没有报告与心动过缓相关的不良事件。在5mg剂量下观察到剂量限制性不良事件,其中3名(50%)受试者经历4AE心动过缓并伴有一级或二级房室(AV)阻滞,导致剂量递增中断。研究中的最大耐受剂量是3mg。研究中没有死亡或严重不良事件。
除了对外周血淋巴细胞计数的预期药理学作用之外,在生命体征、ECG、肺功能测试、检眼镜检查或临床实验室测试方面没有其它临床上显著的安全问题。以3mg和5mg的剂量投与引起外周血B细胞、T细胞、NK细胞以及除TEM细胞外的所有T细胞亚群的绝对数量的剂量响应性下降。投与后2-4小时总外周血淋巴细胞(PBL)计数降低,到第8小时达到最低点,持续24小时,随后4天恢复到基线。在3mg和5mg剂量水平下,PBL计数降低了约40%和约55%。TEM细胞不表达CCR7,且能够独立于S1P受体表达而再循环。因此,这些发现与S1P受体激动剂在临床前研究和人类中的预期药效学作用一致(Gergely等人,《英国药理学杂志(BrJ Pharmacol)》167(5):1035-1047,2012;Brossard等人,《英国临床药理学杂志(Br J ClinPharmacol)》2013年4月18日.doi:10.1111/bcp.12129.[Epub提前打印]PubMed PMID:23594176和Kovarik等人,《临床药理学杂志(J Clin Pharmacol)》44(5):532-537,2004.)
S1P
在一个实施例中,本发明涵盖为S1P
SIP
在一些实施例中,鞘氨醇1-磷酸盐亚型1(S1P
在一些实施例中,S1P
在一些实施例中,S1P
在一些实施例中,S1P
在一些实施例中,S1P
在一些实施例中,S1P
在一些实施例中,S1P
在一些实施例中,S1P
在一些实施例中,S1P
在一些实施例中,S1P
在一些实施例中,S1P
在一些实施例中,S1P
在一些实施例中,S1P
在一些实施例中,S1P
在一些实施例中,S1P
在一些实施例中,S1P
在一些实施例中,S1P
在一些实施例中,S1P
在一些实施例中,S1P
在一些实施例中,S1P
在一些实施例中,S1P
在一些实施例中,S1P
在一些实施例中,S1P
在一些实施例中,S1P1受体相关的病症是心肌缺血-再灌注损伤。
在一些实施例中,S1P
在一些实施例中,S1P
在一些实施例中,标准剂量的量等同于1mg的化合物1。
在一些实施例中,标准剂量的量等同于2mg的化合物1。
在一些实施例中,标准剂量的量等同于3mg的化合物1。
在一些实施例中,每天一次向个体投与标准剂量的化合物1或其药学上可接受的盐、水合物或溶剂合物。
在一些实施例中,口服投与化合物1或其药学上可接受的盐、水合物或溶剂合物。
在一些实施例中,将化合物1或其药学上可接受的盐、水合物或溶剂合物调配为适合于口服投与的胶囊或片剂。
在一些实施例中,化合物1或其药学上可接受的盐、水合物或溶剂合物选自:化合物1;化合物1的钙盐;和化合物1的L-精氨酸盐。在一些实施例中,化合物1或其药学上可接受的盐、水合物或溶剂合物是化合物1的L-精氨酸盐。在一些实施例中,化合物1或其药学上可接受的盐、水合物或溶剂合物是化合物1的L-精氨酸盐的无水、非溶剂化结晶形式。在一些实施例中,化合物1或其药学上可接受的盐、水合物或溶剂合物是化合物1的无水、非溶剂化结晶形式。
在一些实施例中,还向个体投与治疗剂量的口服5-ASA化合物。在一些实施例中,还向个体投与稳定剂量的口服5-ASA化合物。
在一些实施例中,还向个体投与治疗剂量的口服皮质类固醇疗法。在一些实施例中,还向个体投与稳定剂量的口服皮质类固醇疗法。在一些实施例中,皮质类固醇是泼尼松、例如剂量≤10mg/天或20mg/天或等效类固醇的泼尼松。在一些实施例中,皮质类固醇是布地奈德,例如剂量≤9mg/天或等效类固醇。
在一些实施例中,还向个体投与治疗剂量的免疫抑制剂。在一些实施例中,还向个体投与治疗剂量的硫代嘌呤。在一些实施例中,还向个体投与治疗剂量的硫唑嘌呤。在一些实施例中,还向个体投与治疗剂量的6-硫基嘌呤。在一些实施例中,还向个体投与治疗剂量的硫鸟嘌呤(也称为硫代鸟嘌呤或6-硫鸟嘌呤)。
在一些实施例中,还向个体投与治疗剂量的益生菌。在一些实施例中,还向个体投与治疗剂量的康萃乐(Culturelle)。在一些实施例中,还向个体投与治疗剂量的布拉酵母菌(Saccharomyces boulardii)。
在一些实施例中,还向个体投与治疗剂量的止泻药。在一些实施例中,还向个体投与治疗剂量的洛哌丁胺。在一些实施例中,还向个体投与治疗剂量的苯乙呱啶和阿托品。
本发明的一些实施例包括产生用于“组合疗法”的药物组合物的方法,所述方法包含将至少一种根据本文所公开的任何化合物实施例的化合物与至少一种如本文所描述的已知药剂和药学上可接受的载剂混合。
还提供包含标准剂量的化合物1或其药学上可接受的盐、水合物或溶剂合物和任选地一种或多种药学上可接受的载剂的药物组合物。还提供包含化合物1或其药学上可接受的盐、水合物或溶剂合物,任选地一种或多种药学上可接受的载剂的药物组合物。载剂在与调配物的其它成分相容且对受体不过度有害的意义上必须是“可接受的”。
在一些实施例中,化合物1或其药学上可接受的盐、水合物或溶剂合物以原始或纯化学物质(例如以胶囊调配物中的粉末形式)形式投与。
在一些实施例中,将化合物1或其药学上可接受的盐、水合物或溶剂合物调配为进一步包含一种或多种药学上可接受的载剂的药物组合物。
药物组合物可通过任何合适的方法制备,通常通过以所需的比例将活性化合物与液体或细粉状固体载剂或这两者均匀混合,随后,如果需要,将所得混合物形成所需形状。
常规的赋形剂,如粘合剂、填充剂、可接受的湿润剂、压片润滑剂和崩解剂可用于口服投与的片剂和胶囊中。可以使用所属领域的技术人员众所周知的技术将本文所描述的化合物调配到药物组合物中。除了本文提到的那些,合适的药学上可接受的载剂是所属领域已知的;例如,参见《雷明顿:药学的科学与实践(Remington,The Science and Practiceof Pharmacy)》,第20版,2000,Lippincott Williams&Wilkins,(编辑:Gennaro等人)。
对于口服投与,药物组合物可呈例如片剂或胶囊的形式。药物组合物优选以含有特定量活性成分的剂量单位的形式制成。此类剂量单位的实例是胶囊、片剂、粉末、颗粒或悬浮液,其具有常规添加剂如乳糖、甘露醇、玉米淀粉或马铃薯淀粉;具有粘合剂如结晶纤维素、纤维素衍生物、阿拉伯树胶、玉米淀粉或明胶;具有崩解剂如玉米淀粉、马铃薯淀粉或羧甲基纤维素钠;且具有润滑剂如滑石或硬脂酸镁。固体形式制剂包括粉剂、片剂、丸剂、胶囊、扁囊剂、栓剂和可分散颗粒。固体载剂可以是一种或多种物质,其也可充当稀释剂、调味剂、增溶剂、润滑剂、悬浮剂、粘合剂、防腐剂、片剂崩解剂或包封材料。
呈散剂形式时,载剂是细粉状固体,它呈与细粉状活性成分的混合物形式。
呈片剂形式时,活性成分与具有必要结合能力的载剂以合适的比例混合,且被压制成所需的形状和大小。
粉剂和片剂可含有不同百分比量的活性化合物。粉剂或片剂中的代表性量可为活性化合物的0.5%至约90%。然而,技术人员将会知道何时需要超出此范围的量。用于粉剂和片剂的合适载剂包括碳酸镁、硬脂酸镁、滑石、糖、乳糖、果胶、糊精、淀粉、明胶、黄蓍胶、甲基纤维素、羧甲基纤维素钠、低熔点蜡、可可脂等。术语“制剂”包括具有包封材料作为提供胶囊的载剂的活性化合物的调配物,其中具有或不具有载剂的活性组分被载剂包围,所述载剂因此与其结合。类似地,包括扁囊剂和锭剂。片剂、粉剂、胶囊、丸剂、扁囊剂和锭剂可以用作适合于口服投与的固体形式。
医药制剂较佳地呈单位剂型。以此类形式,将制剂再分为含有适量活性成分的单位剂量。单位剂型可以是包装制剂,所述包装含有离散量的制剂,如包装的片剂或胶囊。此外,单位剂型可以是胶囊或片剂本身,或它可以是适当数量的包装形式的这些中的任一种。
其它实施例包括以下实例中所公开的实施例,其不应理解为以任何方式限制。
实例
实例1
如表1所示,制备由含有化合物1的L-精氨酸盐的速释硬明胶胶囊组成的调配物。
表1
实例2
如表2所示,制备由含有化合物1的L-精氨酸盐的速释片剂组成的调配物。
表2
实例3:
化合物1作为细胞色素P450(CYP)的抑制剂的体外评估和UDP-葡糖醛酸基转移酶(UGT)人肝微粒体中的酶
评估化合物1对细胞色素P450(CYP)酶CYP1A2、CYP2B6、CYP2C8、CYP2C9、CYP2C19、CYP2D6、CYP3A4/5(使用两种不同的标记底物)和人肝微粒体中的UDP-葡糖醛酸基转移酶(UGT)酶UGT1A1、UGT1A3、UGT1A4、UGT1A6、UGT1A9、UGT2B7和UGT2B17的潜在抑制作用是为了确定化合物1抑制伴随投与的药物代谢的潜力。
为了评估作为CYP活性的直接和代谢依赖性抑制剂以及作为UGT活性的直接抑制剂的化合物1,在存在或不存在浓度为0、1或10μM的化合物1的情况下,将来自200个个体的集合的人肝微粒体与标记底物一起培育。为了CYP酶的代谢依赖性抑制,在与标记底物一起培育之前,将化合物1与人肝微粒体一起在NADPH再生系统中预先培育30分钟。在适当时,包括CYP和UGT酶的已知代谢依赖性和/或直接抑制剂作为阳性对照。
与rCYP酶培育120分钟后,使用rCYP2C8可以最大程度地观察到化合物1的损失(高达75%),而使用rCYP2C9、rCYP2C19和rCYP3A4则可以观察到12%至36%的损失。在没有化学抑制剂的HLM中,化合物1的总体损失可忽略不计(0至13.0%)。选择性CYP2C8抑制剂(吉非罗齐)分别抑制85%、47%和60%的化合物1向两种氧化代谢物和酮代谢物的有限转化率。CYP2C9抑制剂(替尼酸(tienilic acid))也抑制了化合物1向第二种氧化代谢物的有限转化率(72%)。
在化合物1的存在下,对CYP2C8活性的直接抑制在1μM下为22%,在10μM下为约100%。在浓度高达10μM的化合物1的存在下,几乎没有或没有观察到直接抑制CYP1A2、CYP2B6、CYP2C9、CYP2C19、CYP2D6或CYP3A4/5活性。此外,几乎没有或没有证据表明CYP的代谢依赖性抑制。
发现用化合物1处理培养的人肝细胞对CYP1A2 mRNA没有诱导潜力。此外,未观察到CYP2B6和CYP3A4的诱导潜力,基于诱导标准是在阳性对照中观察到的mRNA增加>2倍,且mRNA增加>20%。
在测试的最高浓度(10μM)下,化合物1直接抑制UGT1A1和UGT1A6的活性分别高达28%和48%。没有证据表明UGT1A3、UGT1A4、UGT1A9、UGT2B7或UGT2B17的直接抑制,因为在浓度高达10μM的化合物1的存在下,观察到抑制率小于8%。
根据两种CYP反应表型方法的结果,确定了CYP2C8和CYP2C9在化合物1向氧化代谢物的转化中起主要作用,且CYP2C8在酮代谢物的形成中也起主要作用。然而,在HLM中化合物1向代谢物的总体转化率可忽略不计,且在存在和不存在直接作用和代谢依赖性选择性CYP抑制剂的情况下观察到的化合物1的损失几乎没有差异。在存在化合物1的情况下,对CYP2C8活性的直接抑制在1μM下是22%,在10μM下是约100%。然而,对于具有化合物1的任何其它CYP,几乎没有观察到或无直接抑制。在测试的最高浓度(10μM)下,化合物1直接抑制UGT1A1和UGT1A6的活性分别高达28%和48%,但没有证据表明UGT1A3、UGT1A4、UGT1A9、UGT2B7或UGT2B17的直接抑制。化合物1(高达10μM)不是CYP1A2、CYP2B6和CYP3A4/5的潜在诱导剂。
实例4
化合物1被评估为人类ABC转运蛋白P-gp、BCRP和BSEP或人类SLC转运蛋白OATP1B1、OATP1B3、OAT1、OAT3、OCT1、OCT2、MATE1和MATE2-K的潜在底物和/或抑制剂。P-gp和BCRP在许多组织的顶膜上表达。P-gp和BCRP在肠细胞的腔膜、脑中的内皮细胞、肾近端小管的刷状缘膜和肝细胞的小管膜中表达,其中它们限制了肠吸收、血脑屏障渗透且有助于排泄到胆汁和尿液中。BSEP主要在肝细胞的小管膜中表达,其中它有助于排泄到胆汁中。OATP1B1、OATP1B3和OCT1在肝细胞的窦状隙膜上表达,且促进内源性和异源性化合物在肝细胞中的积累,以进一步代谢或排泄到胆汁中。OAT1、OAT3和OCT2在肾近端小管的基底外侧膜上表达,且促进化合物在近端小管中的积累,以进一步在尿液中排泄。MATE1和MATE2-K(多药和毒素挤出蛋白)主要在近端管状细胞的内腔(顶端)膜上表达,且被认为在阳离子和两性离子排泄到尿液中起作用。MATE1还在肝脏中在肝细胞的小管膜上表达且介导阳离子药物的胆汁排泄。MATE1和MATE2-K可与在肝细胞小管膜和近端小管的基底外侧膜上表达的OCT转运蛋白协同作用以介导排泄。作为转运蛋白的底物或抑制剂的化合物可为药物-药物相互作用的受害者或肇事者。
用过量表达人类渗透性糖蛋白(P-gp)和乳腺癌抗性蛋白(BCRP)的马丁-达比(Madin-Darby)犬肾细胞(MDCKII)评估作为P-gp和BCRP的底物以及作为BCRP的抑制剂的化合物1。使用衍生自人类结肠癌(Caco 2)细胞的极化细胞系评估作为P-gp抑制剂的化合物1。表达胆汁盐输出泵(BSEP)的膜囊泡用于囊泡转运测定,以评估作为BSEP抑制剂的化合物1。将用含有有机阴离子转运蛋白1B1(OATP1B1)、有机阴离子转运蛋白1B3(OATP1B3)、有机阴离子转运蛋白1(OAT1)、有机阴离子转运蛋白3(OAT3)、有机阳离子转运蛋白1(OCT1)、有机阳离子转运蛋白2(OCT2)的人类转运蛋白cDNA的载体转染的人类胚胎肾细胞(HEK293)和对照细胞(仅用载体转染的HEK293细胞)用于实验,以评估作为OATP1B1、OATP1B3、OAT1、OAT3、OCT1和OCT2的底物和抑制剂的化合物1。用含有肾多药和毒素挤出转运蛋白1和2-K(MATE1、MATE2-K)的载体转染的HEK293细胞用于评估作为MATE1和MATE2-K的抑制剂的化合物1。ABC和SLC转运蛋白的已知底物和抑制剂作为阳性对照包括在所有实验中。
在10μM下不存在P-gp抑制。在100μM化合物1的存在下,地高辛(10μM)穿过Caco-2细胞的流出比降低约50%,指示化合物1是具有约100μM的IC50值的P-gp抑制剂。
在化合物1(10和100μM)的存在下,哌唑嗪(prazosin)(1μM)在MDCKII-BCRP细胞中校正的流出比降低了50%以上;然而,由于测定后的TEER值低于可接受标准,且荧光黄Papp值在10和100μM时均在可接受范围之上,因此无法确定化合物1的抑制潜力。为了确定IC50,使用在0.03至30μM范围内的七种浓度的化合物1进行了第二项实验。所得IC50值是35.7μM。
在化合物1(1和10μM)的存在下,表达BSEP的囊泡中[3H]-牛磺胆酸的ATP依赖性吸收率降低了小于50%,指示化合物1在所评估的浓度下不是BSEP的抑制剂。
1μM化合物1对OATP1B1不存在抑制。在10μM化合物1的存在下,[3H]-雌二醇-17β-葡糖苷酸(50nM)对表达OATP1B1的细胞的吸收率降低了约50%,指示化合物1是OATP1B1的抑制剂,其中IC50值是约10μM。
在化合物1(1和10μM)的存在下,[3H]-雌二醇-17β-葡糖苷酸(50nM)对表达OATP1B3的细胞的吸收率降低了小于50%,指示化合物1在所评估的浓度下不是OATP1B3的抑制剂(IC50>10μM)。
在化合物1(1和10μM)的存在下,表达OAT1的细胞对[3H]-对氨基马尿酸盐(1μM)的吸收率降低了小于50%,指示化合物1在所评估的浓度下不是OAT1的抑制剂(IC50>10μM)。
在化合物1(1和10μM)的存在下,[3H]-雌酮-3-硫酸盐(50nM)对表达OAT3的细胞的吸收率降低了小于50%,指示化合物1在所评估的浓度下不是抑制剂(IC50>10μM)。
在化合物1(1和10μM)的存在下,[14C]-四乙基溴化铵(5μM)对表达OCT1的细胞的吸收率降低了小于50%,指示化合物1在所评估的浓度下不是抑制剂(IC50>10μM)。
在化合物1(1和10μM)的存在下,[14C]-二甲双胍(10μM)在表达OCT2的细胞的吸收率并未降低,指示化合物1在所评估的浓度下不是OCT2的抑制剂(IC50>10μM)。
在P-gp抑制剂伐司扑达(valspodar)(10μM)的存在下,化合物1(1μM)穿过MDCKII-P-gp细胞的流出比是1.12且增加至3.08。化合物1(10μM)穿过MDCKII-P-gp细胞的流出比是3.17,且在伐司扑达的存在下并未降低。这些结果指示化合物1不是P-gp的底物,因为在抑制剂的存在下化合物1的流出比并未降低。应注意,化合物1的回收率低至18%至60%,且可能归因于与板的非特异性结合。
在不存在和存在BCRP抑制剂Ko143(1μM)的情况下,化合物1穿过MDCKII-BCRP细胞的流出比在1μM下小于2,且在10μM下大于2。尽管化合物1在10μM下的流出比是7.36,且在Ko143(1μM)的存在下降低至3.17,但化合物1在1μM下的流出比数据指示化合物1不是BCRP的底物。考虑作为BCRP转运蛋白的底物,化合物1的流出比在较低浓度(1μM)下将明显高于在较高浓度(10μM)下,且随后发生的抑制作用在较低浓度(1μM)下将可能更高。应注意,化合物1的回收率低至27%至48%,且可能归因于与板的非特异性结合。
在不存在和存在OATP1B1抑制剂利福平的情况下,化合物1(1和10μM)对表达OATP1B1的细胞的吸收率小于2。这些结果表明化合物1不是OATP1B1的底物。
在不存在和存在OATP1B3抑制剂利福平的情况下,化合物1(1和10μM)对表达OATP1B3的细胞的吸收率小于2。这些结果表明化合物1不是OATP1B3的底物。
在不存在和存在OAT1抑制剂丙磺舒(100μM)的情况下,化合物1(1和10μM)对表达OAT1的细胞的吸收率小于2。这些结果表明化合物1不是OAT1的底物。
在不存在和存在OAT3抑制剂丙磺舒(100μM)的情况下,化合物1(1和10μM)对表达OAT3的细胞的吸收率低于2。这些结果表明化合物1不是OAT3的底物。
在不存在和存在OCT1抑制剂奎尼丁(100μM)的情况下,化合物1(1和10μM)对表达OCT1的细胞的吸收率低于2。这些结果表明化合物1不是OCT1的底物。
在不存在和存在OCT2抑制剂奎尼丁(300μM)的情况下,化合物1(1和10μM)对表达OCT2的细胞的吸收率低于2。这些结果表明化合物1不是OCT2的底物。
表3.化合物1作为人类ABC和SLC转运蛋白的潜在底物的体外表征
表4.化合物1作为人类ABC和SLC转运蛋白的潜在抑制剂的体外表征
总体来说,此研究的结果表明:
化合物1在所评估的浓度下不是P-gp、BCRP、OATP1B1、OATP1B3、OAT1、OAT3、OCT1和OCT2转运蛋白的底物。化合物1抑制P-gp、BCRP和OATP1B1,其中IC
化合物1抑制P-gp、BCRP和OATP1B1,其中IC50值分别是约100、35.7和约10μM。
化合物1(高达10μM)对所检查的其它转运蛋白(BSEP、OATP1B3、OAT1、OAT3、OCT1、OCT2、MATE1和MATE2-K)引起小于50%的抑制。发现化合物1对BSEP、OATP1B3、OAT1、OAT3、OCT1、OCT2、MATE1和MATE2-K转运蛋白不具有IC50>10μM的抑制潜力。
实例5
评估化合物1以评估禁食状态下2mg片剂和胶囊调配物的单剂量相对口服生物利用度,确定食物对2mg片剂的药代动力学的影响,评估化合物1药代动力学中潜在的性别差异,且评估健康成人受试者的安全性和耐受性。
在健康成人受试者中进行了一项随机、单剂量、开放标签、三期、交叉、1期研究。将总共14名受试者(7名男性;7名女性)1:1随机分成两组,即序列1和序列2。在第一治疗期的禁食条件下,序列1组接受2mg剂量的化合物1硬明胶胶囊调配物(治疗A),序列2组接受2mg剂量的化合物1片剂调配物(治疗B)。经过7天的冲洗后,两组在第二治疗期间在禁食条件下交叉接受替代治疗。在另外7天洗脱后,所有受试者在第三治疗期间接受治疗C(在进食条件下单次2mg剂量的化合物1片剂调配物;即,FDA标准高脂肪高热量餐)。在投与后直至120个小时的预定时间点收集用于确定化合物1的血浆浓度的血液样品。使用经过验证的LC/MS/MS分析对血浆样品中的化合物1进行分析。在PhoenixTM
在禁食条件下,片剂和胶囊调配物的化合物1平均血浆浓度-时间曲线之间没有发现显著差异(图1)。比较片剂与胶囊调配物,化合物1峰值(C
对于在进食和禁食条件下投与的片剂调配物,在化合物1的平均血浆浓度-时间曲线之间没有发现显著差异(图1)。在进食和禁食条件下比较片剂调配物时,化合物1峰值(C
在整个治疗过程中,与男性相比,化合物1的平均血浆浓度和暴露参数在女性中仅适度较高。
在研究过程中,三个受试者共报告了八例不良事件,且与先前在健康志愿者研究中看到的一致。所有不良事件均被认为是治疗紧急事件,只有三例不良事件与研究药物的投与有关。没有严重的不良事件(SAE)或导致受试者停药的不良事件。在ECG或身体检查中未观察到临床上显著的异常。没有与临床上显著超出范围的生命体征相关的不良事件。
发现化合物1胶囊和片剂调配物是生物等效的(可互换的)。对于在禁食和进食条件下投与的片剂调配物,在化合物1的暴露方面无明显差异,且因此可以服用化合物1而无需考虑进餐。认为在PK暴露测量中观察到的适度的性别差异不太可能具有临床意义。总体来说,当在进食和禁食条件下以2mg单剂量投与时,研究产品在健康受试者中耐受良好。
表5.统计分析比较在禁食条件下2mg片剂(治疗B;测试)与在禁食条件下2mg胶囊的全身暴露量(治疗A;参考)
表6.统计分析比较在进食条件下(治疗C;测试)和禁食条件下(治疗B;参考)2mg片剂的全身暴露
实例6
在开放标签、三治疗、随机、固定序列研究中评估化合物1(1mg,2mg),以评估在健康成人受试者中在存在和不存在给与氟康唑(CYP2C9的中度抑制剂)、吉非罗齐(CYP2C8的强抑制剂)或利福平(CYP2C8和CYP2C9的中度诱导剂)的情况下血浆药代动力学(PK)、药效动力学(PD)、安全性和耐受性。
基于完成的体外代谢研究和人体质量平衡临床研究的结果,化合物1似乎通过氧化(CYP2C8和CYP2C9)和结合(UGT1A7,微小程度UGT1A1和UGT1A4)途径被广泛地代谢(总体>25%)。化合物1主要由于新陈代谢和胆汁/粪便排泄而从全身循环中消除。在存在和不存在可能影响一或多种这些代谢/消除途径的共同投与药物的情况下,评估化合物1药代动力学的潜在变化-氟康唑或吉非罗齐(归因于显著的CYP抑制作用)和利福平(归因于显著的CYP诱导作用)。
研究需要三种治疗之一:1)单独的和在稳态氟康唑(400mg负荷剂量,接着是每日口服剂量200mg)的存在下的化合物1(1mg);2)单独的和在稳态吉非罗齐(600mg BID口服剂量)的存在下的化合物1(1mg);和3)单独的和在稳态利福平的存在下的化合物1(2mg)(600mg每日口服剂量)。
每种治疗包括两个阶段:阶段1(单独投与单次口服剂量的化合物1)和阶段2(在存在抑制剂或诱导剂的情况下投与化合物1)。每个阶段间隔7天的洗脱期。
在阶段1的第1天,禁食过夜后,受试者(n=16/治疗组)将接受单次口服剂量的化合物1(对于治疗A和B是1mg;对于治疗C是2mg),且在接下来的7天内评估单次剂量PK。
在阶段2中,每个治疗组的所有16名受试者将接受以下治疗:
在第1天(单独的化合物1)和第12/15天(在稳态CYP2C8/CYP2C9抑制剂或诱导剂的存在下)的剂量投与后评估化合物1的PK和PD曲线。所测量的PK参数将包括以下使用每个比较的协方差模型的分析来评估(即,单独的化合物1对比CYP2C8/CYP2C9抑制剂或诱导剂):
·Cmax:直接由浓度时间曲线确定的最大浓度
·tmax:在直接由浓度-时间曲线确定的药物投与之后达到最大血浆浓度的时间
·t1/2:末端消除半衰期计算为:ln2/λz
·λz:通过在浓度-时间曲线的末端阶段上选择至少3个数据点来确定末端消除速率常数
·AUC0-24:使用线性对数梯形法则计算的零至24小时的浓度-时间曲线下面积(AUC)
·AUC0-168:使用线性对数梯形法则计算的零至168小时的浓度-时间曲线下面积(AUC)
·AUC0-t
·AUC最后:使用线性梯形法则计算的从时间零到最后可定量的浓度时间(t最后)的AUC
·AUC
·CL/F:口服后的全身清除率
·Vz/F:口服后基于末期的表观分布量
·MR:按AUC0-168(代谢物)/AUC0-168(母体)计算的代谢比
初级分析评估化合物1和潜在主要代谢物的PK以评估药物-药物相互作用的程度。
尤其基于对本专利文献的回顾,所公开的方法的其它用途对于所属领域的技术人员将变得显而易见。
- 治疗与S1P1受体相关的病况的方法
- 调整MC5受体活性的方法以及与这种受体相关的病况的治疗