一种嘧啶-2,4-二胺类化合物及其制备方法与应用
文献发布时间:2023-06-19 11:19:16
技术领域:
本发明涉及药物化学技术领域,具体涉及一种嘧啶-2,4-二胺类化合物及其制备方法与应用。
背景技术:
炎症性疾病和自身免疫性疾病的治疗药物开发一直是药物化学领域的重要任务。已经有大量的药物广泛的应用于临床治疗,但是这些药物的安全性一直是难以克服的问题。因此,在现阶段新型抗炎药物和自身免疫性疾病治疗药物的开发过程中,药物的安全性是首要考虑的问题。
严重急性呼吸综合征冠状病毒2(SARS-CoV-2)已经在全世界范围内肆虐。出现SARS-CoV-2感染后严重炎症性疾病的患者的身体状况无法承受传统抗炎药物治疗过程中的副作用。因此,寻找新型、有效和安全的治疗靶点并开发相应的药物,不仅仅是治疗炎症性疾病和自身免疫性疾病的需求,更是治疗SARS-CoV-2感染后严重炎症性疾病的迫切需求。
寻找新型、有效和安全的治疗靶点是突破这一困境的基本出发点。组织蛋白酶C(Cathepsin C),也称为二肽基肽酶1(DPP1,CTSC,EC 3.4.14.1),是一种重要的溶酶体半胱氨酸蛋白酶,来自木瓜蛋白酶家族。组织蛋白酶C通过介导中性粒细胞丝氨酸蛋白酶(NSP)的成熟过程,参与多形核中性粒细胞相关的炎症和免疫调节过程。因此,组织蛋白酶C是治疗炎症性疾病和自身免疫性疾病的有效靶标。
多形核中性粒细胞是最早募集到炎症位点的细胞,并形成了抵御入侵微生物的首道防线。多形核中性粒细胞发挥免疫功能的机制之一是分泌中性粒细胞丝氨酸蛋白酶,包括弹性蛋白酶(NE),组织蛋白酶G,蛋白酶3(PR3)和中性粒细胞丝氨酸蛋白酶(NSP4)。在中性粒细胞成熟的早期,NSPs被合成为在氨基末端含有二肽结构的惰性酶原形式。NSP酶原被组织蛋白酶C激活后,成熟的NSPs与髓过氧化物酶和NADPH氧化酶复合物产生的活性氧结合,以帮助降解吞噬溶酶体内的病原微生物。在某些疾病状态下,由嗜中性粒细胞的积累和活化引起的活性NSPs的过度分泌会导致组织损伤和炎症。NSPs参与多种炎性疾病的进展,例如败血症,急性胰腺炎,类风湿性关节炎,抗中性粒细胞胞浆抗体相关的坏死性新月形肾小球肾炎,慢性阻塞性肺疾病(COPD),支气管扩张和囊性纤维化。此外,最新的证据指出,NSPs分泌到细胞外会导致嗜中性粒细胞胞外陷阱的形成,该现象被认为是严重COVID-19的驱动因素。因此,NSPs被认为是包括COVID-19在内的嗜中性粒细胞相关炎症性疾病治疗有希望的生物学靶标。组织蛋白酶C是通过介导NSPs的成熟来参与调节炎症和免疫过程的。因此,通过靶向组织蛋白酶C发挥抗炎作用的药物必须能够影响体内NSPs的活性。
组织蛋白酶C抑制剂的开发已经经历了三十多年,直到2020年6月,美国食品和药物管理局才授予刚结束II期临床试验的组织蛋白酶C抑制剂brensocatib突破性药物资格,用于治疗成人非囊性纤维化支气管扩张症(NCFBE)。几乎所有报道的组织蛋白酶C抑制剂都含有能与Cys234形成共价键的“战斗部”。亲电子“战斗部”基团通常可以提高针对目标的选择性和效率。但是有时,这些组织蛋白酶C抑制剂的肽性质和亲电子性质与不良代谢稳定性有关。亲电子“战斗部”基团的高反应性有时可能导致选择性差、脱靶效应,这可能带来潜在的安全隐患。这些问题是药物开发中的严重障碍,也可能是组织蛋白酶C抑制剂在临床药物开发中未取得重大进展的主要原因。
由于组织蛋白酶C抑制剂或催化底物的结合环境相对较浅,因此,普遍认为非共价抑制剂带来的相互作用有限而不适合作为小分子组织蛋白酶C抑制剂的开发方向。我们保留对这种观点的评价,因为我们认为,非共价抑制剂同样可以有效抑制组织蛋白酶C的催化活性。尽管Cys234是组织蛋白酶C功能的关键氨基酸,但没有证据表明引入亲电子“战斗部”基团对于开发有效的组织蛋白酶C抑制剂是必要的。亲电子“战斗部”造成的代谢稳定性差和潜在的安全隐患是需要克服的问题。因此,我们决定开发一种新型的“非肽衍生物非共价组织蛋白酶C抑制剂”,希望在避免亲电子“战斗部”基团引起的代谢稳定性差和潜在的安全隐患的同时,这种抑制剂抑制组织蛋白酶C活性和下游NSP的活化,并可以表现出有效的体内抗炎活性。迄今为止,还没有小分子作为“非肽衍生物非共价组织蛋白酶C抑制剂”抑制组织蛋白酶C和下游NSP活化的报道。
本发明通过化合物分子库的筛选和基于结构的药物化学优化,确定了以嘧啶-2,4-二胺为核心的结构骨架进行“非肽衍生物非共价组织蛋白酶C抑制剂”的研发,以期发现具有更佳治疗收益的组织蛋白酶C抑制剂并扩展靶向组织蛋白酶C的小分子库。
发明内容:
本发明所要解决的技术问题在于提供一种嘧啶-2,4-二胺类化合物及其制备方法,该类化合物作为一类“非肽衍生物非共价组织蛋白酶C抑制剂”,可以应用于制备预防或治疗炎症性疾病和自身免疫性疾病的药物中。
本发明所要解决的技术问题采用以下的技术方案来实现:
一种嘧啶-2,4-二胺类化合物,如式I、式II所示:
其中,R
R
R
R
R
所述嘧啶-2,4-二胺类化合物的结构式如下所示(化合物1-71):
所述嘧啶-2,4-二胺类化合物的制备方法,包括以下步骤:
(1)将2,4-二氯嘧啶或者C5取代的2,4-二氯嘧啶类化合物和R
(2)中间体I和R
(3)化合物NO
(4)中间体II发生加氢反应,得到中间体III;
(5)中间体I和中间体III经布赫瓦尔德-哈特维希反应,得到式II所示的嘧啶-2,4-二胺类化合物;
反应方程式如下:
所述嘧啶-2,4-二胺类化合物在制备调控组织蛋白酶催化活性制剂中的应用。
进一步地,所述组织蛋白酶为组织蛋白酶C。
所述嘧啶-2,4-二胺类化合物在制备治疗炎症和自身免疫性疾病药物中的应用。
进一步地,所述炎症和自身免疫性疾病选自关节炎、类风湿性关节炎、败血症、急性胰腺炎、肾炎、慢性阻塞性肺疾病、囊性纤维化和支气管扩张。
本发明的有益效果是:
(1)本发明的嘧啶-2,4-二胺类化合物可用于生物学或药理学现象,组织蛋白酶C参与的信号通路传导研究,以及对于新型组织蛋白酶C抑制剂的评价;
(2)本发明的嘧啶-2,4-二胺类化合物经体外抗组织蛋白酶C活性筛选,结果显示其对组织蛋白酶C表现出较强的抑制活性,同时毒性较低;
(3)本发明的嘧啶-2,4-二胺类化合物经体内抗组织蛋白酶C活性筛选,结果显示其对组织蛋白酶C表现出较强的抑制活性,同时毒性较低;
(4)本发明的嘧啶-2,4-二胺类化合物经体内抗中性粒细胞丝氨酸蛋白酶活性筛选,结果显示其对中性粒细胞丝氨酸蛋白酶表现出较强的抑制活性,同时毒性较低;
(5)本发明的嘧啶-2,4-二胺类化合物经体内抗炎症活性筛选,结果显示其对炎症疾病模型有效的治疗作用,同时毒性较低;
(6)本发明所述的嘧啶-2,4-二胺类化合物结构新颖、合成工艺简单、产品纯度高,具有良好的应用前景。
附图说明:
图1为本发明的化合物1对正常小鼠体内组织蛋白酶C和下游NSPs的抑制作用;
图2为本发明的化合物1在模型组对COPD模型小鼠体内组织蛋白酶C和下游NSPs的抑制作用;
图3为本发明的化合物1对COPD模型小鼠体内炎症因子的影响。
具体实施方式:
为了使本发明实现的技术手段、创作特征、达成目的与功效易于明白了解,下面结合具体实施例和图示,进一步阐述本发明。
实施例1
化合物41的合成:5-氯-N
1.1中间体I-1的制备:5-氯-N
称取0.5g 2,4,5-三氯嘧啶(2.73mmol)加入到50mL圆底烧瓶中,并以5mL二甲基亚砜溶解,然后加入催化量的四丁基碘化铵,搅拌5min。待溶液由无色变为黄色后加入0.28g间三氟甲基苯胺(3.00mmol)和0.3g三乙胺(3.00mmol),在室温下搅拌3h。用薄层色谱法(石油醚:乙酸乙酯=20:1)检测反应终点。待反应结束后,用25mL冰水淬灭反应并搅拌30min后,用乙酸乙酯萃取(3次,每次30mL)混合物。合并萃取的乙酸乙酯层,用饱和氯化钠溶液洗涤三次。乙酸乙酯层用无水硫酸钠上干燥24h后过滤并浓缩乙酸乙酯层,得到的残留物用自动化中压色谱纯化系统进行纯化分离。产物为白色固体;产率为89%(0.58g)。White solid(yield 69%).mp 121-122℃;
1.2中间体III-1的制备:5-(4-甲基哌嗪-1-基)吡啶-2-胺
称取15g 5-溴-2-硝基吡啶(73.9mmol)加入到250mL圆底烧瓶中,并以100mL二甲基亚砜溶解,然后加入碳酸钾15.3g(110.7mmol),在70℃油浴锅上搅拌10min。随后加入12.7g1-甲基-哌嗪(126.1mmol),并继续反应5h。用薄层色谱法(石油醚:乙酸乙酯=10:1)检测反应终点。待反应结束后,用300mL冰水淬灭反应并搅拌30min后,用乙酸乙酯萃取(3次,每次200mL)混合物。合并萃取的乙酸乙酯层,用饱和氯化钠溶液洗涤三次。乙酸乙酯层用无水硫酸钠上干燥24h后过滤并浓缩乙酸乙酯层,得到的黄色残留固体用乙醇重结晶即可获得纯的1-甲基-4-(6-硝基吡啶-3-基)哌嗪即中间体II-1。将1-甲基-4-(6-硝基吡啶-3-基)哌嗪2g(9.0mmol)重新完全溶解在甲醇中,加入10%Pd/C(0.2g)作为催化剂,在高压反应釜中或在氢气氛围的圆底烧瓶中进行催化加氢反应。待高压反应釜中压力不再降低或溶液颜色变得澄清,反应即为结束。将反应液过滤除去催化剂,浓缩液体即可得到纯品中间体III-1。中间体III-1在密闭的惰性气体中保存或现制现用。
1.3化合物41的合成
取25mL封管以氩气进行气体置换。取0.26g中间体I-1(0.83mmol)、0.2g中间体III-1(1.00mmol)、38mg Pd
实施例2
化合物1的合成:5-氯-N
实验步骤同实施例1,仅以中间体I-1替换为中间体I-2,以中间体III-1替换为苯胺。White solid(yield:77%).mp 142-144℃;
实施例3
化合物2的合成:5-氯-N
实验步骤同实施例1,仅以中间体I-1替换为中间体I-3,以中间体III-1替换为苯胺。White solid(yield:59%).mp 183-185℃;
实施例4
化合物3的合成:5-氯-N
实验步骤同实施例1,仅以中间体I-1替换为中间体I-4,以中间体III-1替换为苯胺。White solid(yield:68%).mp 181-183℃;
实施例5
化合物4的合成:5-氯-N
实验步骤同实施例1,仅以中间体I-1替换为中间体I-5,以中间体III-1替换为苯胺。White solid(yield:50%).mp 145-146℃;
实施例6
化合物5的合成:5-氯-N
实验步骤同实施例1,仅以中间体I-1替换为中间体I-6,以中间体III-1替换为苯胺。White solid(yield:64%).mp 147-150℃;
实施例7
化合物6的合成:5-氯-N
实验步骤同实施例1,仅以中间体I-1替换为中间体I-7,以中间体III-1替换为苯胺。White solid(yield:70%).mp 150-153℃;
实施例8
化合物7的合成:N-(3-((5-氯-2-(苯基氨基)嘧啶-4-基)氨基)苯基)乙酰胺(7)
实验步骤同实施例1,仅以中间体I-1替换为中间体I-8,以中间体III-1替换为苯胺。White solid(yield:67%).mp 208-210℃;
实施例9
化合物8的合成:5-氯-N
实验步骤同实施例1,仅以中间体I-1替换为中间体I-9,以中间体III-1替换为苯胺。White solid(yield:40%).mp 181-183℃;
实施例10
化合物9的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为苯胺。White solid(yield:68%).mp160.5-161.2℃;
实施例11
化合物10的合成:5-氯-N
实验步骤同实施例1,仅以中间体I-1替换为中间体I-10,以中间体III-1替换为苯胺。White solid(yield:66%).mp 195-197℃;
实施例12
化合物11的合成:5-氯-N
实验步骤同实施例1,仅以中间体I-1替换为中间体I-11,以中间体III-1替换为苯胺。White solid(yield:30%).mp 193-196℃;
实施例13
化合物12的合成:5-氯-N
实验步骤同实施例1,仅以中间体I-1替换为中间体I-12,以中间体III-1替换为苯胺。White solid(yield:60%).mp 188-190℃;
实施例14
化合物13的合成:5-氯-N
实验步骤同实施例1,仅以中间体I-1替换为中间体I-13,以中间体III-1替换为苯胺。White solid(yield:63%).mp 160-163℃;
实施例15
化合物14的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为环丙胺。White solid(yield:59%).mp138.5-139.1℃;
实施例16
化合物15的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为环戊胺。White solid(yield:61%).mp117.2-118.2℃;
实施例17
化合物16的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为环己胺。White solid(yield:57%).mp 155.7-158.1℃;
实施例18
化合物17的合成:N
实验步骤同实施例1,仅以中间体III-1替换为苄胺。White solid(yield:39%).mp 120.3-121.3℃;
实施例19
化合物18的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为苯乙胺。White solid(yield:35%).mp107-108.2℃;
实施例20
化合物19的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为对甲基苯胺。White solid(yield:53%).mp 160-161.2℃;
实施例21
化合物20的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为对甲氧基苯胺。White solid(yield:39%).mp 184.7-185.2℃;
实施例22
化合物21的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为对乙基苯胺。White solid(yield:48%).mp 155-156.4℃;
实施例23
化合物22的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为对异丙基苯胺。White solid(yield:57%).mp 185.3-186.1℃;
实施例24
化合物23的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为对叔丁基苯胺。White solid(yield:62%).mp 211-211.8℃;
实施例25
化合物24的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为对氟苯胺。White solid(yield:63%).mp 157.6-158℃;
实施例26
化合物25的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为对三氟甲氧基苯胺。White solid(yield:59%).mp 129.2-130.1℃;
实施例27
化合物26的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为对甲酰胺基苯胺。White solid(yield:89%).mp 250.0-250.8℃;
实施例28
化合物27的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为对氨甲酰基苯胺。White solid(yield:40%).mp 225.0-226.0℃;
实施例29
化合物28的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为对甲磺酰基苯胺。White solid(yield:15%).mp 240-241℃;
实施例30
化合物29的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为对氰基苯胺。White solid(yield:12%).mp 216.9-217.3℃;
实施例31
化合物30的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为2-胺甲基吡啶。White solid(yield:34%).mp 135.1-136.1℃;
实施例32
化合物31的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为3-胺甲基吡啶。White solid(yield:45%).mp 171.9-172.2℃;
实施例33
化合物32的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为4-胺甲基吡啶。White solid(yield:42%).mp 163.1-164.2℃;
实施例34
化合物33的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为2-胺乙基吡啶。White solid(yield:43%).mp 120-121.1℃;
实施例35
化合物34的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为3-胺乙基吡啶。White solid(yield:53%).mp 158-159℃;
实施例36
化合物35的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为2-氨基吡啶。White solid(yield:57%).mp 135.7-136.3℃;
实施例37
化合物36的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为3-氨基吡啶。White solid(yield:23%).mp 218.1-219.3℃;
实施例38
化合物37的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为4-氨基吡啶。White solid(yield:35%).mp 223-224℃;
实施例39
化合物38的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为4-氨基嘧啶。White solid(yield:77%).mp 213-214℃;
实施例40
化合物39的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为3-氨基哒嗪。White solid(yield:67%).mp 188-189℃;
实施例41
化合物40的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为III-2。White solid(yield:71%).mp 140-144℃;
实施例42
化合物42的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为III-3。White solid(yield:47%).mp 169-171℃;
实施例43
化合物43的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为III-4。White solid(yield:32%).mp 195-197℃;
实施例44
化合物44的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为III-5。White solid(yield:54%).mp 190-193℃;
实施例45
化合物45的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为III-6。White solid(yield:54%).mp 184-186℃;
实施例46
化合物46的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为III-7。White solid(yield:54%).mp 192-195℃;
实施例47
化合物47的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为III-8。White solid(yield:54%).mp 207-209℃;
实施例48
化合物48的合成:N
实验步骤同实施例1,仅以中间体III-1替换为III-9。White solid(yield:54%).mp 167-170℃;
实施例49
化合物49的合成:1-(4-(6-((5-氯-4-((3-(三氟甲基)苯基)氨基)嘧啶-2-基)氨基]吡啶-3-基)哌嗪-1-基)乙酮(49)
实验步骤同实施例1,仅以中间体III-1替换为III-10。White solid(yield:62%).mp 247-248℃;
实施例50
化合物50的合成:1-(4-(6-((5-氯-4-((3-(三氟甲基)苯基)氨基)嘧啶-2-基)氨基)吡啶-3-基)哌嗪-1-基)丙-1(50)
实验步骤同实施例1,仅以中间体III-1替换为III-11。White solid(yield:54%).mp 241-242℃;
实施例51
化合物51的合成:4-(6-((5-(4-氯-4-((3-(三氟甲基)苯基)氨基)嘧啶-2-基)氨基)吡啶-3-基)哌嗪-1-甲酸叔丁基酯(51)
实验步骤同实施例1,仅以中间体III-1替换为III-12。White solid(yield:64%).mp 233-234℃;
实施例52
化合物52的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为III-13。White solid(yield:50%).mp 158-160℃;
实施例53
化合物53的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为III-14。White solid(yield:26%).mp 205-207℃;
实施例54
化合物54的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为III-15。White solid(yield:75%).mp 149-150℃;
实施例55
化合物55的合成:N
实验步骤同实施例1,仅以中间体III-1替换为III-16。White solid(yield:58%).mp 191-192℃;
实施例56
化合物56的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为III-17。White solid(yield:62%).mp 196-197℃;
实施例57
化合物57的合成:1-(6-((5-氯-4-((3-(三氟甲基)苯基)氨基)嘧啶-2-基)氨基)吡啶-3-基)哌啶-4-腈(57)
实验步骤同实施例1,仅以中间体III-1替换为III-18。White solid(yield:53%).mp202-203℃;
实施例58
化合物58的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为III-19。White solid(yield:58%).mp 210-211℃;
实施例59
化合物59的合成:N
实验步骤同实施例1,仅以中间体III-1替换为III-20。White solid(yield:41%).mp 206-207℃;
实施例60
化合物60的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为III-21。White solid(yield:53%).mp 233-234℃;
实施例61
化合物61的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为III-22。White solid(yield:36%).mp 265-266℃;
实施例62
化合物62的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为III-23。White solid(yield:27%).mp 204-205℃;
实施例63
化合物63的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为III-24。White solid(yield:58%).mp198-199℃;
实施例64
化合物64的合成:5-氯-N
实验步骤同实施例1,仅以中间体III-1替换为III-25。White solid(yield:42%).mp 221-222℃;
实施例65
化合物65的合成:5-氟-N
实验步骤同实施例1,仅以中间体I-1替换为I-3。White solid(yield:43%).mp181-182℃;
实施例66
化合物66的合成:5-甲基-N
实验步骤同实施例1,仅以中间体I-1替换为I-4。White solid(yield:43%).mp204-205℃;
实施例67
化合物67的合成:5-甲基-N
实验步骤同实施例1,仅以中间体I-1替换为I-5。White solid(yield:42%).mp192-193℃;
实施例68
化合物68的合成:N
实验步骤同实施例1,仅以中间体I-1替换为I-2,以中间体III-1替换为III-25。White solid(yield:63%).mp 222-223℃;
实施例69
化合物69的合成:5-氟-N
实验步骤同实施例1,仅以中间体I-1替换为I-3,以中间体III-1替换为III-25。White solid(yield:60%).mp 225-226℃;
实施例70
化合物70的合成:5-甲基-N
实验步骤同实施例1,仅以中间体I-1替换为I-4,以中间体III-1替换为III-25。White solid(yield:60%).mp 198-199℃;
实施例71
化合物71的合成:5-甲氧基-N
实验步骤同实施例1,仅以中间体I-1替换为I-5,以中间体III-1替换为III-25。White solid(yield:58%).mp 202-203℃;
实施例72
体外组织蛋白酶C酶抑制作用评价:
用黑色384孔板测定化合物对人重组Cat C酶的抑制作用。在含有25mM HEPES缓冲液、50mM NaCl、5mM MDTT和0.01%(v/v)Tritonx-100(pH5.0)的测定缓冲液中,将人重组Cat C稀释至浓度为5nM。在孔中加入20μL稀释酶(在测定中对应于2nM的Cat C)和10μL被测化合物或阳性对照(AZD7986),然后在25℃下孵育30min,然后加入20μL底物(h-gly-arg-amc),最终浓度为100μM。反应60min后,在EXλ350nm和EMλ450nm测定AMC的吸收。以上检测结果用SPSS17.0计算IC50值,结果如下表:
实施例73
体外细胞内组织蛋白酶C酶抑制作用评价:
胞内酶活测定在96孔板中进行。将含有U937或THP-1细胞的30μL PBS细胞悬液加入孔中,使每孔含有3×105个细胞,并在井中加入AZD7986或被测化合物10μL,在37℃下孵育1h后,加入h-gly-arg-amc(200μM),并开始反应,在37℃下进一步孵育1h,测定AMC在EXλ350nm和EMλ450nm处的吸收。以上检测结果用SPSS17.0计算IC50值,结果如下表:
“NT”:未测试,因为在0.2μM的浓度下未检测到化合物对Cat C的抑制作用。
实施例74
体内急性毒性评价:
其中优选了化合物1进行了体内活性测试,其对ICR小鼠的选择20只ICR小鼠(约一半,约20g,从安徽医科大学动物系购买),年龄6~8周,进行化合物1的急性毒性试验。将它们随机分为两组并适应性饲养一周。禁食12h后,一次分别口服1500mg/kg(0.5%CMC-Na作为溶剂)化合物1。每天观察并记录小鼠的体重,死亡率和行为,持续一周。随后,将小鼠麻醉并将组织用于HE染色。
急性毒性测试结果为LD
实施例75
体内组织蛋白酶C和NSPs抑制作用测试。
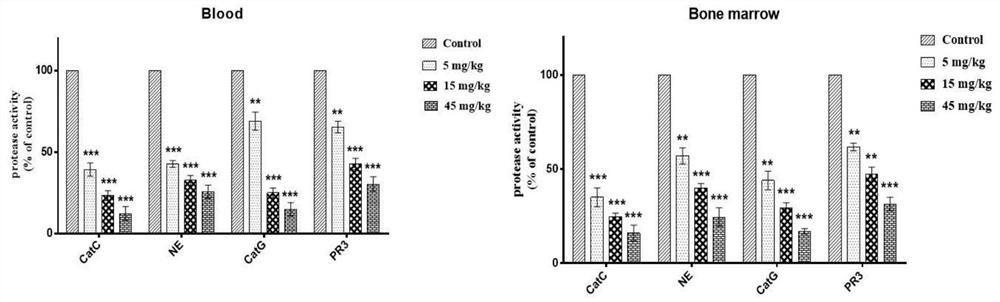
将C57BL/6小鼠(半性,约20g,购自安徽医科大学动物系)随机分为四组(N=6)。化合物1治疗组的小鼠每天口服5、15和45mg/kg化合物1,共6天。对照组每天两次给予等量的生理盐水,共6天。终止时,抽取骨髓和血液分析Cat C和NSPs的活性。将骨髓和血液裂解物添加到384孔板中。合成的肽底物用于分析NSP和Cat C活性。
如图1所示,在骨髓和血液中均观察到Cat C活性和下游NSP活化的明显剂量依赖性降低。
实施例76
体内抗炎活性评估:
采用了熏蒸和LPS(Sigma)相结合的方法。每天给大鼠口服两次盐水(对照组和模型组,N=8)或化合物1,剂量分别为5mg/kg(N=8),15mg/kg(N=8)或45mg/kg(N=8)从研究第1天到研究第13天以及研究第15天到研究第30天(总共28天)。除阴性对照组外,所有大鼠从研究第1天到研究第13天以及研究第15天到研究第30天暴露于香烟烟雾(XIONGSHI)中45min,然后在经历的第0天(研究开始)进行气管滴注LPS(200μg),并在第14天进行气管滴注LPS(100μg)。每三天记录一次体重,每天观察其行为,毛色和呼吸。在第31天,用过量的水合氯醛使动物安乐死。然后收集肺,支气管肺泡灌洗液(BALF)和血液,骨髓样品,并在-80℃下保存直至被检测到。用先前描述的方法检测Cat C和NSP的水平,并通过ELISA试剂盒(MULTI SCIENCES)分析所有样品中的细胞因子水平。
与正常组相比,在开始建模的一周后,模型组的大鼠表现出典型的COPD症状,包括呼吸困难,气道粘液增加,皮毛发黄和食欲不振。与模型组相比,用化合物1处理的组可显着缓解COPD大鼠的肺部炎症和全身炎症。用化合物1治疗的组的大鼠体重缓慢减轻,约20天后略有增加。如图2所示,在骨髓、血液、肺和支气管肺泡灌洗液中均观察到Cat C活性和下游NSP激活的明显剂量依赖性降低。
为了评估化合物1治疗COPD大鼠模型后的组织病理学变化,在肺组织上进行了H&E染色。在模型组中,可以观察到明显的促炎性改变,包括肺泡出血和扩张,部分肺泡融合,炎性细胞浸润和肺泡结构破坏。但是,化合物1以明显的剂量依赖性方式改善了这些组织病理学变化。如图3所示,模型组的大鼠表现出明显的炎症反应,并伴随着细胞因子水平的明显上调(IFN-γ,IL-1β,IL-6,IL-17,TNF-α和GM-CSF)以及不同组织(骨髓,血液,肺和BALF)中IL-10水平的下调。但是,经化合物1治疗的组,促炎细胞因子(IFN-γ,IL-1β,IL-6,IL-17,TNF-α和GM-CSF)的水平降低,抗炎药的浓度降低细胞因子(IL-10)以剂量依赖性方式增加。
以上显示和描述了本发明的基本原理和主要特征和本发明的优点。本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明精神和范围的前提下,本发明还会有各种变化和改进,这些变化和改进都落入要求保护的本发明范围内。本发明要求保护范围由所附的权利要求书及其等效物界定。
- 一种嘧啶-2,4-二胺类化合物及其制备方法与应用
- 一种三氮唑修饰的5-氟-2,4-嘧啶二胺类化合物及其应用