用于多核苷酸空间组织的系统和方法
文献发布时间:2023-06-19 12:16:29
相关申请的交叉引用
本申请要求2018年8月24日提交的美国临时申请第62/722,684号的优先权,其公开通过引用整体并入本文用于所有目的。对根据联邦赞助的研究和开发下完成的发明权利的声明
本发明是在国家卫生研究院授予的资助号EB021240下,在政府的支持下完成的。政府享有本发明的某些权利。
背景
活细胞内多核苷酸的三维(3D)空间组织在调控和维持基因表达,基因组稳定性和细胞功能等过程中起重要作用。例如,与核层或核周边相关的基因组序列通常显示低转录活性,而定位于核内部的那些通常显示相对较高的活性。此外,真核细胞核含有许多无膜核小体,例如Cajal小体、PML小体、核仁和斑点,它们在各种生物学过程中在功能上是重要的。基因组学和细胞生物学的中心目标是理解基因组结构,其在各种核隔室内的组织,和基因表达之间的关系,但是该目标受到目前可用的方法的限制。
通过使用基于显微镜检查的成像(例如FISH)和染色体构象捕获(3C)技术的许多研究已经提出了基因组组织和细胞命运确定之间的相关性。例如,在淋巴细胞发育过程中,在祖细胞中位于核周边的IgH和Igκ基因座通常在前-B细胞中重新定位至核内部,这是与免疫球蛋白基因座的激活和重排同步的过程。类似地,前神经转录因子Asc11的基因组基因座位于未分化胚胎干细胞的核周边,但在神经元分化过程中重新定位到核内部。此外,基于3C的研究已经揭示了在发育和疾病过程中高分辨率染色质相互作用(例如,拓扑相关结构域)的变化。总之,这些是用于绘制基因组组织和测量染色质元件的物理相互作用的强有力的方法,但是它们通常不能提供基因组定位和功能之间的因果联系,并且它们不能测量活细胞中的动态变化。
已经观察到核隔室在基因组组织和功能中起重要作用。提出通过液相-液相分离来组装核小体,所述液相-液相分离是由蛋白质和RNA之间的多价相互作用驱动的。通过蛋白质或RNA组分固定在染色质上,从头的核小体形成可以成核。在核小体中,Cajal小体(CB)对于脊椎动物胚胎发生是必需的,并且在肿瘤细胞和神经元中是丰富的。CB由支架蛋白组分Coilin标记,并且在小核RNA(snRNA)生物发生、核糖核蛋白(RNP)组装和端粒酶生物发生中起重要作用。由肿瘤抑制蛋白PML标记的早幼粒细胞白血病(PML)核小体是与疾病过程(包括肿瘤和病毒感染)相关的丰富的核点结构。然而,核小体和染色质的共定位如何因果地影响基因表达和细胞功能仍然是很难解决的。
为了理解这种因果关系,已经开发了序列特异性DNA-蛋白质相互作用来介导靶向的基因组重组。该技术利用插入到基因组基因座中的LacO重复序列阵列,其当与融合到核膜蛋白的LacI组合时,有助于将相邻的基因组序列束缚到核周边。使用这种技术,一些研究已经报道了将基因重新定位到核周边导致基因抑制。然而,这种技术不适于可编程的基因组靶向,并且是繁琐且难以实施的。例如,建立稳定的含有LacO重复序列的细胞系是该技术的先决条件,其已经涉及许多步骤,例如使大的LacO重复序列阵列随机插入基因组,筛查含有单个插入基因座的细胞,产生稳定的细胞系,以及通过FISH表征基因组插入位点。需要新的工具来以可编程,精确和靶向的方式操纵基因组的空间和时间组织。
原核II类CRISPR-Cas(成簇规律间隔短回文重复序列-CRISPR相关的)系统已经被用作用于基因编辑,基因调节,表观基因组编辑,染色质环形成和活细胞基因组成像的工具箱(例如Cas9和Cpf1)。与转录效应物或表观遗传修饰结构域偶联的核酸酶失活的Cas(dCas)蛋白允许调节邻近单一导向RNA(sgRNA)靶位点的基因的表达。CRISPR-Cas系统是否可用于介导基因组组织和重新定位染色质DNA相对于哺乳动物细胞核内各种核隔室的位置仍然是未知的。
鉴于以上所述,存在对进行靶多核苷酸的空间组织的替代系统和方法的需求。本公开解决了这种需求和其它需求。
发明概述
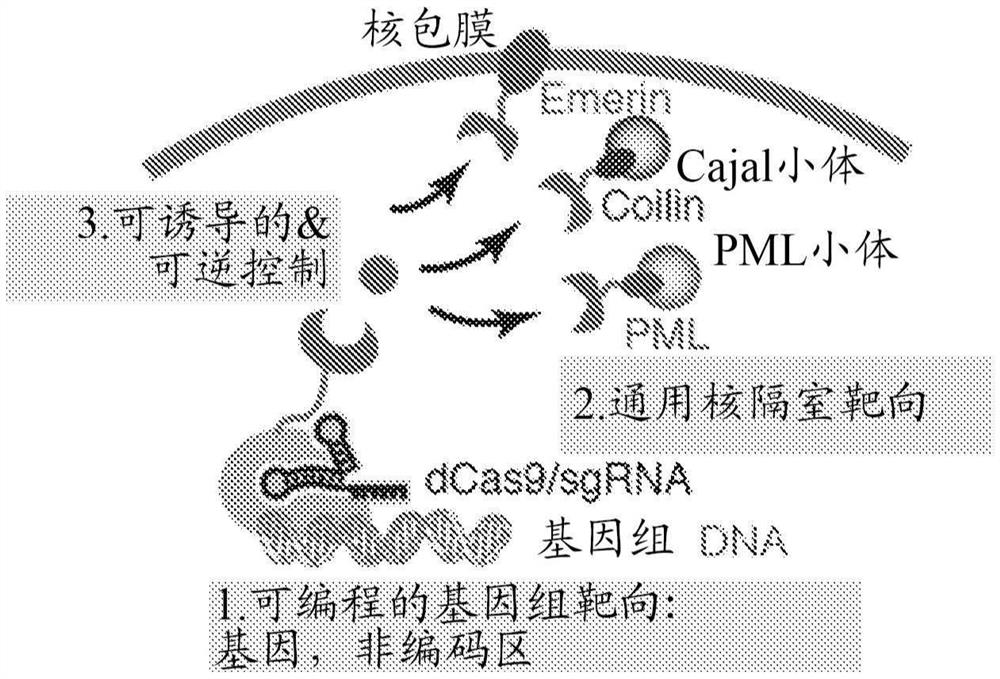
通常,本文提供了用于可编程多核苷酸重组的系统和方法。所述系统和方法可以通过诱导型系统如化学诱导型系统将致动器部分与细胞隔室特异性蛋白连接,并且可以允许将多核苷酸,例如基因组基因座有效,可诱导和动态重新定位到特定的细胞位置,例如核周边,Cajal小体和PML核小体(图1)。所述系统和方法可以扩展现有的多核苷酸编辑和调控工具,提供改进的技术来操纵多核苷酸相对于细胞隔室的3D组织,以及研究大规模空间多核苷酸组织与细胞功能之间的关系。
在一个方面,提供了控制靶多核苷酸在细胞的隔室中的空间定位的系统。所述系统包含与第一二聚化结构域连接(例如融合)的隔室特异性蛋白。所述系统还包含靶向靶多核苷酸的致动器部分,其中致动器部分与第二二聚化结构域连接(例如融合),所述第二二聚化结构域能够与第一二聚化结构域组装成二聚体。在一些实施方案中,细胞是真核细胞。
在一些实施方案中,所述靶多核苷酸包括基因组DNA。在一些实施方案中,所述靶多核苷酸包括RNA。在一些实施方案中,所述致动器部分包括Cas蛋白,并且所述系统还包含与所述致动器部分复合并与所述靶多核苷酸(例如,基因组DNA)杂交的导向RNA。在一些实施方案中,所述致动器部分包括RNA结合蛋白,并且所述系统还包含与所述致动器部分复合并与所述靶多核苷酸(例如,RNA)杂交的导向RNA。在某些情况下,所述系统还包含与所述导向RNA复合的Cas蛋白。在一些实施方案中,RNA结合蛋白是ADAR1或ADAR2,并且导向RNA包括ADAR募集RNA(arRNA)。在一些实施方案中,Cas蛋白基本上缺乏DNA切割活性。在一些实施方案中,Cas蛋白是Cas9蛋白、Cas12蛋白、Cas13蛋白、CasX蛋白或Casy蛋白。在一些实施方案中,Cas12蛋白选自Cas12a、Cas12b、Cas12c、Cas12d和Cas12e。在一些实施方案中,Cas13蛋白选自Cas13a、Cas13b、Cas13c和Cas13d。在某些情况下,Cas13d蛋白是CasRx。在一些实施方案中,所述致动器部分包含与靶多核苷酸杂交的结合蛋白,其中所述结合蛋白是锌指核酸酶或TALE核酸酶。在一些实施方案中,所述致动器部分包含与导向多核苷酸复合的Argonaute蛋白,其中所述导向多核苷酸是导向RNA或导向DNA,并且其中所述导向多核苷酸与所述靶多核苷酸杂交。
在一些实施方案中,隔室特异性蛋白选自:隔室内源性的蛋白、调节蛋白、马达蛋白、DNA修复蛋白、以及以上的组合。在某些情况下,隔室内源性的蛋白是定位于隔室的蛋白质,隔室的组分,隔室内发现的蛋白质和/或与隔室相关的蛋白质。在某些情况下,调节蛋白是基因表达的物或阻抑物。在某些情况下,马达蛋白是促进分子沿着微管或肌动蛋白丝转运的任何蛋白质。在某些情况下,DNA修复蛋白是修复双链断裂的任何蛋白质。
在一些实施方案中,隔室是核隔室(例如,核小体)。在一些实施方案中,核隔室包含内核膜和/或隔室特异性蛋白包含Emerin、Lap2β、核纤层蛋白B或以上的组合。在一些实施方案中,核隔室包含Cajal小体和/或隔室特异性蛋白包含coilin、SMN、Gemin 3、SmD1、SmE或以上的组合。在一些实施方案中,核隔室包含核小点和/或隔室特异性蛋白包含SC35。在一些实施方案中,核隔室包含PML小体和/或隔室特异性蛋白包含PML、SP100或以上的组合。在一些实施方案中,核隔室包含核芯复合物和/或隔室特异性蛋白包含Nup50、Nup98、Nup53、Nup153、Nup62或以上的组合。在一些实施方案中,核隔室包含核仁和/或隔室特异性蛋白包含核仁蛋白B23。在一些实施方案中,核隔室包含异染色质和/或隔室特异性蛋白包含调节蛋白,例如异染色质蛋白1(例如HP1α、HP1β和/或HP1γ,包括截短的和全长的)、Krüppel相关盒-锌指蛋白(KRAB-ZFP)、KRAB相关蛋白1(KAP1)、核小体重塑脱乙酰酶复合物(NuRD)、SET结构域分支型1(SETDB1)、DNA甲基转移酶(例如DNMT3A、DNMT3L、DNMT3B)、组蛋白脱乙酰酶(HDAC)、SUV39H1(截短的,全长的)、G9α(截短的,全长的)、Ezh1/2、EED、Suz12、JARID2、AEBP2、RbAp48、PCL1、RBBP7/4、C17orf96、C10orf12或以上的组合。在一些实施方案中,核隔室包含核小体和/或隔室特异性蛋白包含DNA修复蛋白,例如53BP1、Rad51、Rad52、Ubc9、UBL1、BLM、c-Ab1、BCR/Ab1、BRCA1/2、PALB2、RPA、Rad51AP1、Chk1、Arg、Hop2、Mnd1、DMC1或以上的组合。
在一些实施方案中,隔室是细胞质隔室(例如,细胞体)。在一些实施方案中,细胞质隔室包含P颗粒和/或隔室特异性蛋白包含一种或多种RGG结构域蛋白(例如,PGL-1和PGL-3、死亡盒蛋白(Dead box protein)、GLH-1-4或以上的组合)。在一些实施方案中,细胞质隔室包含GW小体和/或隔室特异性蛋白包含GW182。在一些实施方案中,细胞质隔室包含应激颗粒和/或隔室特异性蛋白包含G3BP(Ras-GAP SH3结合蛋白)、TIA-1(T细胞胞内抗原)、eIF2、eIF4E或以上的组合。在一些实施方案中,细胞质隔室包含海绵体和/或隔室特异性蛋白包含EXu、Btz、Tral、Cup、eIF4E、Me31B、Yps、Gus、Dcp1/2、Sqd、bicC、Hrb27C、Bru或以上的组合。在一些实施方案中,细胞质隔室包含细胞质朊病毒蛋白诱导的核糖核蛋白(CyPrP-RNP)颗粒和/或隔室特异性蛋白包含Dcp1a、DDX6/Rck/p54/Me31B/Dhh1、Dicer或以上的组合。在一些实施方案中,细胞质隔室包含U小体和/或隔室特异性蛋白包含一种或多种富含尿苷的小核核糖核蛋白U1、U2、U4/U6和U5;LSm1-7;运动神经元生存(SMN)蛋白或以上的组合。在一些实施方案中,细胞质隔室包含内质网和/或隔室特异性蛋白包含钙网蛋白、钙联蛋白、PDI、GRP 78、GRP 94或以上的组合。在一些实施方案中,细胞质隔室包含线粒体和/或隔室特异性蛋白包含HIF1A、PLN、Cox1、己糖激酶、TOMM40或以上的组合。在一些实施方案中,细胞质隔室包含质膜和/或隔室特异性蛋白包含钠钾ATP酶、CD98、一种或多种钙粘蛋白、质膜钙ATP酶(PMCA)或以上的组合。在一些实施方案中,细胞质隔室包含高尔基体和/或隔室特异性蛋白包含GM130、MAN2A1、MAN2A2、GLG1、B4GALT1、RCAS1、GRASP65或以上的组合。在一些实施方案中,细胞质隔室包含核糖体和/或隔室特异性蛋白包含AGO2、MTOR、PTEN、RPL26、FBL、RPS3或以上的组合。在一些实施方案中,细胞质隔室包含蛋白酶体和/或隔室特异性蛋白包含PSMA1、PSMB5、PSMC1、PSMD1、PSMD7或以上的组合。在一些实施方案中,细胞质隔室包含核内体和/或隔室特异性蛋白包含CFTR、ADRB1、EGFR、IGF2R、AP2S1、CD4、HLA-A、Coveolin、RAB5、ErbB2或以上的组合。在一些实施方案中,细胞质隔室包含脂质体和/或隔室特异性蛋白包含EEA1、LAMTOR2、LAMTOR4或以上的组合。在一些实施方案中,细胞质隔室包含细胞骨架组分(例如,微管和/或肌动蛋白丝)和/或隔室特异性蛋白包含马达蛋白,例如驱动蛋白、动力蛋白、肌球蛋白或以上的组合。
在一些实施方案中,隔室特异性蛋白进一步与荧光蛋白连接(例如融合)。在一些实施方案中,致动器部分进一步与荧光蛋白连接(例如融合)。在一些实施方案中,第一二聚化结构域和第二二聚化结构域包含仅在配体、光或酶的存在下组装以形成二聚体的诱导型二聚化系统。在一些实施方案中,第一二聚化结构域和第二二聚化结构域各自在配体存在下与配体结合。在一些实施方案中,配体是化学诱导物或光生诱导物。在一些实施方案中,第一二聚化结构域和第二二聚化结构域包含自发二聚化系统。
在一些实施方案中,所述系统包含第一多核苷酸(例如载体)和第二多核苷酸(例如载体),所述第一多核苷酸(例如载体)包含编码与第一二聚化结构域连接的隔室特异性蛋白的核酸序列,所述第二多核苷酸(例如载体)包含编码与第二二聚化结构域连接的致动器部分的核酸序列。
在另一个方面,提供了用于控制靶多核苷酸在细胞的隔室中的空间定位的方法。所述方法包括提供(例如,引入细胞)与第一二聚化结构域连接(例如,融合)的隔室特异性蛋白。所述方法还包括提供(例如,引入细胞)与第二二聚化结构域连接(例如,融合)的致动器部分。所述方法还包括形成包含所述致动器部分和所述靶多核苷酸的复合物。所述方法还包括组装包含第一二聚化结构域和第二二聚化结构域的二聚体,从而将靶多核苷酸定位在隔室中。在一些实施方案中,细胞是真核细胞。
在一些实施方案中,靶多核苷酸不是隔室内源性的。在一些实施方案中,靶多核苷酸的定位包括调节靶多核苷酸的表达。在一些实施方案中,调节包括降低靶多核苷酸的表达。在一些实施方案中,调节包括增加靶多核苷酸的表达。在一些实施方案中,靶多核苷酸的定位还包括调节隔室内源性的一种或多种另外的多核苷酸的表达。在一些实施方案中,靶多核苷酸的定位包括改变细胞功能,细胞命运,细胞生长,细胞凋亡和/或细胞分化,例如通过将靶多核苷酸(例如端粒)重新定位到不同的细胞隔室。在某些情况下,靶多核苷酸(例如,端粒)定位至核隔室(例如,核周边或Cajal小体)增加或降低细胞活力。在一些实施方案中,靶多核苷酸的定位还包括在细胞内产生一个或多个另外的隔室。在一些实施方案中,靶多核苷酸的定位还包括修复DNA断裂。在某些实施方案中,DNA断裂是单链断裂或双链断裂。在一些实施方案中,修复包括引入外源DNA。在一些实施方案中,引入包括重组,非同源末端连接(NHEJ)或同源性定向修复(HDR)。在一些实施方案中,靶多核苷酸的定位诱导相分离以形成隔室。在某些实施方案中,隔室是包含蛋白质、RNA、DNA或以上的组合的人工聚集体。在一些情况下,隔室是核小体(例如Cajal小体)或细胞体。在一些实施方案中,靶多核苷酸的定位诱导促进DNA修复(例如,促进双链断裂的修复)并提高基因编辑效率(例如,增强HDR)的核小体的形成。
在一些实施方案中,靶多核苷酸包括基因组DNA。在一些实施方案中,靶多核苷酸包括RNA。在一些实施方案中,致动器部分包括Cas蛋白,并且所述方法还包括提供与所述致动器部分复合并与所述靶多核苷酸(例如,基因组DNA)杂交的导向RNA。在一些实施方案中,致动器部分包括RNA结合蛋白,并且所述方法还包括提供与所述致动器部分复合并与所述靶多核苷酸(例如,RNA)杂交的导向RNA。在某些情况下,所述方法还包括提供与导向RNA复合的Cas蛋白。在一些实施方案中,RNA结合蛋白是ADAR1或ADAR2,并且导向RNA包括ADAR募集RNA(arRNA)。在一些实施方案中,Cas蛋白基本上缺乏DNA切割活性。在一些实施方案中,Cas蛋白是Cas9蛋白、Cas12蛋白、Cas13蛋白、CasX蛋白或CasY蛋白。在一些实施方案中,Cas12蛋白选自Cas12a、Cas12b、Cas12c、Cas12d和Cas12e。在一些实施方案中,Cas13蛋白选自Cas13a、Cas13b、Cas13c和Cas13d。在某些情况下,Cas13d蛋白是CasRx。在一些实施方案中,致动器部分包含与靶多核苷酸杂交的结合蛋白,其中所述结合蛋白是锌指核酸酶或TALE核酸酶。在一些实施方案中,致动器部分包含与导向多核苷酸复合的Argonaute蛋白,其中所述导向多核苷酸是导向RNA或导向DNA,并且其中所述导向多核苷酸与所述靶多核苷酸杂交。
在一些实施方案中,隔室特异性蛋白选自:隔室内源性的蛋白、调节蛋白、马达蛋白、DNA修复蛋白、以及以上的组合。在某些情况下,隔室内源性的蛋白是位于隔室的蛋白质,隔室的组分,隔室内发现的蛋白质和/或与隔室相关的蛋白质。在某些情况下,调节蛋白是基因表达的激活物或阻抑物。在某些情况下,马达蛋白是促进分子沿着微管或肌动蛋白丝转运的任何蛋白质。在某些情况下,DNA修复蛋白是修复双链断裂的任何蛋白质。
在一些实施方案中,隔室是核隔室(例如,核小体)。在一些实施方案中,核隔室包含内核膜和/或隔室特异性蛋白包含Emerin、Lap2β、核纤层蛋白B或以上的组合。在一些实施方案中,核隔室包含Cajal小体和/或隔室特异性蛋白包含coilin、SMN、Gemin 3、SmD1、SmE或以上的组合。在一些实施方案中,核隔室包含核小点和/或隔室特异性蛋白包含SC35。在一些实施方案中,核隔室包含PML小体和/或隔室特异性蛋白包含PML、SP100或以上的组合。在一些实施方案中,核隔室包含核芯复合物和/或隔室特异性蛋白包含Nup50、Nup98、Nup53、Nup153、Nup62或以上的组合。在一些实施方案中,核隔室包含核仁和/或隔室特异性蛋白包含核仁蛋白B23。在一些实施方案中,核隔室包含异染色质和/或隔室特异性蛋白包含调节蛋白,例如异染色质蛋白1(例如HP1α、HP1β和/或HP1γ,包括截短的和全长的)、Krüppel相关盒-锌指蛋白(KRAB-ZFP)、KRAB相关的蛋白1(KAP1)、核小体重塑脱乙酰酶复合物(NuRD)、SET结构域分支型1(SETDB1)、DNA甲基转移酶(例如DNMT3A、DNMT3L、DNMT3B)、组蛋白脱乙酰酶(HDAC)、SUV39H1(截短的,全长的)、G9α(截短的,全长的)、Ezh1/2、EED、Suz12、JARID2、AEBP2、RbAp48、PCL1、RBBP7/4、C17orf96、C10orf12或以上的组合。在一些实施方案中,核隔室包含核小体和/或隔室特异性蛋白包含DNA修复蛋白,例如53BP1、Rad51、Rad52、Ubc9、UBL1、BLM、c-Abl、BCR/Abl、BRCA1/2、PALB2、RPA、Rad51AP1、Chk1、Arg、Hop2、Mnd1、DMC1或以上的组合。
在一些实施方案中,隔室是细胞质隔室(例如,细胞体)。在一些实施方案中,细胞质隔室包含P颗粒和/或隔室特异性蛋白包含一种或多种RGG结构域蛋白(例如,PGL-1和PGL-3、死盒蛋白、GLH-1-4或以上的组合。在一些实施方案中,细胞质隔室包含GW小体和/或隔室特异性蛋白包含GW182。在一些实施方案中,细胞质隔室包含应激颗粒和/或隔室特异性蛋白包含G3BP(Ras-GAP SH3结合蛋白)、TIA-1(T细胞胞内抗原)、eIF2、eIF4E或以上的组合。在一些实施方案中,细胞质隔室包含海绵体和/或隔室特异性蛋白包含EXu、Btz、Tral、Cup、eIF4E、Me31B、Yps、Gus、Dcp1/2、Sqd、BicC、Hrb27C、Bru或以上的组合。在一些实施方案中,细胞质隔室包含细胞质朊病毒蛋白诱导的核糖核蛋白(CyPrP-RNP)颗粒和/或隔室特异性蛋白包含Dcp1a、DDX6/Rck/p54/Me31B/Dhh1、Dicer或以上的组合。在一些实施方案中,细胞质隔室包含U小体和/或隔室特异性蛋白包含一种或多种富含尿苷的小核核糖核蛋白U1、U2、U4/U6和U5;LSm1-7;运动神经元生存(SMN)蛋白或以上的组合。在一些实施方案中,细胞质隔室包含内质网和/或隔室特异性蛋白包含钙网蛋白、钙联蛋白、PDI、GRP 78、GRP 94或以上的组合。在一些实施方案中,细胞质隔室包含线粒体和/或隔室特异性蛋白包含HIF1A、PLN、Cox1、己糖激酶、TOMM40或以上的组合。在一些实施方案中,细胞质隔室包含质膜和/或隔室特异性蛋白包含钠钾ATP酶、CD98、一种或多种钙粘蛋白、质膜钙ATP酶(PMCA)或以上的组合。在一些实施方案中,细胞质隔室包含高尔基体和/或隔室特异性蛋白包含GM130、MAN2A1、MAN2A2、GLG1、B4GALT1、RCAS1、GRASP65或以上的组合。在一些实施方案中,细胞质隔室包含核糖体和/或隔室特异性蛋白包含AGO2、MTOR、PTEN、RPL26、FBL、RPS3或以上的组合。在一些实施方案中,细胞质隔室包含蛋白酶体和/或隔室特异性蛋白包含PSMA1、PSMB5、PSMC1、PSMD1、PSMD7或以上的组合。在一些实施方案中,细胞质隔室包含核内体和/或隔室特异性蛋白包含CFTR、ADRB1、EGFR、IGF2R、AP2S1、CD4、HLA-A、Coveolin、RAB5、ErbB2或以上的组合。在一些实施方案中,细胞质隔室包含脂质体和/或隔室特异性蛋白包含EEA1、LAMTOR2、LAMTOR4或以上的组合。在一些实施方案中,细胞质隔室包含细胞骨架组分(例如,微管和/或肌动蛋白丝)和/或隔室特异性蛋白包含马达蛋白,例如驱动蛋白、动力蛋白、肌球蛋白或以上的组合。
在一些实施方案中,隔室特异性蛋白进一步与荧光蛋白连接(例如融合)。在一些实施方案中,致动器部分进一步与荧光蛋白连接(例如融合)。在一些实施方案中,第一二聚化结构域和第二二聚化结构域的组装是可诱导的并且仅在配体、光或酶的存在下发生。在一些实施方案中,第一二聚化结构域和第二二聚化结构域各自在配体存在下与配体结合。在一些实施方案中,配体是化学诱导物或光生诱导物。在一些实施方案中,第一二聚化结构域和第二二聚化结构域包含自发二聚化系统。
在一些实施方案中,所述方法包括将本文所述的系统引入细胞中,所述系统包含第一多核苷酸(例如,载体)和第二多核苷酸(例如,载体),所述第一多核苷酸(例如,载体)包含编码与第一二聚化结构域连接的隔室特异性蛋白的核酸序列,所述第二多核苷酸(例如,载体)包含编码与第二二聚化结构域连接的致动器部分的核酸序列。
附图的简要说明
图1是用于将基因组基因座靶向各种核隔室的可编程,可诱导和通用的系统的示意图。dCas9和核隔室特异性蛋白融合到互补的异二聚化结构域对,其仅在化学诱导物存在下组装。基因组靶标由sgRNA序列指定,并且通过将CRISPR-GO与隔室特异性分子融合来编程核隔室。
图2是脱落酸(ABA)诱导型CRISPR-GO系统通过ABI-dCas9和PYL1-GFP-Emerin在人细胞中的共表达将基因组基因座靶向核包膜(NE)的示意图。在ABA、ABI和PYL1二聚化的存在下,导致ABI-dCas9靶向的基因组基因座重新定位到核包膜处的PYL1-GFP-Emerin。去除ABA后,ABI和PYL1解离,基因组基因座不再束缚于NE。
图3是ABA诱导型CRISPR-GO系统与ABI-BFP-dCas9和PYL1-GFP-Emerin在人细胞中共表达的示意图。ABA处理使ABI和PYL1二聚化,并将ABI-BFP-dCas9靶向的基因组基因座重新定位到含有PYL1-GFP-Emerin的核周边。
图4是TMP-HTag诱导型CRISPR-GO系统与dCas9-EGFP-HaloTag和DHFR-Emerin-mCherry在人细胞中共表达的示意图。TMP-HTag处理使DHFR和HaloTag二聚化,并将dCas9-EGFP-HaloTag靶向的基因组基因座重新定位到含有DHFR-Emerin-mCherry的核周边。
图5是使用CRISPR-Cas9成像来可视化活细胞中CRISPR-GO系统靶向的重复基因组基因座的方法的示意图。AB1-dCas9和dCas9-HaloTag都与相同的重复基因组基因座结合。当AB1-dCas9与PYL1-Emerin二聚化以重新定位基因组基因座时,dCas9-HaloTag与细胞可渗透的JF549-HaloTag染料配体结合以使得活细胞中靶向的基因组基因座能够可视化。
图6显示了U2OS细胞的代表性显微图像,其显示AB1-BFP-dCas9、PYL1-GFP-Emerin和dCas9-HaloTag的共表达,没有sgRNA。AB1-BFP-dCas9在没有ABA处理的情况下可能在核仁中积累。ABA处理诱导的异二聚化将AB1-BFP-dCas9重新定位于核包膜(NE)和内质网(ER),如PYL1-GFP-Emerin所标记的。dCas9-HaloTag具有低的表达水平,并且分布均匀于整个核;其定位不受ABA处理的影响。比例尺,10μm。
图7是图8和9中CRISPR-GO靶向的高度重复区域的染色体位置的概述。单一sgRNA结合靶向区域内的多个重复序列(实心灰色框)。与靶向位点相邻的基因以斜体字母显示在灰色框中。
图8显示了高度重复的基因组基因座的CRISPR-GO诱导的基因组重新定位效率的定量图。使用活细胞中的CRISPR-Cas9成像来标记Chr3、Chr13和LacO基因座。端粒被端粒标志物TRF1-mCherry标记。通过GFP-Emerin可视化核包膜。对于每一个基因座,左边的柱状图显示了核周边的基因组基因座的百分比,右边的柱状图显示了含有至少一个核周边相关基因座的细胞的百分比。所分析的基因座和细胞的数目在底部。
图9显示了较少重复的内源性基因组基因座的CRISPR-GO诱导的核重新定位效率的定量图。通过3D-FISH可视化基因组基因座,并通过DAPI染色细胞核。对于每一个基因座,左边的柱状图显示了核周边的基因组基因座的百分比,右边的柱状图显示了含有至少一个核周边相关基因座的细胞的百分比。所分析的基因座和细胞的数目在底部。
图10显示了代表性的显微图像,其比较了通过CRISPR-Cas9成像在有或没有ABA的情况下标记的靶向的基因组基因座(箭头)的定位。PYL1-GFP-Emerin显示定位于核包膜(NE)和内质网(ER)。核周边由虚白线显示,除了邻近束缚的基因组基因座的区域。插图显示了周边束缚的基因组基因座的放大图像。比例尺,10μm。
图11显示了图10中代表性显微图像的各个通道,其比较了在有或没有ABA的情况下靶向的基因组基因座(箭头)和核周边(虚线)的定位。顶行显示了PYL1-GFP-Emerin,其定位于核包膜(NE)和内质网(ER)。核周边由虚白线(底部)显示,除了邻近束缚的基因组基因座的区域。比例尺,10μm。
图12显示了在没有(顶部)和有ABA处理(底部)的情况下,沿着图11顶部的Emerin图像中所示的虚线,标记的Chr3基因座和标记的PYL1-GFP-Emerin的荧光强度的线性扫描图。通过加入JF549-halotag染料通过CRISPR-Cas9成像来标记Chr3基因座。
图13显示了在没有(顶部)和有ABA处理(底部)的情况下,沿着所示的虚线,标记的LacO基因座(FISH,Alexa646)和标记的核(DAPI)的荧光强度的线性扫描图。
图14是图8和9中CRISPR-GO靶向的较少重复的区域的染色体位置的概述。单个sgRNA结合靶向区域内的多个重复序列(实心灰色框)。与靶向位点相邻的基因以斜体字母显示在灰色框中。
图15显示了代表性的显微图像,其比较了在有或没有ABA的情况下通过3D-FISH标记的靶向基因组基因座(箭头)的定位。显示了DAPI标记的细胞核。核周边由虚白线显示,除了邻近束缚的基因组基因座的区域。插图显示了周边束缚的基因组基因座的放大图像。对于单独的通道,参见图11。比例尺,10μm。
图16显示了用非靶向sgRNA转染的CRISPR-GO细胞中核周边定位的基因组基因座(Chr7、ChrX和CXCR4)的百分比的定量图。对于每个基因座,左边的柱状图显示了核周边定位的基因组基因座的百分比,右边的柱状图显示了含有至少一个周边相关基因座的细胞的百分比。
图17显示了图18和19中的CRISPR-GO靶向的非重复区域的染色体位置的概述。设计多种sgRNA以沿着靶向基因(XIST、PTEN、CXCR4)上游或基因主体内的区域展开。sgRNA靶向区域以实灰框显示。顶部灰色框显示正向链内的sgRNA靶标,底部灰色框显示反向链内的sgRNA靶标。与靶向位点相邻的基因以斜体字母以灰色框显示。
图18显示了非重复内源性基因组基因座的CRISPR-GO诱导的核重新定位效率的定量图。与CXCR4相邻的非重复基因座用单一sgRNA或汇集在一起的多种sgRNA靶向。通过3D-FISH可视化基因组基因座,并通过DAPI染色细胞核。对于每一个基因座,左边的柱状图显示了核周边的基因组基因座的百分比,右边的柱状图显示了含有至少一个核周边相关基因座的细胞的百分比。所分析的基因座和细胞的数目在底部。
图19显示了使用单一sgRNA(sgCXCR4-1,左;sgCXCR4-2,中间)或6种sgRNA(右)靶向CXCR4基因座的重新定位效率的比较图。对于每一个基因座,左边的柱状图显示了核周边的基因组基因座的百分比,右边的柱状图显示了含有至少一个核周边相关基因座的细胞的百分比。所分析的基因座和细胞的数目在底部。
图20是通过加入或去除ABA介导的内源性基因座Chr3:q29的可诱导和可逆的重新定位的时间过程图。Y轴显示了周边定位的Chr3:q29基因座的百分比。X轴显示了加入或去除ABA的时间(小时)。数据表示为平均值±SEM。
图21是在加入ABA后的不同时间点S期停滞细胞(+ABA,+HU)和对照细胞(+ABA,-HU)中基因组重新定位效力的比较图。Y轴显示了在不同时间点周围定位的Chr3:q29基因座的百分比。数据表示为平均值±SEM。左边的框表示时程实验的概要。
图22显示了代表性的显微图像,其显示了内源性Chr3:q29基因座(箭头)不依赖有丝分裂束缚至核包膜。Chr3:q29基因座(箭头)在记录的前4小时开始与核包膜分离。核周边束缚发生在4.5小时并且在记录的8小时的其余时间保持稳定。此处的图像是图23中的插图。比例尺,2μm。
图23显示了代表性的显微图像,其显示了内源性基因组基因座不依赖有丝分裂束缚至核周边。插图也在图22中示出。PYL1-GFP-Emerin定位于核包膜(NE)和内质网(ER),并且核包膜由虚线显示。Chr3基因座在记录的前4小时不与核包膜相邻。核周边束缚发生在4.5h处,并在记录的8h的其余时间保持。核旋转发生在10小时至12小时。比例尺,10μm。
图24是显示在不同时间点图22中的基因组基因座与最近的核周边之间的距离的图。每30分钟拍摄一次图像。
图25显示了未束缚(1&2)和束缚(3&4)的Chr3基因座的步长位移(dx,dy)的散点图。通过从新位置减去前一时间点的位置来计算步长位移:dx
图26是未束缚(19个细胞中的1696个步长)和束缚(14个细胞中的1669个步长)的Chr3:q29基因座的平均步长距离的比较图。通过具有不等方差的双侧t检验,p<0.0001。数据表示为平均值±SD。
图27是使用伽马分布的未束缚和束缚的Chr3:q29基因座的步长距离的拟合图。拟合参数:形状参数k=2.4(对于未束缚基因座)和1.9(对于束缚基因座);速率参数β=21.9(对于未束缚基因座)和46.3(对于束缚基因座)。
图28是通过ABI-dCas9和PYL1-GFP-Coilin在人细胞中共表达将基因组基因座靶向CB的ABA诱导型CRISPR-GO系统的示意图。ABA处理使ABI和PYL1二聚化,并将ABI-dCas9靶向的基因组基因座束缚至含有PYL1-GFP-Coilin的CB。
图29显示了代表性的显微图像,其显示了在有或没有ABA的情况下的靶向LacO基因座(上图,通过FISH)和Coilin-GFP-标记的CB(中图)的共定位。
图30显示了LacO基因座的CRISPR-GO诱导的CB束缚效率的定量图。左边的柱状图显示了与Coilin-GFP标记的CB共定位的LacO基因座的百分比,右边的柱状图显示了含有至少一个CB-共定位的LacO基因座的细胞的百分比。在底部标记了所分析的基因座和细胞的数目。数据表示为平均值±SEM。
图31显示了代表性的显微图像,其显示了使用CRISPR-GO系统将LacO基因座束缚到CB,其它CB组分(SMN,原纤维蛋白,Gemin2,通过免疫染色)与LacO基因座(通过FISH)的共定位。
图32显示了代表性的显微图像,其显示了在有或没有ABA的情况下,靶向的Chr3:q29基因座(上图,通过CRISPR-Cas9成像)和Coilin-GFP标记的CB(中图)的共定位。
图33显示了Chr3:q29基因座的CRISPR-GO诱导的CB-束缚效率的定量图。左边的柱状图显示了与CB共定位的Chr3:q29基因座的百分比,右边的柱状图显示了含有至少一个CB-共定位的Chr3:q29基因座的细胞的百分比。基因座和细胞的数目在底部。数据表示为平均值±SEM。
图34是通过ABI-dCas9和PYL1-GFP-PML共表达将基因组基因座靶向PML小体的ABA诱导型CRISPR-GO系统的示意图。
图35显示了代表性的显微图像,其显示了在有或没有ABA的情况下,靶向的Chr3:q29基因座(上图,通过CRISPR-Cas9成像)和PML-GFP标记的PML小体(中图)的共定位。
图36显示了CRISPR-GO诱导的PML小体对靶向的Chr3:q29基因座的束缚效率的定量图。左边的柱状图显示了与PML小体共定位的Chr3:q29基因座的百分比,右边的柱状图显示了含有至少一个PML小体共定位的Chr3:q29基因座的细胞的百分比。基因座和细胞的数目在底部。数据表示为平均值±SEM。
图37显示了代表性的显微图像,其显示了在使用CRISPR-GO将Chr3:q29基因座束缚到PML小体之后,另一种PML小体标志物SP100(免疫染色)与Chr3:q29基因座(通过CRISPR-Cas9成像)的共定位。比例尺,10μm。
图38是通过加入ABA快速诱导的染色质-CB结合的图。Y轴显示了CB-共定位的LacO基因座的百分比。数据表示为平均值±SEM。
图39是显示去除ABA后染色质-CBs解离的动力学的曲线图。Y轴显示了CB-共定位的LacO基因座的百分比。X轴显示了从ABA去除后的时间(小时)。数据表示为平均值±SEM。
图40显示了用ABA处理(顶部)和ABA去除后6小时(底部两行)的细胞中靶向的LacO基因座的GFP-Coilin荧光的比较。显示了具有变暗的CB的细胞(中间)或其中GFP-CoilinCB已经消失的细胞(底部)的两个代表性的显微图像。线性扫描(右)测量GFP-Coilin和LacO基因座沿左边所示的虚线的原始荧光强度。
图41显示了代表性的实时显微图像,其显示了由CRISPR-GO介导的靶向的LacO位点处的从头CB(Coilin)的快速形成。在ABA处理之前(~150s)首先对选择的细胞成像。在-150s至0s将ABA加入到培养基中,并且0s表示在加入ABA后立即拍摄的相同细胞的第一个图像。
图42显示了Cajal小体共定位对邻近靶向的基因座和跨长距离的内源性基因表达的抑制。左:将Chr3:q29基因座共定位于U2OS细胞中的CB的CRISPR-GO系统的示意图。ACAP2位于sgRNA靶位点上游~35kb,PPP1R2位于sgRNA靶位点下游~36kb。右:在+/-ABA条件下,使用CRISPR-GO将Chr3:q29基因座共定位于CB的ACAP2和PPP1R2基因表达的比较图(通过RT-qPCR测量)。对于对照参见图43。
图43显示了使用CRISPR-GO将内源性Chr3基因座与CB共定位的对照图。左:在有和没有ABA的情况下,用CRISPR-GO系统但没有靶向sgRNA来测量ACAP2和PPP1R2 mRNA表达;右:在有和没有ABA的情况下,用ABI-dCas9和Chr3靶向sgRNA,但没有PYL1-GFP-Coilin来测量ACAP2和PPP1R2 mRNA表达。在不同条件下使用RT-qPCR测量mRNA。
图44是在图41所示的靶向的LacO基因座处Coilin-GFP荧光强度的定量图。在加入ABA前~150s的荧光强度设定为0(背景)。
图45显示了实时显微图像,其显示了CRISPR-GO介导的现有CB(Coilin,箭头)与相邻靶向的LacO基因座的共定位。在ABA处理之前(~200s)对选择的细胞成像。在-200s至0s将ABA加入到培养基中,并且0s表示在加入ABA后立即拍摄的第一个图像。比例尺,10μm。
图46显示了通过将靶向的染色质DNA重新定位到核周边而抑制的相邻报告基因表达。左:将LacO重复序列阵列重新定位到U2OS2-6-3细胞中的核周边的CRISPR-GO系统的示意图,其插入到驱动CFP-SKL报告基因的强力霉素(Dox)诱导的TRE-miniCMV启动子附近。右:使用CRISPR-GO系统在+/-Dox和+/-ABA条件下将LacO基因座重新定位到核周边的CFP报告基因表达水平的比较图。数据表示为平均值±SD。对于代表性直方图和对照参见图47。
图47显示了代表性的流式细胞术直方图,其比较了在不同处理下使用CRISPR-GO将LacO基因座束缚至核周边的CFP报告基因表达的荧光强度。统计图显示于图46中。右图显示了对于+/-Dox,在有或没有ABA处理的情况下,使用非靶向sgRNA的相对CFP荧光的定量。
数据表示为平均值±SD。
图48显示了当使用CRISPR-GO系统将Chr3基因座重新定位到核周边时,ACAP2和PPP1R2基因表达的比较图。在不同条件下使用RT-qPCR测量mRNA。非靶向sgRNA(sgNT)转染的细胞用作对照。
数据表示为平均值±SD。
图49显示了通过Cajal小体共定位抑制的靶向的基因座相邻的报告基因表达。左:将LacO重复序列阵列共定位到U2OS 2-6-3细胞中的CBCRISPR-GO系统的示意图。右:对于+/-Dox和+/-ABA条件,使用CRISPR-GO系统将LacO基因座共定位于CB的CFP报告基因表达的比较图。对于代表性直方图和对照参见图50。
图50显示了代表性的流式细胞术直方图,其比较了在不同处理下,使用CRISPR-GO将LacO基因座束缚到CB的CFP报告基因表达的荧光强度。统计图显示于图49中。右图显示了对于+/-Dox,在有或没有ABA处理的情况下,使用非靶向sgRNA的相对CFP荧光的定量。使用非靶向sgRNA的情况下,ABA处理导致CFP报告基因表达的轻微但不显著的降低(p>0.05)。数据表示为平均值±SD。
图51显示了在用或不用ABA处理的示例细胞中,在间期期间,端粒和最近的核被膜点之间的距离的直方图。
图52是在使用CRISPR-GO系统将端粒重新定位到核包膜后,通过Alamar蓝测定测量的相对细胞活力的比较图。数据表示为平均值±SD。
图53显示了使用CRISPR-GO将端粒重新定位到核周边的细胞的细胞周期分析。细胞用ABA处理3天。顶部:代表性的流式细胞术直方图(左)比较端粒靶向ABA处理的细胞(处理的)和非靶向sgRNA对照细胞中的荧光Hoechst 33342染色的DNA组分。底部:三个实验重复的定量。数据表示为平均值±SD。
图54显示了在有或没有ABA的情况下,使用CRISPR-GO将端粒(TRF1-mCherry,顶部)和CB(GFP-Coilin,中部)共定位的U2OS细胞的代表性显微图像。比例尺,10μm。
图55显示了在有或没有ABA的情况下,使用CRISPR-GO将端粒(TRF1-mCherry,顶部)和CB(GFP-Coilin,中部)共定位的HeLa细胞的代表性显微图像。比例尺,10μm。
图56是在有或没有ABA的情况下,通过使用CRISPR-GO系统将端粒靶向CB的Alamar蓝测定测量的相对U2OS细胞活力的比较图。细胞用ABA处理两天。数据表示为平均值±SD。
图57是在有或没有ABA的情况下,通过U2OS细胞的Alamar蓝测定测量的相对细胞活力的比较图。细胞用ABA处理两天。数据表示为平均值±SD。
图58显示了CRISPR-GO系统,其能够相对于其它核隔室对3D基因组组织进行可编程控制,从而扩展了用于基因组工程的CRISPR-Cas工具箱。CRISPR-GO方法允许3D基因组定位的可编程控制和靶向的染色质基因座相对于不同的核隔室的组织。这扩展了CRISPR-Cas工具箱的用途,超出了诸如基因编辑,转录调节,表观遗传修饰的应用。
图59是通过ABI-dCas9和PYL1-GFP-HP1α在人细胞中共表达将基因组基因座靶向异染色质的ABA诱导型CRISPR-GO系统的示意图。还呈现了显示ABA处理使ABI和PYL1二聚化并且使ABI-dCas9靶向的基因组基因座共定位于PYL1-GFP-HP1α的代表性显微图像。比例尺,10μm。
图60是每个人类染色体的重复序列(四个或更多个)的分布及其相对坐标的图。
图61是全基因组生物信息学分析的图,其揭示了位于与相邻重复序列的给定距离内的人类基因的百分比。
图62显示了CRISPR-GO系统3D基因组组织平台的概述。
图63显示了在将端粒重新定位到核周边后,通过RNA测序比较基因表达变化的图。
图64显示了在将端粒与Cajal小体共定位后,通过RNA测序比较基因表达变化的图。
图65A-65C显示了募集DNA修复组分(例如53BP1)的CRISPR编辑产生促进DNA修复和更好的基因编辑结果的核小体。
详细描述
I.引言
真核细胞是能够在空间和时间上协调许多生化反应的复杂结构。这种协调的关键是多核苷酸如基因组的3D组织,以及将细胞内空间细分为功能隔室。可以通过围绕细胞器并充当物理屏障的细胞内膜来实现隔室化。此外,细胞已经开发出了复杂的机制来以严格调节的方式分隔它们的内部物质。最近的研究提供了引人注目的证据,即无膜隔室化可以通过液体分离(在液-液相分离中进行的过程)以及相界的形成来实现。
本发明人已经令人惊奇地发现了能够有效地控制多核苷酸相对于功能隔室(包括核隔室,例如核周边,Cajal小体和早幼粒细胞白血病(PML)小体)的空间定位的通用系统和方法。所述系统和方法还可用于通过形成在细胞内组织或分配的蛋白质,RNA和/或DNA分子的超分子组装体来产生合成的相分离。本文公开的系统和方法可用于操纵细胞核/细胞质中基因组DNA和RNA组分的时空组织和调节不同的细胞功能。所提供的系统和方法还可以用于空间基因组组织的可编程控制,以及用于应用该组织以影响多核苷酸调节和细胞功能,以及介导靶向的多核苷酸和不同细胞隔室之间的相互作用动力学。所公开的系统可用于例如实现亚细胞空间的动态重组,作为在包括癌症和神经变性的疾病中操纵病理蛋白组装的框架。
所公开的系统可以是化学诱导的和可逆的,使得能够探查例如染色质与活细胞中的核隔室的相互作用的实时动力学。作为进一步的实例,基因组基因座到核周边的可诱导的重新定位可以允许剖析依赖有丝分裂和不依赖有丝分裂的重新定位事件,探查基因位置和表达之间的关系,以及理解端粒重新定位对细胞生长的影响。本文所述的系统可介导Cajal小体在靶染色质基因座处的快速从头形成,并引起跨长距离(>30kb)的相邻内源性基因表达的显著抑制。因此,所提供的系统提供了以靶向方式研究大规模空间多核苷酸组织和功能的新平台。
在一些实施方案中,使用不同的sgRNA允许系统被编程以灵活地靶向不同的基因组序列。基因组基因座到核周边的重新定位可以以依赖有丝分裂和不依赖有丝分裂的方式实现。通过快速从头形成Cajal小体或通过将靶DNA重新定位到现有的Cajal小体,可以触发靶DNA与Cajal小体的共定位。使用所提供的系统和方法将基因组基因座靶向核周边或Cajal小体也可以抑制相邻的报告基因表达。重要的是,基因组基因座与Cajal小体的共定位也可以抑制相邻的内源性基因(>30kb)的表达。此外,使用本公开的方面将端粒隔离至核周边可负面地影响细胞生长。
II.定义
如本文所用,除非另有说明,否则以下术语具有赋予它们的含义。
如说明书和权利要求书中所使用的,单数形式“一个”、“一种”和“所述”包括复数引用,除非上下文另有明确规定。例如,术语“一个细胞”包括多个细胞。
术语“隔室”是指细胞隔室,其包括被单层或双层脂质层膜包围的膜封闭区域和无膜区域,例如通过相分离和相边界的形成实现的核小体和细胞体。隔室包括核隔室和细胞质隔室。核隔室的非限制性实例包括核周边,内核膜,核孔复合物和异染色质,以及核小体,例如Cajal小体,早幼粒细胞白血病(PML)小体,核小点和核仁。细胞质隔室的非限制性实例包括膜结合和非膜结合的细胞器,例如线粒体、叶绿体、过氧化物酶体、溶酶体、内质网、高尔基体、囊泡、液泡、溶酶体、核内体、核糖体、蛋白酶体、中心粒和细胞骨架,以及细胞体,例如,P颗粒、GW小体、应激颗粒、海绵体、CyPrP-RNP颗粒和U小体。
术语“隔室特异性蛋白”是指能够将靶多核苷酸定位在隔室中,诱导或调节包含靶多核苷酸的隔室的形成或定位,和/或将靶多核苷酸递送至细胞内的特定位置的蛋白。将靶多核苷酸定位在隔室中的隔室特异性蛋白通常是该隔室的内源性组分。诱导或调节包含靶多核苷酸的隔室的形成或定位的隔室特异性蛋白通常是调节蛋白如基因激活物或阻抑物。将靶多核苷酸递送到细胞内特定位置的隔室特异性蛋白通常是马达蛋白或参与细胞内转运的蛋白质。
如本文所用,“细胞”通常可指生物细胞。细胞可以是活生物体的基本结构,功能和/或生物单元。细胞可源自具有一个或多个细胞的任何生物体。一些非限制性实例包括:原核细胞、真核细胞、细菌细胞、古细菌细胞、单细胞真核生物的细胞、原生动物细胞、来自植物的细胞(例如,来自植物作物、水果、蔬菜、谷物、大豆、玉米(corn)、玉蜀黍(maize)、小麦、种子、西红柿、稻、木薯、甘蔗、南瓜、干草、马铃薯、棉花、大麻、烟草、开花植物、针叶树、裸子植物、蕨类植物、石松类植物(clubmosses)、角苔类植物、苔类植物(liverworts)、苔藓植物的细胞)、藻类细胞(例如,布朗葡萄藻(botryococcus braunii)、莱茵衣藻(Chlamydomonas reinhardtii)、微拟球藻(nannochloropsis gaditana)、蛋白核小球藻(chlorella pyrenoidosa)、展枝马尾藻圆干变种(sargassum patens C.agardh)等),海藻(例如,海带)、真菌细胞(例如,酵母细胞、来自蘑菇的细胞)、动物细胞、来自无脊椎动物(例如,果蝇、刺胞动物、棘皮动物、线虫等)的细胞、来自脊椎动物(例如,鱼类、两栖动物、爬行动物、鸟类、哺乳动物)的细胞、来自哺乳动物(例如,猪、牛、山羊、绵羊、啮齿动物、大鼠、小鼠、非人类灵长类动物、人类等)的细胞等。有时细胞不起源于天然生物体(例如,细胞可以是合成的,有时称为人造细胞)。
术语“多核苷酸”、“寡核苷酸”和“核酸”可互换使用,是指任何长度的核苷酸(或者是脱氧核糖核苷酸,或者是核糖核苷酸,或者它们的类似物)的聚合形式,或者是单链,双链或多链形式。多核苷酸可以是细胞的外源性或内源性的。多核苷酸可存在于无细胞环境中。多核苷酸可以是基因或其片段。多核苷酸可以是DNA。多核苷酸可以是RNA。多核苷酸可以具有任何三维结构,并且可以执行已知或未知的任何功能。多核苷酸可包含一种或多种类似物(例如,改变的主链,糖或核碱基)。如果存在,可以在聚合物组装之前或之后对核苷酸结构进行修饰。类似物的一些非限制性实例包括:5-溴尿嘧啶、肽核酸、异源核酸、吗啉、锁核酸、二醇核酸、苏糖核酸、双脱氧核苷酸、虫草素、7-脱氮-GTP、氟磷(例如与糖连接的罗丹明或荧光素)、含硫醇的核苷酸、生物素连接的核苷酸、荧光碱基类似物、CpG岛、甲基-7-鸟苷、甲基化核苷酸、肌苷、硫苷、假尿嘧啶核苷、二氢尿苷、辫苷和怀俄苷。多核苷酸的非限制性实例包括基因或基因片段的编码或非编码区、由连锁分析定义的多个基因座(一个基因座)、外显子、内含子、信使RNA(mRNA)、转移RNA(tRNA)、核糖体RNA(rRNA)、短干扰RNA(siRNA)、短发夹RNA(shRNA)、微RNA(miRNA)、核酶、cDNA、重组多核苷酸、分支多核苷酸、质粒、载体、任何序列的分离DNA、任何序列的分离RNA、包括无细胞DNA(cfDNA)和无细胞RNA(cfRNA)的无细胞多核苷酸、核酸探针和引物。核苷酸序列可以被非核苷酸组分中断。
术语“靶多核苷酸”是指由本公开的致动器部分靶向的多核苷酸或核酸。靶多核苷酸可以是DNA。靶多核苷酸可以是RNA。靶多核苷酸可指染色体序列或染色体外序列(例如,附加体序列、小环序列、线粒体序列、叶绿体序列等)。靶多核苷酸可以是核酸序列,其可以通过单核苷酸取代而与核酸样品中的任何其它序列不相关。靶多核苷酸可以是核酸序列,其可以通过至少2、3、4、5、6、7、8、9或10个核苷酸取代而与核酸样品中的任何其它序列不相关。在一些实施方案中,取代可以不在靶多核苷酸的5’端的5、10、15、20、25、30或35个核苷酸内发生。在一些实施方案中,取代可以不在靶多核苷酸的3’端的5、10、15、20、25、30、35个核苷酸内发生。通常,术语“靶序列”是指靶多核苷酸单链上的核酸序列。靶序列可以是基因、调控序列、基因组DNA、包括cfDNA和/或cfRNA的无细胞核酸、cDNA、融合基因和包括mRNA、miRNA、rRNA等的RNA的一部分。
本文所用的术语“致动器部分”是指可以调节基因的表达或活性和/或编辑核酸序列(无论是外源性的还是内源性的)的部分。致动器部分可以在转录水平和/或翻译水平调节基因的表达。致动器部分可以在转录水平上调节基因表达,例如通过调节从DNA(例如染色体DNA或cDNA)产生mRNA。在一些实施方案中,致动器部分募集至少一种结合特定DNA序列的转录因子,从而控制遗传信息从DNA到mRNA的转录速率。致动器部分本身可以结合DNA并通过物理障碍调节转录,例如通过防止蛋白质如RNA聚合酶和其它相关蛋白质装配在DNA模板上。致动器部分可以在翻译水平上调节基因的表达,例如,通过调节从mRNA模板产生蛋白质。在一些实施方案中,致动器部分通过影响mRNA转录物的稳定性来调节基因表达。在一些实施方案中,致动器部分通过编辑核酸序列(例如,基因组的区域)来调节基因的表达。在一些实施方案中,致动器部分通过编辑mRNA模板来调节基因的表达。在某些情况下,编辑核酸序列可以改变基因表达的基础模板。
本文所指的Cas蛋白可以是任何类型的蛋白质或多肽。Cas蛋白可以指核酸酶。Cas蛋白可以指核糖核酸酶。Cas蛋白可以指Cas蛋白的任何修饰的(例如,缩短的,突变的,延长的)多肽序列或同源物。Cas蛋白可以是密码子优化的。Cas蛋白可以是Cas蛋白的密码子优化的同源物。Cas蛋白可以是无酶促活性的、部分活性的、组成型活性的、完全活性的、可诱导活性的和/或更有活性的(例如,超过蛋白质或多肽的野生型同源物)。Cas蛋白可以是Cas9。Cas蛋白可以是Cas12a(Cpf1)。Cas蛋白可以是Cas13a(C2c2)。Cas蛋白(例如,变异的,突变的,无酶促活性的和/或有条件无酶促活性的定点多肽)可结合靶多核苷酸。Cas蛋白(例如,变异的,突变的,无酶促活性的和/或有条件无酶促活性的内切核糖核酸酶)可以结合靶RNA或DNA。
本文所述的蛋白质或多肽可以通过接头(例如肽或多肽接头)或通过肽键彼此“连接”。肽或多肽接头可含有天然氨基酸,非天然氨基酸或其组合。在一些实施方案中,肽或多肽接头可以是柔性接头,例如含有氨基酸如Gly、Asn、Ser、Thr、Ala等。这种接头使用已知参数设计,并且可以是任何长度,并且含有任何长度的任何数目的重复序列单元(例如,Gly和Ser残基的重复序列单元)。例如,接头可以具有重复序列,例如两个,三个,四个,五个或更多个Gly
III.CRISPR-GO系统
CRISPR-Cas系统已经被重新用作灵活的基因组工程平台,并且已经用于诸如基因编辑、转录调节、表观遗传修饰、DNA环形成和基因组成像的应用。本文提供了对呈多核苷酸组织系统形式的CRISPR-Cas工具箱的进一步扩展,其使得能够对细胞隔室内的靶向的多核苷酸定位进行可编程控制。在某些方面,靶向的多核苷酸包含基因组DNA,并且所述系统被称为CRISPR-GO(图58),其中GO是指基因组组织。本文公开的系统和方法可以有效地将多核苷酸(例如,内源性基因组基因座)靶向各种细胞隔室(例如,核周边、Cajal小体和PML小体)。所提供的系统可以是可诱导的和可逆的,允许例如对靶向的染色质DNA和核隔室之间的相互作用动力学进行探查。利用该特征,已经实现了基因组基因座到核周边的依赖有丝分裂和不依赖有丝分裂的重新定位,并且已经证明了Cajal小体在靶基因座处的从头形成和现有Cajal小体与靶向的染色质基因座的共定位。使用本文公开的系统和方法将基因组基因座与核周边或Cajal小体共定位已被用于影响相邻基因的表达。值得注意的是,使用所提供的系统和方法将内源性基因座与Cajal小体共定位可以显著抑制附近的基因表达,即使这些基因远离(>30kb)靶位点。最后,已经发现用本文公开的系统和方法将端粒重新定位到核周边可破坏端粒动力学并降低细胞活力。所提供的方法提供了用于可编程控制多核苷酸(例如基因组DNA)与各种细胞(例如核)隔室相互作用的平台,其可以促进对时空多核苷酸组织在调节、稳定性和细胞功能方面的功能作用的更深入的理解。
细胞生物学的主要目标是了解基因组与不同核隔室的相互作用如何影响基因表达,染色质构象和细胞功能。CRISPR-GO系统可以有效地将特定基因组位点靶向核周边,Cajal小体和PML小体,并且还具有扩展到其他核隔室如核仁,核孔复合物和核小点的潜力。可以通过将CRISPR-GO与不同的隔室特异性蛋白(例如异染色质蛋白1α(HP1α))偶联来实现将基因组基因座靶向其它核隔室(图59)。类似地,本文所公开的系统和方法提供了一种通用的模块化平台,其可应用于各种蜂窝隔室的研究。
所提供的系统(例如CRISPR-GO)允许多核苷酸(例如基因组基因座)以精确和靶向的方式可编程地重新定位。例如,CRISPR-GO系统可以有效地将位于不同染色体上的重复和非重复染色质基因座靶向核隔室。与LacI-LacO系统不同,CRISPR-GO系统的基因组靶标可以通过sgRNA与靶DNA序列之间的碱基配对相互作用灵活地限定,并且简单地改变sgRNA上的~20nt区域允许靶向不同的基因组基因座。该可编程特征可允许使用CRISPR-GO靶向各种基因组元件,包括蛋白质编码基因,非编码RNA基因和调节元件。相比之下,LacO-LacI技术不适于可编程的基因组靶向,因为它只能在含有高度重复的LacO阵列的良好表征的细胞系上进行。建立和表征有用的含LacO细胞系是困难且费力的。通常将LacO阵列随机插入到基因组中,之后选择含有单拷贝插入的细胞以在通过FISH和其它方法表征精确的基因组整合位点之前构建稳定的细胞系。此外,基因组中大LacO阵列的整合可能改变局部染色质构象。总之,在此公开的系统和方法的通用性提供了比研究细胞组织的常规方法更大的技术优势。
用本文公开的系统和方法靶向多核苷酸的新基因座的总的便利性可以促进对3D多核苷酸组织中的扰动与细胞表型变化之间的关系的更广泛的研究。例如,不同的sgRNA设计策略可用于靶向重复和非重复基因组基因座。使用在确定的基因组区域内具有多个靶标的单一sgRNA可以容易地靶向重复的基因组基因座。人类基因组具有丰富的重复序列或重复序列衍生的序列,其中许多可能具有重要的基因组组织作用。这些重复序列是大规模筛查实验的候选序列,为研究基因组相对于核隔室的组织和细胞表型之间的关系开辟了更高通量方法的大门。此外,可以使用多种sgRNA或使用单一sgRNA靶向非重复的基因组基因座。为了靶向非重复的基因座,可以使用展开sgRNA的库作为起始点。
所提供的系统和方法也可用于研究多核苷酸重新定位的实时动力学以及细胞隔室与活细胞中特定区域的缔合和解离。在CRISPR-GO系统中,基因组基因座通过化学诱导的dCas9结合的基因组基因座和隔室特异性蛋白之间的物理相互作用靶向期望的隔室。CRISPR-GO的可诱导的和可逆的特征防止了将染色质DNA连续地重新定位到给定的核隔室的潜在的不利影响。
作为一个实例,通过组合使用CRISPR-Cas9活细胞基因组成像和CRISPR-GO,已显示内源性基因组基因座重新定位至核周边以依赖有丝分裂和不依赖有丝分裂的方式发生。在有丝分裂过程中,核膜在前中期瓦解,然后在末期重新形成。有丝分裂过程中染色质和核结构的显著变化可以促进基因组基因座和核膜之间的相互作用以产生核包膜束缚。在间期期间,尽管染色质结构保持相对稳定,但基因组基因座与核周边在其紧密接近时仍可形成相互作用。在间期期间的核周边束缚可依赖于靶向的基因座和核周边之间的接近度,并且位于核周边远端的基因组基因座不太可能通过不依赖有丝分裂的方式束缚。
一些提供的实施方案的化学诱导方法还允许研究靶多核苷酸基因座和活细胞中的细胞隔室之间的实时关联。例如,与相对较慢地重新定位到核周边(在数小时内)相比,基因组基因座和Cajal小体之间的共定位以快得多的速率(在数分钟内)发生,这可能是因为Cajal小体组分更多地扩散到整个细胞核中。使用所公开的系统和方法,已经观察到CB和靶基因组基因座之间的共定位可以以两种方式发生:一种是在基因组基因座处快速形成从头Cajal小体,另一种是现有的CB与靶基因组基因座的重新定位,这是一种以前没有报道过的现象。先前的工作已经提出Cajal小体是通过相分离形成的。CRISPR-GO将核小体组分(例如CB的Coilin)募集至靶向的基因组基因座可在靶染色质基因座处产生合成的相分离。
所提供的方法和系统还被用于在将基因组基因座重新定位到核周边时观察相邻荧光报告基因的抑制。先前的工作报道了在将LacO基因座束缚到核周边后对基因表达的不同影响。特别地,早期的研究已经观察到LacO重复序列被LacI-Lamin B募集到核周边后转录没有变化,并且已经显示通过LacI-Emerin将LacO重复序列束缚到核周边导致相邻基因的抑制。本文公开的系统已经显示将报告基因重新定位到Emerin引起基因抑制(约59%)。
本文公开的系统和方法还被用于在CRISPR-GO介导的染色质基因座共定位于CB之后抑制相邻的报告基因和内源性基因。重要的是,Cajal小体与内源性基因座的靶向共定位抑制了跨长距离(>30kb)的相邻基因的表达。在将基因组基因座靶向CB后的这种观察到的基因抑制尚未报道。相比之下,CRISPRi/a方法通过募集转录效应物起作用,所述转录效应物主要影响靶位点周围几千碱基内的局部基因的表达。因此,所提供的方法和系统提供了一种重要的用于调节长距离内的多核苷酸的表达的新方法。所述方法和系统还提供了以系统方式控制靶多核苷酸到不同细胞隔室的重新定位以研究细胞效应和程序多核苷酸调节的能力。
在一些实施方案中,CRISPR-GO系统可以被编程以募集调节蛋白(例如,活化或抑制效应物)用于基因(例如,靶多核苷酸)表达调节。调节蛋白的非限制性实例包括异染色质蛋白1(例如HP1α、HP1β和/或HP1γ)、Krüppel相关盒-锌指蛋白(KRAB-ZFP)、KRAB相关蛋白1(KAP1)、核小体重塑脱乙酰酶复合物(NuRD)、SET结构域分支型1(SETDB1)、DNA甲基转移酶(例如DNMT3A、DNMT3L、DNMT3B)、组蛋白脱乙酰酶(HDAC)、SUV39H1、G9α、Ezh1/2、EED、Suz12、JARID2、AEBP2、RbAp48、PCL1、RBBP7/4、C17orf96、C10orf12,其截短形式,其片段及以上的组合。
在一些实施方案中,CRISPR-GO系统可以被编程以改变细胞功能、细胞命运、细胞生长、细胞凋亡和/或细胞分化,这可以通过将发育调节基因组区域和RNA重新定位到不同的细胞隔室来实现。这用作使用基于介质的方法来诱导细胞命运改变或使用转录因子混合物来改变细胞命运的替代方法。如实施例12所述,将端粒靶向核周边导致细胞活力降低,引起包括细胞凋亡基因、分化基因和细胞功能基因在内的基因表达水平的系统变化,而将端粒靶向Cajal小体导致伴随基因表达变化如生长基因和细胞功能基因的上调的细胞活力增加。
在大多数不对称细胞的细胞质中,使用诸如驱动蛋白,动力蛋白和肌球蛋白的马达蛋白作为隔室特异性蛋白,mRNA沿着微管和肌动蛋白丝转运。在一些实施方案中,CRISPR-GO系统可以被编程用于使用这些马达蛋白沿着细胞骨架重新定位mRNA。如实施例13所述,使用诸如驱动蛋白-1重链(KIF5B)的马达蛋白,例如没有货物结合尾结构域,可以将mRNA重新定位到微管的正端(MT+),或者使用诸如Bicaudal D2(例如N-端片段)的马达蛋白(其诱导动力蛋白介导的货物转运),可以将mRNA重新定位到微管的负端(MT-),或使用诸如肌球蛋白5a(MYO5A)的马达蛋白可以沿着肌动蛋白丝(AF)重新定位mRNA。
在一些实施方案中,CRISPR-GO系统可以被编程以形成核隔室,例如促进DNA修复(例如,促进复合物的形成以修复DNA双链断裂(DSB))并导致改进的基因编辑结果(例如,增强的同源性定向修复(HDR))的核小体。可促进核小体形成的隔室特异性蛋白的非限制性实例包括DNA修复基因,例如53BP1、Rad51、Rad52、Ubc9、UBL1、BLM、c-Ab1、BCR/Ab1、BRCA1/2、PALB2、RPA、Rad51AP1、Chk1、Arg、Hop2、MnD1、DMC1或以上的组合。在某些情况下,寡聚化53BP1(截短的,例如,氨基酸1207-1711,或全长的)可用于促进复合物的形成以修复DNA双链断裂(DSB)。在某些情况下,Rad51、Rad52、Ubc9、UBL1、BLM、c-Abl、BCR/Abl、BRCA1/2、PALB2、RPA、Rad51AP1、Chk1、Arg、Hop2、Mnd1和/或DMC1可用于增强同源性定向修复(HDR)。如实施例14中所述,在基因编辑时观察到53BP1焦点形成,其可促进CRISPR介导的基因编辑后的DSB分辨和DNA修复。
在一些实施方案中,本文公开的系统和方法与内源性或合成寡聚化蛋白一起使用,所述内源性或合成寡聚化蛋白自聚集以形成人工蛋白质/RNA/DNA聚集体,其可具有一种或多种独特的化学、物理或生物特性(例如特定蛋白、RNA或DNA的选择性扩散;与其它分子缔合或解离;基因调节机制的促进或抑制;或DNA重组或稳定机制的促进或抑制)。这种聚集体在本文中被称为合成细胞期(SCP)。这些聚集体可以对基因调节具有强的影响。在一些实施方案中,蛋白质,蛋白质结构域,RNA,RNA结构域或其组合与所提供的系统偶联,以在所需染色质DNA或RNA周围特异性地形成所需的SCP。在一些实施方案中,所提供的系统可用于操纵细胞核和/或细胞质中基因组DNA和RNA组分的时空组织,以及用于调节不同的细胞功能。
在一些实施方案中,所述系统和方法包括诱导型二聚化,其中所述二聚化是化学诱导的二聚化、光(例如,光生或化学-光生)诱导的二聚化或酶催化的蛋白质连接。二聚化可以包括相同二聚化域的均二聚化或两个不同二聚化域的异二聚化。
在某些实施方案中,二聚化是由分子配体(例如化学诱导物)介导的化学诱导的二聚化。在某些方面,二聚化系统选自ABA诱导的ABI/PYL1二聚化系统、赤霉素(GA)诱导的GID1/GAI二聚化系统、雷帕霉素诱导的FRB/FKBP二聚化系统、TMP-HTag诱导的HaloTag/DHFR二聚化系统、FK1012诱导的FKBP/FKBP二聚化系统、FK506诱导的FKBP/钙调磷酸酶A(CNA)二聚化系统、FKCsA诱导的FKBP/CyP-Fas二聚化系统、香豆霉素诱导的GyrB/GyrB二聚化系统、HaXS诱导的SnapTag/HaloTag二聚化系统和ABT-737诱导的BCL-xL/Fab(AZ1)二聚化系统。也考虑其它化学诱导的二聚化系统。
在某些实施方案中,二聚化是光诱导的二聚化。光诱导的二聚化的非限制性实例包括光生和化学-光生二聚化系统。光生二聚化系统通常使用在照射时经历构象变化的光敏蛋白,并因此诱导蛋白质相互作用。化学-光生二聚化系统通常使用光可激活和/或可裂解的小分子二聚体,从而可通过光诱导和/或破坏接近度。参见,例如,Klewer等人,“Light-Induced Dimerization Approaches to Control Cellular Processes,”Chem.Eur.J.(2019)25:1-13。也考虑其它光诱导的二聚化系统。
在某些实施方案中,使用酶催化的反应例如酶催化的蛋白质连接实现二聚化。作为非限制性实例,二聚化可以通过由肽连接酶如枯草蛋白酶(subtiligase)或其变体催化的二聚化结构域的连接来介导。参见,例如Henager,S.,“Enzyme-catalyzed expressedprotein ligation,”Nat Methods(2016)13(11):925-927。也考虑使用酶催化反应的其它二聚化系统。
在一些实施方案中,所提供的系统和方法的靶向多核苷酸包含DNA,例如基因组DNA。在一些实施方案中,靶多核苷酸包含RNA,例如mRNA、微RNA、siRNA或非编码RNA。适用于所提供的系统和方法的致动器部分和相关靶向系统包括例如CRISPR-Cas(包括所有类型的CRISPR,I型、II型、III型、IV型、V型、VI型,例如Cas9、Cas12、Cas13);Argonaute介导的靶向或锌指靶向;TALE(转录激活因子样效应物);LacO-LacI或TetO-TetR;以及DNA相互作用蛋白或RNA结构域的特异性对。Cas9和Cas13也可以以序列依赖性的方式靶向RNA,并且可以以这种方式与所提供的系统一起用于将RNA分子重新定位到不同的细胞隔室。Cas蛋白可缺乏DNA切割活性。靶向系统可包括序列特异性导向RNA或导向DNA。
致动器部分可以包含核酸酶(例如DNA核酸酶和/或RNA核酸酶)、与野生型核酸酶相比,核酸酶缺陷型的或具有降低的核酸酶活性的修饰的核酸酶(例如DNA核酸酶和/或RNA核酸酶)、其衍生物、其变体或其片段。致动器部分可以调节基因的表达或活性和/或编辑核酸(例如基因和/或基因产物)的序列。在一些实施方案中,致动器部分包含DNA核酸酶,例如经改造(例如,可编程或可靶向)的DNA核酸酶,以诱导靶DNA序列的基因组编辑。在一些实施方案中,致动器部分包含RNA核酸酶,例如经改造(例如,可编程或可靶向)的RNA核酸酶,以诱导靶RNA序列的编辑。在一些实施方案中,致动器部分具有降低的或最小的核酸酶活性。具有降低的或最小的核酸酶活性的致动器部分可以通过靶多核苷酸的物理阻塞或有效抑制或增强靶多核苷酸表达的其它因子的募集来调节基因的表达和/或活性。在一些实施方案中,致动器部分包含来源于DNA核酸酶的无核酸酶的DNA结合蛋白,其可诱导靶DNA序列的转录激活或抑制。在一些实施方案中,致动器部分包含来源于RNA核酸酶的无核酸酶的RNA结合蛋白,其可诱导靶RNA序列的转录激活或抑制。在一些实施方案中,致动器部分是核酸引导的致动器部分。在一些实施方案中,致动器部分是DNA引导的致动器部分。在一些实施方案中,致动器部分是RNA引导的致动器部分。致动器部分可以调节基因的表达或活性和/或编辑核酸序列,无论是外源性的还是内源性的。
任何合适的核酸酶可用于致动器部分。合适的核酸酶包括但不限于CRISPR-相关(Cas)蛋白或Cas核酸酶,包括I型CRISPR-相关(Cas)多肽、II型CRISPR-相关(Cas)多肽、III型CRISPR-相关(Cas)多肽、IV型CRISPR-相关(Cas)多肽、V型CRISPR-相关(Cas)多肽和VI型CRISPR相关(Cas)多肽;锌指核酸酶(ZFN);转录激活因子样效应物核酸酶(TALEN);大范围核酸酶;RNA结合蛋白(RBP);CRISPR-相关的RNA-结合蛋白;重组酶;翻转酶(flippases);转座酶;Argonaute(Ago)蛋白(例如原核Argonaute(pAgo)、古细菌Argonaute(aAgo)和真核Argonaute(eAgo));其任何衍生物;其任何变体;及其任何片段。
在一些实施方案中,致动器部分包含CRISPR-相关(Cas)蛋白或Cas核酸酶,其在非天然存在的CRISPR(成簇规律间隔短回文重复序列)/Cas(CRISPR-相关)系统中起作用。在细菌中,该系统可以提供针对外源DNA的适应性免疫(Barrangou,R.等人,“CRISPRprovides acquired resistance against viruses in prokaryotes,”Science(2007)315:1709-1712;Makarova,K.S.等人,“Evolution and classification of the CRISPR-Cas systems,”Nat Rev Microbiol(2011)9:467-477;Garneau,J.E.等人,“The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA,”Nature(2010)468:67-71;Sapranauskas,R.等人,“The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli,”Nucleic Acids Res(2011)39:9275-9282)。
在包括不同的哺乳动物,动物,植物和酵母在内的多种生物体中,CRISPR/Cas系统(例如,修饰的和/或未修饰的)可用作基因组工程改造工具。CRISPR/Cas系统可包含与Cas蛋白复合的导向核酸,如导向RNA(gRNA),用于靶向调节基因表达和/或活性或核酸编辑。RNA导向的Cas蛋白(例如,Cas核酸酶,如Cas9核酸酶)可以以序列依赖性方式特异性结合靶多核苷酸(例如,DNA)。Cas蛋白,如果具有核酸酶活性,可以切割DNA(Gasiunas,G.等人,“Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage foradaptive immunity in bacteria,”Proc Natl Acad Sci USA(2012)109:E2579-E2 86;Jinek,M.等人,“A programmable dual-RNA-guided DNA endonuclease in adaptivebacterial immunity,”Science(2012)337:816-821;Sternberg,S.H.等人,“DNAinterrogation by the CRISPR RNA-guided endonuclease Cas9,”Nature(2014)507:62;Deltcheva,E.等人,“CRISPR RNA maturation by trans-encoded small RNA and hostfactor RNase III,”Nature(201 1)471:602-607),并且已被广泛用于在各种生物体和模型系统中进行可编程的基因组编辑(Cong,L.等人,“Multiplex genome engineeringusing CRISPR Cas systems,”Science(2013)339:819-823;Jiang,W.等人,“RNA-guidedediting of bacterial genomes using CRISPR-Cas systems,”Nat.Biotechnol.(2013)31:233-239;Sander,J.D.&Joung,J.K,“CRISPR-Cas systems for editing,regulatingand targeting genomes,”Nature Biotechnol.(2014)32:347-355)。
在一些情况下,Cas蛋白被突变和/或修饰以产生核酸酶缺陷蛋白或相对于野生型Cas蛋白具有降低的核酸酶活性的蛋白质。核酸酶缺陷蛋白可保留结合DNA的能力,但可能缺乏或具有降低的核酸切割活性。包含Cas核酸酶(例如,保留野生型核酸酶活性,具有降低的核酸酶活性,和/或缺乏核酸酶活性)的致动器部分可以在CRISPR/Cas系统中起作用以调节靶基因或蛋白质的水平和/或活性(例如,降低,增加或消除)。Cas蛋白可结合靶多核苷酸并通过物理障碍阻止转录或编辑核酸序列以产生非功能性基因产物。
在一些实施方案中,致动器部分包含与导向核酸如导向RNA形成复合物的Cas蛋白。在一些实施方案中,致动器部分包含与单一导向核酸如单一导向RNA(sgRNA)形成复合物的Cas蛋白。在一些实施方案中,致动器部分包含任选地与导向核酸如导向RNA(例如sgRNA)复合的RNA-结合蛋白(RBP),所述导向核酸能够与Cas蛋白形成复合物。在一些实施方案中,致动器部分包含来源于DNA核酸酶的无核酸酶的DNA结合蛋白,其可诱导靶DNA序列的转录激活或抑制。在一些实施方案中,致动器部分包含来源于RNA核酸酶的无核酸酶的RNA结合蛋白,其可诱导靶RNA序列的转录激活或抑制。
可以使用任何合适的CRISPR/Cas系统。CRISPR/Cas系统可以使用各种命名系统来提及。在以下当中提供了示例性的命名系统:Makarova,K.S.等人,“An updatedevolutionary classification of CRISPR-Cas systems,”Nat Rev Microbiol(2015)13:722-736和Shmakov,S.等人,“Discovery and Functional Characterization of DiverseClass 2CRISPR-Cas Systems,”Mol Cell(2015)60:1-13。CRISPR/Cas系统可以是I型、II型、III型、IV型、V型、VI型系统或任何其它合适的CRISPR/Cas系统。本文使用的CRISPR/Cas系统可以是1类、2类或任何其它适当分类的CRISPR/Cas系统。1类或2类的确定可以基于编码效应物模块的基因。1类系统通常具有多亚单位crRNA-效应物复合物,而2类系统通常具有单一蛋白,例如Cas9、Cpf1、C2c1、c2c2、C2c3或crRNA-效应物复合物。1类CRISPR/Cas系统可以使用多种Cas蛋白的复合物来实现调节。1类CRISPR/Cas系统可以包括例如I型(例如,I、IA、IB、IC、ID、IE、IF、IU)、III型(例如,III、IIIA、IIIB、IIIC、IIID)和IV型(例如,IV、IVA、IVB)CRISPR/Cas类型。2类CRISPR/Cas系统可以使用单一大Cas蛋白来实现调节。2类CRISPR/Cas系统可以包括例如II型(例如,II、IIA、IIB)和V型CRISPR/Cas类型。CRISPR系统可以彼此互补,和/或可以反式提供功能单元以促进CRISPR基因座靶向。
包含Cas蛋白的致动器部分可以是1类或2类Cas蛋白。Cas蛋白可以是I型、II型、III型、IV型、V型Cas蛋白或VI型Cas蛋白。Cas蛋白可包含一个或多个结构域。结构域的非限制性实例包括导向核酸识别和/或结合结构域、核酸酶结构域(例如DNase或RNase结构域、RuvC、HNH)、DNA结合结构域、RNA结合结构域、解旋酶结构域、蛋白质-蛋白质相互作用结构域和二聚化结构域。导向核酸识别和/或结合结构域可以与导向核酸相互作用。核酸酶结构域可包含用于核酸切割的催化活性。核酸酶结构域可缺乏防止核酸切割的催化活性。Cas蛋白可以是与其它蛋白或多肽融合的嵌合Cas蛋白。Cas蛋白可以是各种Cas蛋白的嵌合体,例如包含来自不同Cas蛋白的结构域。
CAS蛋白的非限制性实例包括c2c1、C2c2、c2c3、Casl、CaslB、Cas2、Cas3、Cas4、Cas5、Cas5e(CasD)、Cas6、Cas6e、Cas6f、Cas7、Cas8a、Cas8al、Cas8a2、Cas8b、Cas8c、Cas9(Csnl或Csxl2)、Cas10、Cas10d、CaslO、CaslOd、CasF、CasG、CasH、Cpf1、Csyl、Csy2、Csy3、Csel(CasA)、Cse2(CasB)、Cse3(CasE)、Cse4(CasC)、Cscl、Csc2、Csa5、Csn2、Csm2、Csm3、Csm4、Csm5、Csm6、Cmrl、Cmr3、Cmr4、Cmr5、Cmr6、Csbl、Csb2、Csb3、Csxl7、Csxl4、CsxlO、Csxl6、CsaX、Csx3、Csxl、Csxl5、Csfl、Csf2、Csf3、Csf4和Cul966及其同系物或修饰形式。
Cas蛋白可来自任何合适的生物体。非限制性实例包括化脓链球菌(Streptococcus pyogenes)、嗜热链球菌(Streptococcus thermophilus)、链球菌(Streptococcus sp.)、金黄色葡萄球菌(Staphylococcus aureus)、达松维尔拟诺卡氏菌(Nocardiopsis dassonvillei)、始旋链霉菌(Streptomyces pristinae spiralis)、绿色产色链霉菌(Streptomyces viridochromo genes)、绿色产色链霉菌(Streptomycesviridochromogenes)、玫瑰链孢囊菌(Streptosporangium roseum)、玫瑰链孢囊菌、酸热脂环酸杆菌(AlicyclobacHlus acidocaldarius)、假真菌样芽孢杆菌(Bacilluspseudomycoides)、还原硒酸盐芽孢杆菌(Bacillus selenitireducens)、西伯利亚微小杆菌(Exiguobacterium sibiricum)、德氏乳杆菌(Lactobacillus delbrueckii)、唾液乳杆菌(Lactobacillus salivarius)、海洋微颤蓝细菌(Microscilla marina)、伯克霍尔德氏菌目细菌(Burkholderiales bacterium)、萘降解极地单胞菌(Polaromonasnaphthalenivorans)、极地单胞菌(Polaromonas sp.)、瓦氏鳄球藻(Crocosphaerawatsonii)、蓝丝菌(Cyanothece sp.)、铜绿微囊蓝细菌(Microcystis aeruginosa)、铜绿假单胞菌(Pseudomonas aeruginosa)、聚球蓝细菌(Synechococcus sp.)、阿拉伯糖醋盐杆菌(Acetohalobium arabaticum)、Ammonifex degensii、Caldicelulosiruptor becscii、Candidatus Desulforudis、肉毒梭菌(Clostridium botulinum)、艰难梭菌(Clostridiumdifficile)、大芬戈尔德菌(Finegoldia magna)、嗜热盐碱厌氧菌(Natranaerobiusthermophilus)、喜温发酵菌(Pelotomaculum thermopropionicum)、喜温嗜酸硫杆菌(Acidithiobacillus caldus)、氧化亚铁嗜酸硫杆菌(Acidithiobacillusferrooxidans)、Allochromatium vinosum、海杆菌(Marinobacter sp.)、嗜盐亚硝化球菌(Nitrosococcus halophilus)、Nitrosococcus watsoni、Pseudoalteromonashaloplanktis、消旋纤线杆菌(Ktedonobacter racemifer)、Methanohalobiumevestigatum、多变鱼腥蓝细菌(Anabaena variabilis)、产泡沫节球蓝细菌(Nodulariaspumigena)、念珠蓝细菌(Nostoc sp.)、最大节螺蓝细菌(Arthrospira maxima)、盘状节螺蓝细菌(Arthrospira platensis)、节螺蓝细菌(Arthrospira sp.)、鞘丝蓝细菌(Lyngbyasp.)、原型微鞘藻(Microcoleus chthonoplastes)、颤蓝细菌(Oscillatoria sp.)、Petrotoga mobilis、非洲栖热腔菌(Thermosipho africanus)、深海单细胞蓝藻(Acaryochloris marina)、Leptotrichia shahii和新凶手弗朗西丝氏菌(Francisellanovicida)。在一些方面,生物体是化脓链球菌(S.pyogenes)。在一些方面,生物体是金黄色葡萄球菌(S.aureus)。在一些方面,生物体是嗜热链球菌(S.thermophilus)。
Cas蛋白可衍生自多种细菌物种,包括但不限于非典型韦荣氏球菌(Veillonellaatypical)、具核梭杆菌(Fusobacterium nucleatum)、龈沟产线菌(Filifactor alocis)、Solobacterium moorei、尖锐粪球菌(Coprococcus catus)、齿垢密螺旋体(Treponemadenticola)、Peptoniphilus duerdenii、Catenibacterium mitsuokai、变异链球菌(Streptococcus mutans)、无害利斯特氏菌(Listeria innocua)、伪中间葡萄球菌(Staphylococcus pseudintermedius)、肠氨基酸球菌(Acidaminococcus intestine)、Olsenella uli、北原酒球菌(Oenococcus kitaharae)、双歧双歧杆菌(Bifidobacteriumbifidum)、鼠李糖乳杆菌(Lactobacillus rhamnosus)、加氏乳杆菌(Lactobacillusgasseri)、大芬戈尔德菌(Finegoldia magna)、运动支原体(Mycoplasma mobile)、鸡败血支原体(Mycoplasma gallisepticum)、羊肺炎支原体(Mycoplasma ovipneumoniae)、犬支原体(Mycoplasma canis)、关节液支原体(Mycoplasma synoviae)、直肠假杆菌(Eubacterium rectale)、嗜热链球菌、长真杆菌(Eubacterium dolichum)、棒状乳杆菌极取亚种(Lactobacillus coryniformis subsp.Torquens)、多养型泥杆菌(Ilyobacterpolytropus)、白色瘤胃球菌(Ruminococcus albus)、阿克曼氏菌(Akkermansiamuciniphila)、解纤维热酸菌(Acidothermus cellulolyticus)、长双歧杆菌(Bifidobacterium longum)、齿双歧杆菌(Bifidobacterium dentium)、白喉棒杆菌(Corynebacterium diphtheria)、Elusimicrobium minutum、Nitratifractorsalsuginis、Sphaerochaeta globus、产琥珀酸丝状杆菌产琥珀酸亚种(Fibrobactersuccinogenes subsp.Succinogenes)、脆弱拟杆菌(Bacteroides fragilis)、黄褐二氧化碳嗜纤维菌(Capnocytophaga ochracea)、血色红假单胞菌(Rhodopseudomonaspalustris)、彩虹普雷沃菌(Prevotella micans)、栖瘤胃普雷沃氏菌(Prevotellaruminicola)、柱状黄杆菌(Flavobacterium columnare)、少食氨基单胞菌(Aminomonaspaucivorans)、深红红螺菌(Rhodospirillum rubrum)、Candidatus Puniceispirillummarinum、Verminephrobacter eiseniae、Ralstonia syzygii、Dinoroseobacter shibae、固氮螺菌属(Azospirillum)、汉堡硝化杆菌(Nitrobacter hamburgensis)、慢生根瘤菌属(Bradyrhizobium)、产琥珀酸沃林氏菌(Wolinella succinogenes)、空肠弯曲杆菌空肠亚种(Campylobacter jejuni subsp.Jejuni)、鼬鼠螺杆菌(Helicobacter mustelae)、蜡状芽孢杆菌(Bacillus cereus)、Acidovorax ebreus、产气荚膜梭菌(Clostridiumperfringens)、食清洁剂细小棒菌(Parvibaculum lavamentivorans)、Roseburiaintestinalis、脑膜炎奈瑟氏球菌(Neisseria meningitidis)、多杀巴斯德氏菌属多杀亚种(Pasteurella multocida subsp.Multocida)、华德萨特菌(Sutterellawadsworthensis)、变形菌(proteobacterium)、侵肺军团菌(Legionella pneumophila)、Parasutterella excrementihominis、产琥珀酸沃林氏菌和新凶手弗朗西丝氏菌。
本文所用的Cas蛋白可以是Cas蛋白的野生型或修饰形式。Cas蛋白可以是野生型或修饰的Cas蛋白的活性变体,无活性变体或片段。相对于野生型形式的Cas蛋白,Cas蛋白可包含氨基酸改变,例如缺失、插入、取代、变体、突变、融合、嵌合体或以上的任何组合。Cas蛋白可以是与野生型示例性Cas蛋白具有至少约5%、10%、20%、30%、40%、50%、60%、70%、80%、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%或100%序列同一性或序列相似性的多肽。Cas蛋白可以是与野生型示例性Cas蛋白具有至多约5%、10%、20%、30%、40%、50%、60%、70%、80%、90%、100%序列同一性和/或序列相似性的多肽。变体或片段可包含与野生型或修饰的Cas蛋白或其部分至少约5%、10%、20%、30%、40%、50%、60%、70%、80%、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%或100%的序列同一性或序列相似性。变体或片段可以靶向与导向核酸复合的核酸基因座,同时缺乏核酸切割活性。
Cas蛋白可包含一个或多个核酸酶结构域,如DNase结构域。例如,Cas9蛋白可包含RuvC样核酸酶结构域和/或HNH样核酸酶结构域。RuvC和HNH结构域可以各自切割双链DNA的不同链以在DNA中产生双链断裂。Cas蛋白可仅包含一个核酸酶结构域(例如,Cpf1包含RuvC结构域但缺乏HNH结构域)。
Cas蛋白可包含与野生型Cas蛋白的核酸酶结构域(例如,RuvC结构域、HNH结构域)具有至少约5%、10%、20%、30%、40%、50%、60%、70%、80%、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%或100%序列同一性或序列相似性的氨基酸序列。
可以修饰Cas蛋白以优化基因表达的调节。Cas蛋白可被修饰以增加或降低核酸结合亲和力,核酸结合特异性和/或酶促活性。Cas蛋白也可以被修饰以改变蛋白的任何其它活性或性质,例如稳定性。例如,Cas蛋白的一个或多个核酸酶结构域可以被修饰,缺失或失活,或者Cas蛋白可以被截短以去除对于蛋白质的功能不是必需的结构域或优化(例如,增强或降低)Cas蛋白的调节基因表达的活性。
Cas蛋白可以是融合蛋白。例如,Cas蛋白可与切割结构域,表观遗传修饰结构域,转录激活结构域或转录阻抑结构域融合。Cas蛋白也可以与提供增加或降低的稳定性的异源多肽融合。融合的结构域或异源多肽可以位于Cas蛋白的N-末端、C-末端或内部。
Cas蛋白可以任何形式提供。例如,Cas蛋白可以以蛋白质的形式提供,例如单独的Cas蛋白或与导向核酸复合的Cas蛋白。Cas蛋白可以以编码Cas蛋白的核酸的形式提供,例如RNA(例如信使RNA(mRNA))或DNA。可以对编码Cas蛋白的核酸进行密码子优化,以便在特定的细胞或生物体中有效地翻译成蛋白质。
编码Cas蛋白的核酸可以稳定地整合到细胞的基因组中。编码Cas蛋白的核酸可以与细胞中有活性的启动子可操作地连接。编码Cas蛋白的核酸可以与表达构建体中的启动子可操作地连接。表达构建体可以包括能够指导基因或其它感兴趣的核酸序列(例如Cas基因)的表达并且可以将这种感兴趣的核酸序列转移到靶细胞的任何核酸构建体。
在一些实施方案中,Cas蛋白是死亡的Cas蛋白。死亡的Cas蛋白可以是缺乏核酸切割活性的蛋白质。
Cas蛋白可以包含野生型Cas蛋白的修饰形式。野生型Cas蛋白的修饰形式可以包含降低Cas蛋白的核酸切割活性的氨基酸改变(例如,缺失,插入或取代)。例如,Cas蛋白的修饰形式可以具有小于90%、小于80%、小于70%、小于60%、小于50%、小于40%、小于30%、小于20%、小于10%、小于5%或小于1%的野生型Cas蛋白(例如,来自化脓链球菌的Cas9)的核酸切割活性。Cas蛋白的修饰形式不具有实质性的核酸切割活性。当Cas蛋白是没有实质性核酸切割活性的修饰形式时,它可以被称为无酶促活性的和/或“死亡的”(缩写为“d”)。死亡的Cas蛋白(例如,dCAS、dCas9)可以结合靶多核苷酸,但不可以切割靶多核苷酸。在一些方面,死亡的Cas蛋白是死亡的Cas9蛋白。
dCas9多肽可与单一导向RNA(sgRNA)结合以激活或抑制靶DNA的转录。可以将sgRNAs引入表达工程化嵌合受体多肽的细胞中。在一些情况下,这样的细胞含有一种或多种靶向相同核酸的不同sgRNA。在其它情况下,sgRNA靶向细胞中的不同核酸。由导向RNA靶向的核酸可以是在细胞如免疫细胞中表达的任何核酸。靶向的核酸可以是参与免疫细胞调节的基因。在一些实施方案中,核酸与癌症相关。与癌症相关的核酸可以是细胞周期基因,细胞应答基因,细胞凋亡基因或吞噬基因。重组导向RNA可被CRISPR蛋白,无核酸酶的CRISPR蛋白,其变体或其衍生物识别。
无酶促活性的可指能以序列特异性方式与多核苷酸中的核酸序列结合,但不能切割靶多核苷酸的多肽。无酶促活性的定点多肽可以包含无酶促活性的结构域(例如核酸酶结构域)。无酶促活性的可以指无活性。无酶促活性的可以指基本上没有活性。无酶促活性的可以指基本上没有活性。无酶促活性的可指与野生型示例性活性(例如核酸切割活性、野生型Cas9活性)相比小于1%、小于2%、小于3%、小于4%、小于5%、小于6%、小于7%、小于8%、小于9%或小于10%的活性。
Cas蛋白的一个或多个核酸酶结构域(例如RuvC、HNH)可以被缺失或突变,使得它们不再有功能或包含降低的核酸酶活性。例如,在包含至少两个核酸酶结构域的Cas蛋白(例如,Cas9)中,如果核酸酶结构域之一被缺失或突变,则所得Cas蛋白(称为切口酶)可在双链DNA内的CRISPR RNA(crRNA)识别序列处产生单链断裂,但不产生双链断裂。这种切口酶可以切割互补链或非互补链,但不能切割两者。如果Cas蛋白的所有核酸酶结构域(例如,Cas9蛋白中的RuvC和HNH核酸酶结构域;Cpf1蛋白中的RuvC核酸酶结构域)被缺失或突变,则所得Cas蛋白可具有降低的切割双链DNA的两条链的能力或不能切割双链DNA的两条链的能力。可以将Cas9蛋白转化为切口酶的突变的实例是来自化脓链球菌的Cas9的RuvC结构域中的D10A(在Cas9的第10位的天冬氨酸至丙氨酸)突变。来自化脓链球菌的Cas9的HNH结构域中的H939A(在氨基酸位置839处的组氨酸至丙氨酸)或H840A(在氨基酸位置840处的组氨酸至丙氨酸)可以将Cas9转化为切口酶。可以将Cas9蛋白转化为死亡的Cas9的突变的实例是来自化脓链球菌的Cas9的RuvC结构域中的D10A(在Cas9的第10位的天冬氨酸至丙氨酸)突变和HNH结构域中的H939A(在氨基酸位置839处的组氨酸至丙氨酸)或H840A(在氨基酸位置840处的组氨酸至丙氨酸)。
死亡的Cas蛋白相对于野生型形式的蛋白可包含一个或多个突变。突变可导致小于90%、小于80%、小于70%、小于60%、小于50%、小于40%、小于30%、小于20%、小于10%、小于5%或小于1%的野生型Cas蛋白的多个核酸切割结构域的一个或多个中的核酸切割活性。突变可导致多个核酸切割结构域中的一个或多个保留切割靶核酸的互补链的能力,但降低其切割靶核酸的非互补链的能力。突变可导致多个核酸切割结构域中的一个或多个保留切割靶核酸的非互补链的能力,但降低切割靶核酸的互补链的能力。突变可导致多个核酸切割结构域中的一个或多个缺乏切割靶核酸的互补链和非互补链的能力。核酸酶结构域中待突变的残基可对应于核酸酶的一个或多个催化残基。例如,野生型示例性化脓链球菌Cas9多肽中的残基如Asp10、His840、Asn854和Asn856可以被突变以灭活多个核酸切割结构域(例如核酸酶结构域)中的一个或多个。Cas蛋白的核酸酶结构域中待突变的残基可以对应于野生型化脓链球菌Cas9多肽中的残基Asp10、His840、Asn854和Asn856,例如,通过序列和/或结构比对确定的。
作为非限制性实例,残基D10、G12、G17、E762、H840、N854、N863、H982、H983、A984、D986和/或A987(或任何Cas蛋白的相应突变)可以被突变。例如,D10A、G12A、G17A、E762A、H840A、N854A、N863A、H982A、H983A、A984A和/或D986A。除了丙氨酸取代以外的突变也是合适的。
D10A突变可以与H840A、N854A或N856A突变中的一种或多种组合以产生基本上缺乏DNA切割活性的Cas9蛋白(例如,死亡的Cas9蛋白)。H840A突变可以与D10A、N854A或N856A突变中的一种或多种组合以产生基本上缺乏DNA切割活性的定点多肽。N854A突变可以与H840A、D10A或N856A突变中的一种或多种组合以产生基本上缺乏DNA切割活性的定点多肽。N856A突变可以与H840A、N854A或D10A突变中的一种或多种组合以产生基本上缺乏DNA切割活性的定点多肽。
在一些实施方案中,Cas蛋白是2类Cas蛋白。在一些实施方案中,Cas蛋白是II型Cas蛋白。在一些实施方案中,Cas蛋白是Cas9蛋白,Cas9蛋白的修饰形式,或衍生自Cas9蛋白。例如,Cas9蛋白缺乏切割活性。在一些实施方案中,Cas9蛋白是来自化脓链球菌的Cas9蛋白(例如,SwissProt登记号Q99ZW2)。在一些实施方案中,Cas9蛋白是来自金黄色葡萄球菌的Cas9(例如,SwissProt登记号J7RUA5)。在一些实施方案中,Cas9蛋白是来自化脓链球菌或金黄色葡萄球菌的Cas9蛋白的修饰形式。在一些实施方案中,Cas9蛋白衍生自来自化脓链球菌或金黄色葡萄球菌的Cas9蛋白。例如,化脓链球菌或金黄色葡萄球菌Cas9蛋白缺乏切割活性。
Cas9通常可以指与野生型示例性Cas9多肽(例如来自化脓链球菌的Cas9)具有至少约5%、10%、20%、30%、40%、50%、60%、70%、80%、90%、100%序列同一性和/或序列相似性的多肽。Cas9可以指与野生型示例性Cas9多肽(例如来自化脓链球菌)具有至多约5%、10%、20%、30%、40%、50%、60%、70%、80%、90%、100%序列同一性和/或序列相似性的多肽。Cas9可以指野生型或修饰形式的Cas9蛋白,其可以包含氨基酸改变,例如缺失、插入、取代、变体、突变、融合、嵌合体或以上的任何组合。
在一些实施方案中,致动器部分包含与导向RNA复合的RNA结合蛋白,所述导向RNA与靶多核苷酸杂交。RNA结合蛋白的非限制性实例包括ADAR1或ADAR2,而导向RNA的非限制性实例包括ADAR募集RNA(arRNA)(Qu,L.等人,“Programmable RNA editing byrecruiting endogenous ADAR using engineered RNAs,”Nat Biotechnol.(2019)Jul15.doi:10.1038/s41587-019-0178-z)。
在一些实施方案中,致动器部分包含“锌指核酸酶”或“ZFN”。ZFN是指切割结构域(例如FokI的切割结构域)与能结合多核苷酸(例如DNA和RNA)的至少一个锌指基序(例如至少2、3、4或5个锌指基序)之间的融合。两个单独的ZFN以某方向和间隔在多核苷酸的某位置处的异二聚化可以导致多核苷酸的切割。例如,与DNA结合的ZFN可诱导DNA中的双链断裂。为了允许两个切割结构域二聚化和切割DNA,两个单独的ZFN可以以其C末端相隔一定距离结合DNA的相对链。在一些情况下,锌指结构域和切割结构域之间的接头序列可需要每个结合位点的5’边缘被分开约5-7个碱基对。在一些情况下,切割结构域与每个锌指结构域的C末端融合。示例性ZFN包括但不限于以下描述的那些:Urnov等人,Nature ReviewsGenetics,2010,11:636-646;Gaj等人,Nat Methods,2012,9(8):805-7;美国专利号6,534,261;6,607,882;6,746,838;6,794,136;6,824,978;6,866,997;6,933,113;6,979,539;7,013,219;7,030,215;7,220,719;7,241,573;7,241,574;7,585,849;7,595,376;6,903,185;6,479,626;以及美国申请公开号2003/0232410和2009/0203140。
在一些实施方案中,包含ZFN的致动器部分可在靶多核苷酸(例如DNA)中产生双链断裂。DNA中的双链断裂可导致DNA断裂修复,其允许引入基因修饰(例如,核酸编辑)。DNA断裂修复可以通过非同源末端连接(NHEJ)或同源性定向修复(HDR)发生。在HDR中,可以提供含有靶DNA的同源臂侧翼位点的供体DNA修复模板。在一些实施方案中,ZFN是锌指切口酶,其诱导位点特异性单链DNA断裂或缺口,从而导致HDR。锌指切口酶的描述可见于例如Ramirez等人,Nucl Acids Res,2012,40(12):5560-8;Kim等人,Genome Res,2012,22(7):1327-33。在一些实施方案中,ZFN结合多核苷酸(例如DNA和/或RNA),但不能切割多核苷酸。
在一些实施方案中,包含ZFN的致动器部分的切割结构域包含野生型切割结构域的修饰形式。切割结构域的修饰形式可以包含降低切割结构域的核酸切割活性的氨基酸改变(例如,缺失、插入或取代)。例如,切割结构域的修饰形式可以具有小于90%、小于80%、小于70%、小于60%、小于50%、小于40%、小于30%、小于20%、小于10%、小于5%或小于1%的野生型切割结构域的核酸切割活性。切割结构域的修饰形式可以不具有显著的核酸切割活性。在一些实施方案中,切割结构域是无酶促活性的。
在一些实施方案中,致动器部分包含“TALEN”或“TAL效应物核酸酶”。TALEN是指工程化的转录激活因子样效应物核酸酶,其通常含有DNA结合串联重复序列的中心结构域和切割结构域。可以通过将TAL效应物DNA结合结构域与DNA切割结构域融合来生产TALEN。在一些情况下,DNA结合串联重复序列包含33-35个氨基酸的长度,并且在位置12和13处含有两个可识别至少一个特定DNA碱基对的高变氨基酸残基。转录激活因子样效应物(TALE)蛋白可与核酸酶如野生型或突变的FokI内切核酸酶或FokI的催化结构域融合。已经对FokI进行了几种突变,以用于TALEN,其例如提高切割特异性或活性。可以对这种TALEN进行工程改造以结合任何所需的DNA序列。通过在靶DNA序列中产生双链断裂,其依次经历NHEJ或HDR,可以使用TALEN来产生基因修饰(例如,核酸序列编辑)。在一些情况下,提供单链供体DNA修复模板以促进HDR。TALEN及其基因编辑用途的详细描述可参见,例如,美国专利号8,440,431;8,440,432;8,450,471;8,586,363和8,697,853;Scharenberg等人,Curr Gene Ther,2013,13(4):291-303;Gaj等人,Nat Methods,2012,9(8):805-7;Beurdeley等人,NatCommun,2013,4:1762;以及Joung和Sander,Nat Rev Mol Cell Biol,2013,14(1):49-55。
在一些实施方案中,为降低核酸酶活性而工程改造TALEN。在一些实施方案中,TALEN的核酸酶结构域包含野生型核酸酶结构域的修饰形式。核酸酶结构域的修饰形式可以包含降低核酸酶结构域的核酸切割活性的氨基酸改变(例如,缺失、插入或取代)。例如,核酸酶结构域的修饰形式可具有小于90%、小于80%、小于70%、小于60%、小于50%、小于40%、小于30%、小于20%、小于10%、小于5%或小于1%的野生型核酸酶结构域的核酸切割活性。核酸酶结构域的修饰形式可以不具有实质性的核酸切割活性。在一些实施方案中,核酸酶结构域是无酶促活性的。
在一些实施方案中,转录激活因子样效应物(TALE)蛋白与可调节转录且不包含核酸酶的结构域融合。在一些实施方案中,转录激活因子样效应物(TALE)蛋白被设计成起转录激活物的作用。在一些实施方案中,转录激活因子样效应物(TALE)蛋白被设计成起转录阻抑物的作用。例如,转录激活物样效应物(TALE)蛋白的DNA结合结构域可以与一个或多个转录激活结构域,或与一个或多个转录阻抑结构域融合(例如,连接)。转录激活结构域的非限制性实例包括单纯疱疹VP16激活结构域和VP16激活结构域的四聚体重复,例如VP64激活结构域。转录阻抑结构域的非限制性实例包括Krüppel相关的盒结构域。
在一些实施方案中,致动器部分包含大范围核酸酶。大范围核酸酶通常是指可以是高度特异性的稀有切割内切核酸酶或归巢内切核酸酶。大范围核酸酶可以识别长度范围为至少12个碱基对,例如长度为12到40个碱基对,12到50个碱基对,或12到60个碱基对的DNA靶位点。大范围核酸酶可以是模块化DNA结合核酸酶,例如包含内切核酸酶的至少一个催化结构域和至少一个DNA结合结构域的任何融合蛋白,或指定核酸靶序列的蛋白质。DNA结合结构域可以含有至少一个识别单链或双链DNA的基序。大范围核酸酶可以是单体的或二聚体的。在一些实施方案中,大范围核酸酶是天然存在的(在自然界中发现的)或野生型的,并且在其它情况下,大范围核酸酶是非天然的,人工的,工程改造的,合成的,合理设计的或人造的。在一些实施方案中,本公开的大范围核酸酶包括I-CreI大范围核酸酶、I-CeuI大范围核酸酶、I-MsoI大范围核酸酶、I-SceI大范围核酸酶、其变体、其衍生物及其片段。有用的大范围核酸酶及其在基因编辑中的应用的详细描述参见例如Silva等人,Curr GeneTher,2011,11(1):11-27;Zaslavoskiy等人,BMC Bioinformatics,2014,15:191;Takeuchi等人,Proc Natl Acad Sci USA,2014,111(11):4061-4066以及美国专利号7,842,489;7,897,372;8,021,867;8,163,514;8,133,697;8,021,867;8,119,361;8,119,381;8,124,36和8,129,134。
在一些实施方案中,大范围核酸酶的核酸酶结构域包含野生型核酸酶结构域的修饰形式。核酸酶结构域的修饰形式可以包含降低核酸酶结构域的核酸切割活性的氨基酸改变(例如,缺失、插入或取代)。例如,核酸酶结构域的修饰形式可具有小于90%、小于80%、小于70%、小于60%、小于50%、小于40%、小于30%、小于20%、小于10%、小于5%或小于1%的野生型核酸酶结构域的核酸切割活性。核酸酶结构域的修饰形式可以不具有实质性的核酸切割活性。在一些实施方案中,核酸酶结构域是无酶促活性的。在一些实施方案中,大范围核酸酶可结合DNA但不能切割DNA。
在一些实施方案中,致动器部分与一个或多个转录阻抑结构域、激活结构域、表观遗传结构域、重组酶结构域、转座酶结构域、翻转酶结构域、切口酶结构域或以上的任何组合融合。激活结构域可以包括位于酶的羧基末端的一个或多个串联激活结构域。在其它情况下,致动器部分包括位于蛋白质羧基末端的一个或多个串联阻抑结构域。非限制性的示例性激活结构域包括GAL4、单纯疱疹病毒激活结构域VP16、VP64(单纯疱疹病毒激活结构域VP16的四聚体)、NF-κB p65亚基、Epstein-Barr病毒R反式激活因子(Rta),并且描述于Chavez等人,Nat Methods,2015,12(4):326-328和美国专利申请公开号20140068797。非限制性的示例性阻抑结构域包括Kox1的KRAB(Krüppel相关盒)结构域,Mad mSin3相互作用结构域(SID)、ERF阻抑结构域(ERD),并且描述于Chavez等人,Nat Methods,2015,12(4):326-328和美国专利申请公开号20140068797。致动器部分也可以与提供增加或降低的稳定性的异源多肽融合。融合的结构域或异源多肽可以位于致动器部分的N-末端、C-末端或内部。
致动器部分可包含易于追踪或纯化的异源多肽,例如荧光蛋白、纯化标签或表位标记。荧光蛋白的实例包括绿色荧光蛋白(例如,GFP、GFP-2、tagGFP、turboGFP、eGFP、EmeraId、Azami Green、单体Azami Green、CopGFP、AceGFP、ZsGreen1)、黄色荧光蛋白(例如,YFP、eYFP、Citrine、Venus、YPet、PhiYFP、ZsYellowl)、蓝色荧光蛋白(例如eBFP、eBFP2、Azurite、mKalamal、GFPuv、Sapphire、T-sapphire)、青色荧光蛋白(例如eCFP、Cerulean、CyPet、AmCyanl、Midoriishi-Cyan)、红色荧光蛋白(mKate、mKate2、mPlum、DsRed单体、mCherry、mRFPl、DsRed-Express、DsRed2、DsRed-单体、HcRed-Tandem、HcRedl、AsRed2、eqFP611、mRaspberry、mStrawberry、Jred)、橙色荧光蛋白(mOrange、mKO、Kusabira-Orange、单体Kusabira-Orange、mTangerine、tdTomato)和任何其它合适的荧光蛋白。标签的实例包括谷胱甘肽-S-转移酶(GST)、几丁质结合蛋白(CBP)、麦芽糖结合蛋白、硫氧还蛋白(TRX)、聚(NANP)、串联亲和纯化(TAP)标签、myc、AcV5、AU1、AU5、E、ECS、E2、FLAG、血凝素(HA)、nus、Softag 1、Softag 3、Strep、SBP、Glu-Glu、HSV、KT3、S、SI、T7、V5、VSV-G、组氨酸(His)、生物素羧基载体蛋白(BCCP)和钙调蛋白。
任何合适的递送方法可用于将包含多肽和/或编码多肽的核酸的本公开的系统引入细胞。系统组分(例如,与第一二聚化结构域连接的隔室特异性蛋白、与第二二聚化结构域连接的致动器部分)可以同时或分开时间递送。遗传修饰方法的选择可以取决于被转化的细胞的类型和/或转化发生的环境(例如,体外、离体或体内)。
递送方法可包括将一种或多种多核苷酸引入细胞(或细胞群),所述多核苷酸包含编码本公开的系统组分(例如,与第一二聚化结构域连接的隔室特异性蛋白、与第二二聚化结构域连接的致动器部分)的核酸序列。包含编码本公开的系统组分的核酸序列的合适的多核苷酸可以包括表达载体,其中包含编码本公开的一个或多个系统组分(例如,与第一二聚化结构域连接的隔室特异性蛋白、与第二二聚化结构域连接的致动器部分)的核酸序列的表达载体是重组表达载体。
递送方法或转化的非限制性实例包括病毒或噬菌体感染、转染、缀合、原生质体融合、脂质体转染、电穿孔、磷酸钙沉淀、聚乙烯亚胺(PEI)介导的转染、DEAE-葡聚糖介导的转染、脂质体介导的转染、粒子枪技术、磷酸钙沉淀、直接显微注射、细胞可渗透肽的使用和纳米颗粒介导的核酸递送。
在一些实施方案中,本公开提供了方法,其包括将如本文所述的一种或多种多核苷酸,寡核苷酸或载体,或其一种或多种转录物,和/或由其转录的一种或多种蛋白质递送至细胞。在一些实施方案中,本公开还提供通过这样的方法产生的细胞,以及包含这样的细胞或由这样的细胞产生的生物体(例如动物、植物或真菌)。在某些实施方案中,通过这样的方法产生的细胞包含编码与第一二聚化结构域连接的隔室特异性蛋白和与第二二聚化结构域连接的致动器部分的多核苷酸(例如,载体)。
与细胞相容的任何合适的载体可以与本公开的方法一起使用。用于真核细胞的载体的非限制性实例包括pXT1、pSG5(Stratagene
在一些实施方案中,编码系统组分(例如,与第一二聚化结构域连接的隔室特异性蛋白、与第二二聚化结构域连接的致动器部分)的多核苷酸序列与控制元件,例如转录控制元件,如启动子可操作地连接。转录控制元件可以在真核细胞(例如哺乳动物细胞)或原核细胞(例如细菌或古细菌细胞)中起作用。在一些实施方案中,编码系统组分的多核苷酸序列与多个控制元件可操作地连接,所述控制元件允许所述多核苷酸序列在原核和/或真核细胞中表达。
可用于本公开的系统和方法的启动子包括,例如,在真核,哺乳动物,非人哺乳动物或人细胞中有活性的启动子。启动子可以是诱导型或组成型活性启动子。可选地或另外地,启动子可以是组织特异性的或细胞特异性的。
合适的真核启动子(即在真核细胞中起作用的启动子)的非限制性实例可以包括来自以下的那些:巨细胞病毒(CMV)立即早期、单纯疱疹病毒(HSV)胸苷激酶、早期和晚期SV40、来自逆转录病毒的长末端重复序列(LTR)、人延伸因子-1启动子(EF1)、包含与鸡β-活性启动子(CAG)融合的巨细胞病毒(CMV)增强子的杂合构建体、鼠干细胞病毒启动子(MSCV)、磷酸甘油酸激酶-1基因座启动子(PGK)和小鼠金属硫蛋白-1。启动子可以是真菌启动子。启动子可以是植物启动子。可以找到植物启动子的数据库(例如PlantProm)。表达载体还可以含有用于翻译起始的核糖体结合位点和转录终止子。表达载体还可以包括用于扩增表达的适当序列。
在一些实施方案中,靶多核苷酸通过所提供的系统和方法定位在内核膜中。适于靶向内核膜的隔室特异性蛋白包括但不限于Emerin、Lap2β和核纤层蛋白B。
在一些实施方案中,靶多核苷酸通过所提供的系统和方法定位在Cajal小体中。适于靶向Cajal小体的隔室特异性蛋白包括但不限于Coilin、SMN、Gemin 3、SmD1和SmE。
在一些实施方案中,靶多核苷酸通过所提供的系统和方法定位在核小点中。适于靶向核小点的隔室特异性蛋白包括但不限于SC35。
在一些实施方案中,靶多核苷酸通过所提供的系统和方法定位在PML小体中。适于靶向PML小体的隔室特异性蛋白包括但不限于PML和SP100。
在一些实施方案中,靶多核苷酸通过所提供的系统和方法定位在核孔复合物中。适于靶向核孔复合物的隔室特异性蛋白包括但不限于Nup50、Nup98、Nup53、Nup153和Nup62。
在一些实施方案中,靶多核苷酸通过所提供的系统和方法定位在核仁中。适于靶向核仁的隔室特异性蛋白包括但不限于核蛋白B23。
在一些实施方案中,靶多核苷酸通过所提供的系统和方法定位在P颗粒中。适于靶向P颗粒的隔室特异性蛋白包括但不限于RGG结构域蛋白(例如,PGL-1和PGL-3)、死盒蛋白和GLH-1-4。
在一些实施方案中,靶多核苷酸通过所提供的系统和方法定位在GW小体中。适于靶向GW小体的隔室特异性蛋白包括但不限于GW182。
在一些实施方案中,靶多核苷酸通过所提供的系统和方法定位在应激颗粒中。适于靶向应激颗粒的隔室特异性蛋白包括但不限于G3BP(Ras-GAP SH3结合蛋白)、TIA-1(T-细胞胞内抗原)、eIF2和eIF4E。
在一些实施方案中,靶多核苷酸通过所提供的系统和方法定位在海绵体中。适于靶向海绵体的隔室特异性蛋白包括但不限于EXu、Btz、Tral、Cup、eIF4E、Me31B、Yps、Gus、Dcp1/2、Sqd、BicC、Hrb27C和Bru。
在一些实施方案中,靶多核苷酸通过所提供的系统和方法定位在细胞质朊病毒蛋白诱导的核糖核蛋白(CyPrP-RNP)颗粒中。适于靶向CyPrP-RNP颗粒的隔室特异性蛋白包括但不限于Dcp1a、DDX6/Rck/p54/Me31B/Dhh1和Dicer。
在一些实施方案中,靶多核苷酸通过所提供的系统和方法定位在U小体中。适于靶向U小体的隔室特异性蛋白包括但不限于一种或多种富含尿苷的小核核糖核蛋白U1、U2、U4/U6和U5;LSm1-7;以及运动神经元生存(SMN)蛋白。
在一些实施方案中,靶多核苷酸通过所提供的系统和方法定位在内质网中。适于靶向内质网的隔室特异性蛋白包括但不限于钙网蛋白、钙联蛋白、PDI、GRP 78和GRP 94。
在一些实施方案中,靶多核苷酸通过所提供的系统和方法定位在线粒体中。适于靶向线粒体的隔室特异性蛋白包括但不限于HIF1A、PLN、Cox1、己糖激酶和TOMM40。
在一些实施方案中,靶多核苷酸通过所提供的系统和方法定位在质膜中。适于靶向质膜的隔室特异性蛋白包括但不限于钠钾ATP酶、CD98、钙粘蛋白和质膜钙ATP酶(PMCA)。
在一些实施方案中,靶多核苷酸通过提供的系统和方法定位在高尔基体中。适于靶向高尔基体的隔室特异性蛋白包括但不限于GM130、MAN2A1、MAN2A2、GLG1、B4GALT1、RCAS1和GRASP65。
在一些实施方案中,靶多核苷酸通过所提供的系统和方法定位在核糖体中。适于靶向核糖体的隔室特异性蛋白包括但不限于AGO2、MTOR、PTEN、RPL26、FBL和RPS3。
在一些实施方案中,靶多核苷酸通过所提供的系统和方法定位在蛋白酶体中。适于靶向蛋白酶体的隔室特异性蛋白包括但不限于PSMA1、PSMB5、PSMC1、PSMD1和PSMD7。
在一些实施方案中,靶多核苷酸通过所提供的系统和方法定位在核内体中。适于靶向核内体的隔室特异性蛋白包括但不限于CFTR、ADRB1、EGFR、IGF2R、AP2S1、CD4、HLA-A、Coveolin、RAB5和ErbB2。
在一些实施方案中,靶多核苷酸通过所提供的系统和方法定位在脂质体中。适于靶向脂质体的隔室特异性蛋白包括但不限于EEA1、LAMTOR2和LAMTOR4。
可用本文公开的系统和方法靶向的其它细胞隔室包括RNP小体、有丝分裂纺锤体、组蛋白基因座体、异染色质区和细胞骨架。也考虑另外的隔室。
靶多核苷酸可以是其所定位的细胞隔室内源性的或外源性的。靶多核苷酸可以是细胞内源性或外源性的。靶多核苷酸可以是人或非人的。靶多核苷酸可以是病毒来源的、质粒、核糖核蛋白、或合成的RNA或DNA链。
本文公开的方法和系统适用于多重过程,其中多种多核苷酸被重新定位到相同或不同的细胞隔室。
在一些实施方案中,所提供的系统和方法用于介导靶向的多核苷酸(例如,基因组)基因座处的从头细胞隔室(例如,核小体)形成,从而提供了经由液-液相分离引发无膜细胞器形成的潜在方法。亚细胞空间的无膜隔室化通过液-液相分离发生。异型协同弱相互作用使得能够在液体隔室内快速重排。固有无序蛋白质由于其结构可塑性和朊病毒样性质而在相变中起重要作用。细胞动态控制相变的范围和持续时间。分子种子如DNA、RNA或聚(ADP-核糖)(PAR)可以以刺激和背景特异性方式触发相转变。伴侣蛋白,分解酶机制(disintegrase machineries)和翻译后修饰协同控制相变。存在聚集倾向的连续区,并且细胞在蛋白质组装中采用不可预料的宽范围的材料状态。这些可以发展成与神经变性疾病相关的病理学聚集体。
可以使用本文公开的系统和方法形成的合成相的实例包括但不限于可以在病毒防御和端粒维持中起作用的合成PML小体,可以是应激诱导的抗凋亡结构的合成核小点和旁斑(paraspeckle),可以是参与神经变性的因子的中枢的合成宝石,可以种子核小体的合成结构RNA。合成核仁,合成异染色质或常染色质,合成组蛋白基因座体(其可以是FLASH积累的位点并增强组蛋白mRNA加工),合成染色质包装系统(其可以涉及使用Xist使整个染色体顺式沉默),合成表观遗传相,合成(细胞质)P小体,合成应激体,合成胚粒(其可以在发育的胚胎中减数分裂时产生有性细胞),神经变性疾病中的合成mRNP颗粒,可调节无膜细胞器结构和动力学的合成翻译后修饰(PTM),形成聚集体的合成IDP(固有无序蛋白质),以及合成朊病毒样结构域(PLD)或富含RGG的低复杂度结构域(LCD)。可以定位多核苷酸的其它非内源性蛋白质/RNA聚集体包括β-淀粉样蛋白体、mRNA聚集体、Xist包装复合物等。
如本文所述的多核苷酸的受控定位可用于调节、修饰或影响,例如,DNA与RNA聚合酶、转录因子、先驱因子、介体、DNA环分子和其它DNA相关蛋白的相互作用;表观遗传修饰标记或常染色质/异染色质调节酶(例如HP1);染色质致密度和其它生物物理学/生化性质;基因编辑,包括重组、NHEJ或HDR;基因组稳定性和癌症;DNA修复过程;通过剪接、降解、翻译、甲基化、定位、和与其他伴侣蛋白和RNA结合蛋白相互作用,实现mRNA的代谢。
本文公开的方法和系统可用于建立可诱导的且可逆的疾病模型以理解疾病机制。例如,所提供的系统和方法可用于研究由蛋白质/RNA错误折叠或聚集引起的疾病。蛋白质组失衡与老化有关,并且经常涉及超过溶解度并倾向于形成细胞内和细胞外聚集体的丰富蛋白质。老化是几种蛋白质错误折叠病症(PMD),特别是进行性神经变性发作的危险因素。蛋白质聚集是神经变性的主要标志,包括阿尔茨海默病(AD)中的淀粉样蛋白β(Ab)和tau聚集、帕金森病(PD)和多系统萎缩中的细胞内α-突触核蛋白聚集、亨廷顿病(HD)中的多Q-驱动蛋白聚集、朊病毒病中的PrPSc以及肌萎缩性侧索硬化(ALS)和额颞痴呆(FTD)中的TDP-43和FET蛋白聚集,只是列出了几个实例。尽管参与斑块形成的蛋白质的化学性质和(病理)生理拓扑结构不同,但控制它们聚集的原理令人惊奇地相似,并且所提供的方法和系统可用于将靶多核苷酸定位在这些斑块或聚集体上。
本文公开的系统和方法可用于通过将关键驱动基因重新定位到不同的核隔室中来控制细胞分化。所述系统和方法可用于通过控制内VD(J)基因座的重组速率来增强抗体产生。所述系统和方法可用于通过消除错误折叠蛋白体的形成来减轻阿尔茨海默病。
本文公开的系统和方法可广泛地应用于所有的生命领域,包括植物、细菌、古细菌、酵母、鱼类、昆虫、鸟类、哺乳动物、小鼠、猪和人。所述系统和方法可用于活的整个生物体或组织或细胞中。
IV.实施例
给出以下实施例用于说明本公开的各实施方案的目的,并且不意味着以任何方式限制本公开。本发明的实施例以及本文所述的方法是优选实施方案的代表,是示例性的,并不旨在限制本公开的范围。
为了实现诱导型CRISPR介导的染色质重新定位系统,测试了两种化学诱导型异二聚化系统。第一种是脱落酸(ABA)诱导的ABI/PYL1系统,第二种是TMP-Htag(甲氧苄氨嘧啶-卤配体)诱导的DHFR/HaloTag系统。对于这两种系统,将化脓链球菌dCas9(D10A&H840A)蛋白与一种异二聚体融合,并将内核包膜(NE)蛋白Emerin与同源异二聚体融合(图2-4)。由EMD基因编码的Emerin是在核内膜介导染色质组织的一组LEM(LAP2、Emerin、MAN1)结构域蛋白中的。Emerin在细胞质中合成,插入内质网(ER)中,然后通过在连续的ER/NE膜内扩散而易位至NE(Berk等人,2013)。使用稳定表达每种二聚化系统的慢病毒转导产生U2OS人骨骨肉瘤上皮细胞系。在这些细胞系中,由于其与PYL1-GFP-Emerin的二聚化,ABA的加入引起ABI-BFP-dCas9蛋白从核内部到NE和ER的空间重新定位(图5和6)。相比之下,TMP-Htag诱导的系统对dCas9-EGFP-HaloTag和DHFR-mCherry-Emerin与配体的共定位没有明显影响。因此,将ABA诱导的ABI/PYL1异源二聚化系统用于以后的实验。
为了测试ABA诱导的CRISPR-GO系统是否能够改变染色体的位置,靶向染色体3(Chr3)上的内源性基因座。将靶向染色体3(3q29)内高度重复(~500x)区的sgRNA经慢病毒转导到稳定表达ABI-BFP-dCas9和PYL1-GFP-Emerin的U2OS细胞系中(图2和7)。考虑到AB1-BFP-dCas9在ABA处理后主要被募集到PYL-GFP-Emerin定位(NE和ER),加入另一种独立的CRISPR-Cas9成像组分,dCas9-HaloTag融合蛋白以可视化靶向的Chr3基因组基因座的位置(图5和6)。在sgChr3的存在下,将JF549-HaloTag染料加入到培养基中以结合dCas9-HaloTag,并使得能够可视化活细胞中的靶向的Chr3:q29基因座。sgChr3通过靶向同一Chr3:q29基因组区域内的多个重复序列而介导CRISPR-Cas9成像(通过dCas9-HaloTag)和CRISPR-GO基因组重新定位(通过AB1-dCas9)。还证实,在不存在sgRNA的情况下,dCas9-HaloTag定位不受AB1-BFP-dCas9和PYL-GFP-Emerin之间的ABA介导的异二聚化的影响(图6)。
在ABA处理2天后,与没有ABA处理的细胞相比,观察到束缚到核周边的靶向的Chr3基因座显著增加(图8和10-12)。在没有ABA处理的情况下,19%的dCas9-HaloTag标记的Chr3基因座位于由PYL1-GFP-Emerin标记的核周边,而大部分标记的基因座保留在核内部(分析的142个基因座,图8)。相比之下,87%的标记的Chr3基因座在ABA处理的细胞中重新定位到核周边(163个基因座,图8)。ABA处理还将显示至少一个位于核膜的Chr3基因座的细胞的百分比从27%(77个细胞)增加到95%(76个细胞,图8)。用化学处理使重新定位的基因组基因座(p<0.0001)和细胞(p<0.0001)都显著增加表明本文公开的系统在细胞中重新定位高度重复的多核苷酸如内源性基因组基因座是有效的。
除了Chr3:q29基因座外,还测试了将其它高度重复的内源性基因组基因座,包括Chr13基因座和端粒重新定位到核周边。使用靶向染色体13q34(Chr13)上的重复区域(~350x重复序列)的sgRNA(图7),束缚的Chr13基因座的百分比从13%(n=103)增加到69%(n=157,p<0.0001),并且含有至少一个周边定位的基因座的细胞的百分比从34%(n=30)增加到94%(n=53,图8,p<0.0001)。类似地,转导具有端粒靶向sgRNA的含有CRISPR-GO的细胞以测试端粒是否也可用我们的系统重新定位。TRF1-mCherry(一种端粒标志物)也被共表达以可视化端粒。在这种情况下,周边定位的端粒基因座的百分比从26%(n=1255)增加到65%(n=491,图8,p<0.0001)。
位于染色体1p36处的合成整合的LacO阵列也靶向于先前用于研究染色体重新定位的U2OS 2-6-3报道细胞系中(图7)。使用靶向LacO序列的sgRNA,活细胞中的CRISPR-Cas9成像揭示了核周边束缚的靶向基因组基因座的百分比从4%(n=73)增加到60%(n=161,p<0.0001),并且含有至少一个周边定位的基因座的细胞的百分比从4.5%(n=66)增加到65%(n=133)(图8,1G,p<0.0001)。用DAPI在固定的细胞中进行荧光原位杂交(FISH)染色进一步证实在ABA处理后大部分LacO基因座位于核周边(图13)。
接下来测试CRISPR-GO系统在重新定位较少重复(<100个重复序列)的序列中的效率。选择含有~71个sgRNA-可靶向重复序列的Chr7 q36.3上的基因组区域和含有~15个sgRNA-可靶向重复序列的ChrX p21.2上的基因组区域作为靶标(图14)。使用3D-FISH可视化靶向的基因组基因座的位置,并通过DAPI对细胞核进行染色(图15)。在ABA处理3天后,对于Chr7,周边定位基因座的百分比从28%(n=97)增加到68%(n=142,p<0.0001),并且对于ChrX,从33%(n=230)增加到62%(n=123,p<0.0001)。对于Chr7,含有至少一个周边相邻基因座的细胞的百分比从32%(n=68)增加到79%(n=76,p<0.0001),并且对于ChrX,从41%(n=136)增加到76%(n=74,p<0.0001)(图9)。当使用非靶向sgRNA作为对照时,在核周边的Chr7和ChrX的百分比与用非ABA处理的样品观察到的那些相似,并且在ABA处理后保持不变(图16)。总之,这些结果提示本文提供的化学诱导系统在将高度重复和较少重复的序列重新定位到细胞隔室如核周边方面是有效的。
尽管重复序列大量存在于人类基因组中,但如果CRISPR-GO系统能够重新定位非重复的基因组基因座,则其是进一步关注的。位于ChrX q13.2的非重复基因XIST首先被靶向,并且设计了展开XIST基因组区域的13种sgRNA(图17)。将所有构建体经慢病毒转导到稳定表达CRISPR-GO系统的U2OS细胞中。使用ABA处理,周边定位的XIST基因座的百分比从39%(n=83)增加到79%(n=71,p<0.0001),并且含有周边定位的基因座的细胞的百分比从59%(n=39)增加到90%(n=33)(图18,1H,p=0.0028)。使用靶向Chr10的基因PTEN附近和内部区域的9种sgRNA的库(图17),CRISPR-GO系统将周边定位的PTEN基因座的百分比从39%(n=128)增加到61%(n=308,p<0.0001),并且含有至少一个周边相邻基因座的细胞的百分比从62%(n=60)增加到88%(n=106,p=0.0002)(图15和18)。
还测试了靶向非重复区域的单一sgRNA是否足以重新定位基因组基因座。使用靶向Chr2上CXCR4基因座的单一sgRNA(sgCXCR4-1),周边定位的CXCR4基因座的百分比从20%(n=241)增加到50%(n=425,p<0.0001),并且含有周边定位的基因座的细胞的百分比从52%(n=69)增加到85%(n=131,p<0.0001)(图19)。类似地,另一单一sgRNA(sgCXCR4-2)将定位的CXCR4基因座从25%(n=202)增加到47%(n=284),并且将细胞从49%(n=74)增加到82%(n=84,p<0.0001)(图19)。比较了使用单一sgRNA和6中sgRNA的库靶向CXCR4基因座的效率。当使用多种sgRNA时,CRISPR-GO系统分别将定位的CXCR4基因座的百分比从32%(n=270)增加到62%(n=402,p<0.0001)和细胞的百分比从46%(n=170)增加到90%(n=168,p<0.0001)(图15和19)。当使用非靶向sgRNA作为对照时,在核周边的CXCR4基因座的百分比与用非ABA处理的样品所观察到的相似,并且在ABA处理后保持不变(图16)。这些结果一起证实了本文公开的系统可以介导非重复基因座到细胞隔室如核周边的有效重新定位。
所提供的系统和方法的一个优点是能够通过向培养基中加入或去除化学诱导物而容易地打开或关闭多核苷酸重新定位。进行化学诱导和去除实验以研究ABA诱导的CRISPR-GO系统的动态和可逆性(图20)。为了研究化学诱导,用ABA处理含有靶向Chr3基因座的CRISPR-GO系统的U2OS细胞,并在不同的时间点检查。对于化学逆转,首先用ABA处理含有靶向Chr3基因座的CRISPR-GO系统的U2OS细胞2天,然后切换到不含ABA的培养基。ABA诱导的基因组重新定位发生相对较快,因为ABA加入16h内,束缚到核膜的Chr3基因座的百分比由19%(n=142)增加到75%(n=93,p<0.0001),并且在ABA加入72h后达到91%(n=160)。ABA去除后,束缚到核膜的Chr3基因座的百分比在24小时后从82%(n=163)降低到45%(n=84,p<0.0001),并且在48小时和72小时后分别进一步降低到27%(n=146)和26%(n=159)。ABA去除48小时和72小时后,在核周边的Chr3的百分比与同时成像的没有ABA处理的对照样品(25%,n=106,p=0.77)没有区别,这表明ABA去除在48小时内完全逆转了由CRISPR-GO系统介导的基因组重新定位效应(图20)。这些结果表明,通过本文公开的系统和方法介导的核重新定位可以容易地通过例如加入或去除化学诱导物如ABA而打开和关闭。
CRISPR-GO系统用于靶向内源性Chr3基因座。通过血清饥饿和羟基脲(HU)处理使含有Chr3靶向的sgRNA的CRISPR-GO细胞同步并停滞在S期,然后用ABA处理用于化学诱导(图21)。有趣的是,在ABA处理24小时后,在HU处理的S期停滞细胞中,核周边束缚的Chr3基因座的百分比从17%(n=175)增加到33%(n=177,p=0008),这显著低于非同步细胞中的百分比(75%,n=251,p=0.0001)。在ABA处理48小时后,HU处理的S期停滞细胞中的核周边束缚的Chr3基因座的百分比增加到54%(n=177,p<0.0001),这也低于非同步细胞中的百分比(83%,n=178,p<0.0001)。因此,在HU处理的S期停滞细胞中,仍观察到核周边重新定位,但与经历有丝分裂的细胞相比,其程度较低。这些结果表明,使用本文公开的系统和方法将内源性基因座重新定位到细胞隔室如核周边可通过依赖有丝分裂和不依赖有丝分裂的机制发生。
不依赖有丝分裂的周边重新定位据我们所知尚没有被报道。为了探查在间期期间基因组基因座是否可以被束缚到核周边,使用活细胞CRISPR-Cas9成像来追踪核周边束缚的动态。检测了内源性Chr3基因座在间期期间束缚到核周边的动态过程。在图22和23所示的代表性示例中,Chr3:q29基因座(箭头)在记录的最初4小时期间开始与核周边(GFP-Emerin)分离,在4.5小时时被束缚到核周边,然后在记录的剩余8小时内保持束缚,甚至在核经历10小时至12小时的旋转时也是如此。我们随时间对该Chr3基因座与最近的核周边之间的距离进行定量,发现在4.5小时发生束缚后,该距离保持为0(图24)。这些结果表明,尽管在间期期间染色质结构相对稳定地组织,但使用本文公开的系统和方法,位于核周边附近的基因组基因座能够以不依赖有丝分裂的方式合成地束缚到核周边。
将CRISPR-GO系统与活细胞内CRISPR-Cas9成像技术相结合,研究了基因组束缚后基因组位点的短时运动动力学。与未束缚的基因座相比,检查被束缚在核周边的Chr3基因座的短期动态(图25)。在共聚焦显微镜下每4-6s拍摄图像。我们观察到未束缚的Chr3基因座比束缚的Chr3基因座移动更多(图25)。我们在二维空间(dx
接下来测试CRISPR-GO系统是否可介导染色质基因座与无膜核小体的共定位。选择基因组基因座以募集至Cajal小体(CB)。为此,通过将PYL1与Cajal小体的标志物Coilin融合来设计Cajal小体靶向的CRISPR-GO系统。通过慢病毒转导将PYL1-GFP-Coilin和ABI-dCas9引入U2OS细胞(图28)。我们测试了在含有插入Chr1:p36的LacO重复序列阵列的U2OS2-6-3细胞中的募集效率。
使用靶向LacO序列的sgRNA,我们在ABA处理2天后使用3D-FISH可视化LacO阵列的空间定位和GFP-Coilin可视化CB的位置(图29)。与GFP-Coilin-标记的CB共定位的LacO基因座的百分比从没有ABA处理的9%(n=78)增加到ABA处理后的64%(n=84),并且含有与LacO基因座共定位的至少一个CB的细胞的百分比从10%(n=68)增加到65%(n=77)(图30,p<0.0001)。联合用FISH的免疫染色显示,在ABA处理20小时后,其它CB组分(SMN,原纤维蛋白和Gemin2)也与LacO基因座共定位(图31),这证实了CRISPR-GO介导的靶向基因组基因座与CB的共定位。
为了使内源性基因组基因座靶向CB,将Chr3:q29靶向的sgRNA引入表达Cajal小体靶向的CRISPR-GO系统的U2OS细胞中。ABA处理24小时后,观察到Chr3基因座(用CRISPR-Cas9成像可视化)和CB(用GFP-Coilin可视化)之间显著的共定位(图32)。与CB共定位的Chr3基因座的百分比从ABA处理前的2%(n=149)增加到ABA处理后的94%(n=229,p<0.0001),并且含有与Chr3基因座共定位的至少一个CB的细胞的百分比从6%(n=50)增加到96%(n=101,p<0.0001)(图33)。
还测试了CRISPR-GO是否可介导染色质基因座与PML核小体的共定位。为此,通过将PYL1与PML小体的支架蛋白PML基因融合来设计靶向PML小体的CRISPR-GO系统。为了使内源性基因组基因座靶向PML小体,将Chr3:q29靶向的sgRNA引入表达PYL1-GFP-PML和ABI-dCas9的细胞中,通过CRISPR-Cas9成像可视化Chr3基因座的定位,并通过GFP-PML可视化PML小体的位置(图34和35)。有趣的是,在没有ABA处理的情况下,高百分比(52.6%,n=300)的Chr3:q29基因座与PML小体共定位,这可能提示天然的Chr3:q29-PML小体共定位(图35和36)。在ABA处理后,与PML小体共定位的靶Chr3基因座的百分比增加到94%(n=196,p<0.0001),并且含有与Chr3基因座共定位的至少一个PML小体的细胞的百分比从75%(n=100)增加到96%(n=69,p=0.0003)(图35和36)。免疫染色也证实Chr3基因座与SP100(另一种PML小体标志物)共定位(图37)。
进行化学诱导和去除实验以研究CRISPR-GO介导的染色质与CB共定位的动态和可逆性。以U2OS 2-6-3细胞中Chr1:p36所插入的LacO基因座为例,我们观察到LacO基因座与GFP-Coilin标记的CB之间的关联迅速发生:在ABA加入后30分钟内,与CB共定位的LacO基因座的百分比从2.6%(n=78)增加到89%(n=85,p<0.0001)(图38)。
在用ABA预处理1天的细胞中,观察到ABA去除导致CB从LacO基因座解离。ABA去除后,与CB共定位的靶向LacO基因座的百分比在6小时后从89%(n=85)降低到22%(n=60,p<0.0001),在24小时后进一步降低到4.6%(n=45,p<0.0001)(图39)。在ABA去除后6小时,在仍然具有LacO-共定位GFP-Coilin的细胞群(22%)中,剩余的GFP-Coilin强度比进行持续ABA处理的细胞(图40)更暗,这可能表明ABA去除后CB的逐步分解过程。
为了进一步表征CRISPR-GO介导的Cajal小体与靶向基因组基因座的关联的动态,在ABA处理之前和之后进行个体细胞的时间推移显微成像。理论上,基因组基因座和核小体之间的共定位可以通过在基因组基因座处核小体的从头形成,或者通过将基因组基因座重新定位到现有的核小体来发生。先前使用LacO-LacI束缚系统的报道表明Cajal小体在靶向DNA位点处从头形成。
使用CRISPR-GO系统使LacO基因座靶向Cajal小体,在加入ABA后在大多数分析的细胞中在LacO基因座处观察到快速(在几分钟内)从头CB形成(图41)。例如,在没有ABA的LacO基因座处没有初始GFP-Coilin积累的细胞中(图41,-150s),ABA处理(在-150s至0s加入,图42)迅速将GFP-Coilin募集到LacO基因座(图41,150s),导致Cajal小体的从头形成。在加入ABA后10分钟内,LacO基因座处的GFP-Coilin荧光强度接近饱和(图44)。
有趣的是,如果靶向染色质基因座与现有CB最初在空间上彼此接近,则观察到两者的动态重新定位。例如,在其中现有CB与没有ABA处理(图45,-200s)的LacO基因座相邻的细胞中,ABA处理(在-200s至0s加入,图45)导致现有的CB和LacO基因座的快速共定位,这表明本文公开的系统也可以介导多核苷酸(例如基因组基因座)和细胞隔室(例如现有的核小体)之间的直接关联,这是一种以前尚未报道的现象。
先前的研究提供了关于基因组重新定位到核周边对基因表达的影响的不同证据。一些研究表明,使用LacI-Emerin或LacI-Lap2β将LacO重复序列束缚到核周边引起相邻基因的抑制。其它研究显示,在U2OS 2-6-3细胞中使用LacI-核纤层蛋白B1融合蛋白将LacO阵列重新定位到核周边,但是在相邻基因表达中没有观察到明显的变化。CRISPR-GO提供了另一种研究这一问题的方法,因为它更容易测试将不同染色体基因座募集到核周边的效果。
检测CRISPR-GO介导的LacO阵列到核周边的重新定位是否可以影响基因表达。U2OS 2-6-3细胞中的LacO基因座位于驱动CFP报告基因表达的强力霉素(Dox)诱导的TRE(四环素应答元件)-CMV启动子的上游(图46)。通过流式细胞术测量ABA处理和未处理细胞中相邻CFP报道基因的表达,观察到与未处理细胞相比,ABA处理细胞显示出一致降低的报告基因表达(降低59%,图46和47)。该基因抑制作用类似于使用LacI-Emerin融合蛋白将LacO基因座重新定位到核周边。作为证实基因抑制是靶标特异性的对照,我们还测试了非靶向sgRNA,并且观察到报告基因表达没有降低(图47)。
接下来测试将内源性基因组基因座重新定位到核周边是否可以改变基因表达。将Chr3的XIST和CXCR4基因座分别重新定位于核周边,并进行RT-qPCR检测相邻基因表达的变化(Chr3:ACAP2&PPP1R2;CXCR4;XIST)。令人惊讶的是,这些基因(例如,图48中的ACAP2和PPP1R2)没有观察到基因表达变化的证据。因此,它提出了这样的问题:基因重新定位至核周边附近是否足以引起内源性基因表达变化,并且重新定位诱导的基因表达变化仍然可能是基因座依赖性的。
接下来测试在U2OS 2-6-3细胞系中使用CRISPR-GO系统将LacO基因座共定位到CB是否足以影响相邻基因的表达(图49)。用ABA处理细胞2天,用Dox诱导1天,并通过流式细胞术测量CFP表达。与未处理的细胞相比,在ABA处理的细胞中观察到报告基因表达持续降低(平均降低45%,图49和50,p<0.0001)。为了证实该基因抑制作用是靶标特异性的,测试了非靶向sgRNA,并且观察到报告基因表达的轻微但不显著的降低(p>0.05)(图50)。
接下来测试将内源性基因组基因座共定位于CB是否可以改变相邻基因的表达。CRISPR-GO系统用于诱导Chr3:q29与CB的共定位(图32和33),然后进行RT-qPCR以检测ABA处理4天后相邻基因表达,(Chr3:ACAP2&PPP1R2)的变化。令人惊奇的是,与未处理的细胞相比,观察到两个相邻基因的显著抑制。位于Chr3上CB-靶向基因座上游约35kb的ACAP2在ABA处理后表现出3.3倍的抑制(p<0.0001,图42),而位于Chr3上CB-靶向基因座下游约36kb的PPP1R2表现出7.7倍的抑制(p<0.0001,图42)。证实没有sgRNA或没有PYL-GFP-Coillin组分的细胞在ACAP2和PPP1R2基因表达上没有变化(图43)。总之,这些结果显示,给定多核苷酸(例如基因组基因座)与细胞隔室(例如CB)的靶向共定位能够抑制相邻多核苷酸的表达。例如,使用本文公开的系统和方法的基因表达扰动介导的长距离功效与CRISPRi或CRISPRa相反,CRISPRi或CRISPRa仅引起距dCas9结合位点相对短距离的基因表达的扰动。所提供的方法和系统能够介导长距离多核苷酸表达扰动的观察可以提供有用的新方法,例如基因调节。
CRISPR-GO系统用于研究端粒重组至核隔室如何影响细胞表型。在所有测试的基因组基因座中,端粒的动态是研究的最佳的,并且显示出与细胞周期的某些阶段的核周边和CB相关。考虑到端粒对基因组完整性的重要作用,它们与核隔室的相互作用可能具有功能意义。例如,在细胞周期期间,当核膜在有丝分裂后的细胞中重新组装时,端粒被动态地束缚到核包膜,然后在G1期期间被重新定位到核的内部,在此它们保持细胞周期的其余部分。端粒束缚至核包膜和与其解束缚的循环对于染色质组织和细胞周期/活力可能是重要的。
为了测试这一点,使用CRISPR-GO系统在细胞周期期间破坏端粒解束缚过程并在间期期间保留端粒至核隔室(图51)。使用通过测量细胞的代谢活性来定量细胞增殖的Alamar蓝细胞活力测定,当与未处理的细胞相比时,发现通过CRISPR-GO维持端粒在核周边在ABA处理6天后导致细胞活力的显著降低(图52,平均72%的降低,p<0.0001)。在ABA处理3天后,细胞周期分析表明,将端粒束缚到核周边增加了G0/G1期的细胞百分比,并降低了S期和G2/M期的细胞百分比,可能表明G0/G1期停滞(图53)。图63显示了在将端粒重新定位到核周边之后通过RNA测序比较基因表达变化的图,并显示将端粒重新定位到核周边引起基因表达的许多变化,这降低了细胞活力。还检测了端粒与CB共定位的效果,证实了CRISPR-GO系统能够诱导端粒和CB的共定位,并且发现当将用ABA处理2天的细胞与未处理的细胞进行比较时,共定位增加了细胞活力(平均50%增加,图54-56)。图64显示了在将端粒与Cajal小体共定位后通过RNA测序比较基因表达变化的图,并显示将端粒与Cajal小体共定位引起基因表达的许多变化,这改变了细胞活力。作为对照,单独的ABA处理对U2OS细胞中的细胞活力没有影响(图57)。总之,这些观察结果表明多核苷酸(例如端粒)相对于各种细胞隔室(例如核隔室)的空间组织在细胞功能中起重要作用。
CRISPR-GO系统可用于使mRNA沿着细胞骨架与马达蛋白如驱动蛋白,动力蛋白和肌球蛋白重新定位。为了将mRNA重新定位到微管的正端(MT+),可以构建表达PYL1-EGFP标记的驱动蛋白-1重链(KIF5B)而没有货物结合尾结构域的质粒(Kapitein等人,2010)。为了将mRNA重新定位到微管的负端(MT-),可以构建表达PYL1-EGFP标记的Bicaudal D2 N-末端部分(BICDN)的质粒,其诱导动力蛋白-介导的货物运输(Hoogenraad等人,2003)。为了沿着肌动蛋白丝(AF)重新定位mRNA,可以构建表达PYL1-EGFP标记的肌球蛋白5a(MYO5A)的质粒。MYO5A是三种V类肌球蛋白中表征最佳的,并且在mRNA沿着肌动蛋白丝向钩端(barbedend)转运中起作用(Gross等人,2007;McCaffrey和Lindsay,2012)。可以将表达ABI-BFP-dCas13的质粒和表达PYL1-EGFP-KIF5/BICDN/MYO5A的质粒转导到MS2-MCP(MS2-结合蛋白)细胞中。随后对细胞分选BFP和EGFP阳性细胞以产生MS2-MCP-CRISPR-GO-MT+/MT-/AF稳定细胞系。稳定的细胞可以用表达靶向MS2-标记的RNA的gRNA的慢病毒转导,并且用嘌呤霉素选择gRNA-阳性细胞。所选择的细胞可以用ABA处理并进行活细胞荧光成像以追踪mCherry的定位,其表示靶向RNA的位置。
CRISPR-GO系统可用于形成促进DNA修复并导致改进的基因编辑结果的核小体。图65A-65C显示CRISPR介导的基因编辑后53BP1病灶的形成。这些数据表明,CRISPR基因编辑募集DNA修复蛋白形成核小体,以促进CRISPR介导的基因编辑后的双链断裂(DSB)分辨和DNA修复。
通过用PYL1替换pHR-SFFV-scFv-sfGFP质粒(Tanenbaum等人,2014)中的scFv序列并在sfGFP后插入Emerin,克隆了pHR-SFFV-PYL1-sfGFP-Emerin。Emerin(由EMD基因编码)克隆自Emerin pEGFP-C1(637),其为来自Eric Schirmer的礼物(Zuleger等人,2011)(Addgene质粒61993)。通过将pHR-SFFV-PYL1-sfGFP-Emerin质粒中的Emerin替换为Coilin,克隆了pHR-SFFV-PYL1-sfGFP-Coilin。Coilin克隆自pEGFP-Coilin(Addgene质粒36906),其为来自Greg Matera博士的礼物。通过用PGK启动子替换pHR-SFFV-PYL1-sfGFP-Coilin质粒中的SFFV启动子,克隆了pHR-PGK-PYL1-sfGFP-Coilin。通过用TRE3G启动子替换PGK启动子,并用PML或HP1a替换pHR-PGK-PYL1-sfGFP-Coilin质粒中Coilin,克隆了pHR-TRE3G-PYL1-sfGFP-PML或pHR-TRE3G-PYL1-sfGFP-HP1a。PML克隆自pLPC-Flag-PML-IV(addgene质粒62804),其为来自Gerardo Ferbeyre的礼物(Vernier等人,2011)。HP1a克隆自GFP-HP1a(Addgene质粒17652),其为来自Tom Misteli的礼物(Cheutin等人,2003)。
先前描述了pHR-SFFV-ABI-tagBFP-dCas9(Gao等人,2016)。通过用PGK启动子pHR-SFFV-ABI-tagBFP-dCas9替换SFFV启动子克隆了pHR-SFFV-ABI-tagBFP-dCas9。通过用PGK启动子替换SFFV,删除tagBFP,并在dCas9 pHR-SFFV-ABI-tagBFP-dCas9中加入P2A-mCherry或P2A-Puro,克隆了pHR-PGK-ABI-dCas9-P2A-Cherry或pHR-PGK-ABI-dCas9-P2A-Puro。ABI和PYL1克隆自Addgene质粒38247(Liang等人,2011),其为来自J.Crabtree,Stanford博士的礼物。
通过用HaloTag替换质粒pHR-TRE3G-dCas9-HA-SunTag10-P2A-mCherry(Tanenbaum等人,2014)中的SunTag10-P2A-mCherry,克隆了pHR-TRE3G-dCas9-HaloTag。通过将HaloTag插入pHR-TRE3G-dCas9-EGFP的EGFP之后,克隆了pHR-TRE3G-dCas9-EGFP-HaloTag(Chen等人,2013)。通过用mCherry-DHFR替换pHR-SFFV-PYL1-sfGFP-Emerin中的PYL1-sfGFP序列,克隆了pHR-SFFV-DHFR-mCherry-Emerin。HaloTag和mCherry-DHFR克隆自pERB221,其为来自David Chenoweth&Michael Lampson的礼物(Ballister等人,2014)(Addgene质粒61502)。
将所有sgRNA克隆到去除P2A-mCherry后的pHR-U6-sgTel-CMV-puro-P2A-mCherry载体中(Chen等人,2013)。将TRF1-mCherry克隆到pHR-U6-sgTel-CMV-puro-P2A-mCherry载体中代替mCherry。TRF1克隆自pLPC-NFLAG TRF1,其为来自Titia de Lange博士的礼物(Smogorzewska和de Lange,2002)(Addgene质粒#16058)。
U2OS(人骨肉瘤上皮,雌性)细胞和Hela细胞(雌性)在含有GlutaMAX的DMEM(LifeTechnologies)中在10%Tet系统批准的FBS(Life Technologies)中培养。U2OS 2-6-3细胞系是来自冷泉港实验室的David L.Spector博士的礼物,并且在相同条件下培养(Kumaran和Spector,2008)。将所有细胞在37℃和5%CO2下在加湿培养箱中培养。
为了产生将内源性基因座靶向核隔室的稳定的CRISPR-GO细胞系,提前1天将U2OS细胞铺板到24孔板中,达到50%汇合,然后用慢病毒混合物转导。通过表达PYL1-sfGFP-Emerin,PYL1-sfGFP-Coilin,PYL1-sfGFP-PML,或PYL1-sfGFP-HP1a和ABI-tagBFP-dCas9的慢病毒转导的细胞通过荧光激活细胞分选(FACS)在Stanford共享FACS设备上对BFP和GFP阳性细胞进行分选以产生稳定的细胞系。对于核周边束缚,选择高BFP和GFP表达水平的细胞。对于其它核隔室束缚,选择高BFP和GFP表达水平的细胞。在用表达靶向sgRNA的慢病毒转导CRISPR-GO细胞系后,用2μg/ml的嘌呤霉素选择sgRNA阳性细胞。
为了靶向U2OS 2-6-3细胞系中的LacO基因座(Kumaran和Spector,2008),通过含有PYL1-sfGFP-Emerin或PYL1-sfGFP-Coilin和ABI-dCas9-P2A-mCherry的慢病毒混合物转导细胞。对含有PYL1-sfGFP-Coilin和ABI-dCas9-P2A-mCherry的细胞分选GFP和mCherry阳性细胞以产生稳定的细胞系。用2μg/ml的嘌呤霉素选择sgRNA阳性细胞。
为了通过CRISPR成像定量LacO核周边重新定位的功效,用编码ABI-dCas9-P2A-Puro而不是ABI-dCas9-P2A-mCherry的慢病毒转导U2OS 2-6-3细胞,并且用2μg/ml的嘌呤霉素选择。
测试靶向U2OS细胞中不同染色体区域的CRISPR-GO系统的功效。对重复区域和非重复基因进行测试(图7、14和17)。内源性重复区域包括Chr3.q29:195478324-195506987;CH13.q34:112,277485-112,319169;Ch7:q36.3:158,122,661-158,135,328;ChX p21.2:30,806,671-30,824,818和端粒。插入U2OS 2-6-3细胞的Chr1.p36区域的合成LacO重复序列也用于靶向。对于重复区域,使用单一sgRNA设计来靶向靶向区域内的多个重复序列(表1)。非重复基因包括位于Chr2.q22.1的CXCR4,位于ChrX.q13.2的XIST和位于Chr10.q23.31的PTEN。为了靶向每个非重复基因,设计了靶向其基因主体和上游区域的多种sgRNA(表2)。
表1.靶向重复区域的sgRNA
表2靶向非重复区域的sgRNA
为了产生慢病毒,用目的pHR构建体和包装质粒pCMV-dR8.91和pMD2.G瞬时转染HEK293T细胞。转染后72小时通过用0.45μm滤膜过滤上清液收集慢病毒。必要时,可以使用Lenti-X浓缩器在4℃下将病毒上清液浓缩过夜,并且在4℃下在1500g下离心30分钟以收集病毒沉淀。将沉淀悬浮在冷培养基中,直接加入细胞中或在-80℃冷冻。
进行CRISPR成像以可视化Chr3,Chr13和LacO基因座在活细胞中的定位(图5)。对于活细胞CRISPR成像,用编码dCas9-HaloTag和靶向sgRNA的慢病毒在ibidi 24孔微孔板(Ibidi.inc)中转导表达CRISPR-GO组分的稳定细胞系。用dCas9-HaloTag标记靶向的基因组基因座,并用JF549-HaloTag配体以0.1-0.5μM在37℃下在培养基中染色15min。染色后,用培养基洗涤细胞两次,然后在显微镜检查过程中在无酚红的培养基中孵育。JF549-HaloTag是在Janelia Research Campus的Luke D.Lavis博士的礼物(Grimm等人,2015)。端粒基因座通过表达端粒结合蛋白TRF1-mCherry在活细胞中被标记。
其它基因组基因座在固定细胞中被DNA FISH标记。使细胞在具有可移除的12孔硅酮室的ibidi室载玻片中生长,并用4%PFA固定20分钟。使用合成的荧光核苷酸探针(Integrated DNA Technologies,Redwood City,CA),根据所描述的FISH方案(Takei等人,2017)标记LacO、Chr7和ChrX基因座)。用Alexa Fluor 647标记的FISH探针5'-TTGTTATCCGCTCACAATTCCACATGTGGCCACAAA-3'以10nM浓度标记LacO基因座。用200nM的Cy3标记的FISH探针5’-Cy3-CCCACACTCTCACCATAAGAGC-3’标记Chr7基因座,并用200nM的5-Cy3-TTGCCTTGTGCCTTGCCTTGC-3’标记ChrX基因座。CXCR4 FISH探针购自Empire Genomics。PTEN和XIST FISH探针购自Cell Line Genetics。根据商人的方案进行FISH。
为了检测Cajal小体标志物和靶向LacO基因座之间的共定位,在第0天用编码PGK-ABI-dCas9-P2A-Puro和sgLacO的慢病毒转染表达低水平PYL1-sfGFP-Coilin的U2OS 2-6-3细胞,在第1天用嘌呤霉素和3mM ABA处理。在第2天在ABA处理20小时后固定。使用AlexaFluor 647标记的FISH探针在固定样品中进行FISH以检测LacO基因座,然后使用小鼠单克隆抗SMN,抗纤维蛋白和抗Gemin2抗体,以及驴抗小鼠Alex Fluor 594第二抗体进行免疫染色。
为了检测PML小体标志物和靶向Chr3基因座之间的共定位,在第0天用编码dCas9-HaloTag(用于CRISPR成像)和sgChr3的慢病毒转染表达PYL1-sfGFP-Coilin和PGK-ABI-dCas9的U2OS细胞,在第1天用嘌呤霉素和3mM ABA处理。通过JF549-HaloTag染色并在第3天固定在4%多聚甲醛(PFA)中。用兔多克隆抗SP100和驴抗兔Alex Fluor 647第二抗体在固定样品中进行免疫染色。
对于免疫染色,固定的样品在透化缓冲液(PBS,1%Triton-X100)中透化15分钟,在封闭缓冲液(PBS,0.3%Triton-X 100,5%驴正常血清)中封闭1小时,与在封闭缓冲液中稀释的第一抗体在4℃孵育过夜,在PBS中洗涤三次,然后与第二抗体在室温孵育1-2小时,在PBS中洗涤4次。
对于重新定位实验,在成像或固定之前,用3mM脱落酸(ABA,Sigma-Aldrich,A1049)处理含有化学诱导重新定位系统和sgRNA的U2OS细胞2天。
对于将Chr3靶向核周边的时程化学诱导实验,用或不用3mM ABA处理含有CRISPR-GO和CRISPR成像系统和靶向Chr3的sgRNA的U2OS细胞,用JF549-HaloTag染色,并在不同的时间点固定。对于时程逆转实验,Chr3靶向U2OS细胞用3mM ABA预处理2天,洗涤5次,并转换到无ABA的培养基中。细胞通过用于CRISPR成像的JF549-HaloTag配体染色,并在不同时间点在4%多聚甲醛中固定20分钟。
对于将LacO靶向Cajal小体的的时程化学诱导实验,在第0天用编码PGK-ABI-BFP-dCas9和sgLacO的慢病毒转染表达低水平PYL1-sfGFP-Coilin的U2OS 2-6-3细胞,在第1天用嘌呤霉素处理,在第2天用或不用3mM ABA处理,并在ABA处理30分钟后固定。对于时程逆转实验,细胞用3mM ABA预处理2天,洗涤5次,并转换到无ABA的培养基中。在不同的时间点将细胞固定在4%多聚甲醛中20分钟。
为了分析基因组重新定位的有丝分裂依赖效应,将含有CRISPR-GO和CRISPR成像系统和靶向Chr3的sgRNA的U2OS细胞用于该实验。在第-3天,使细胞在培养基的0.5%FBS中饥饿2天。在第-1天,将细胞转换至具有10%FBS的正常生长培养基,并用2mM羟基脲(HU)处理用于G1/S期阻断1天。在第0天,在保持HU处理的同时,用或不用ABA处理细胞。对照细胞以相同方式处理,但不用HU。细胞通过用于CRISPR成像的JF549-HaloTag染色并在ABA处理后24小时或48小时固定在4%多聚甲醛中。
除了图22外,所有显微镜检查在Nikon TiE倒置共聚焦显微镜上进行,该显微镜配备有100×PLAN APO油物镜(NA=1.49),60×PLAN APO油物镜(NA=1.40)或60×PLAN APOIR水物镜(NA=1.27),Andor ixon Ultra-897 EM-CCD照相机和405nm、488nm、561nm和642nm激光器。使用NIS Elements 4.60版软件通过时间推移显微镜检查以0.2-μm或0.4-μm的步长使用Z堆叠拍摄图像。对于活细胞成像,将细胞在37℃和5%CO2下保持在加湿室中。
对于图22所示的长期活细胞成像,在配备有63×HC PLAN APO油物镜(NA=1.40),Leica DFC9000 CT照相机和Lumoncor SOLA SM II 405光源的Leica DMI8倒置显微镜中进行显微镜检查。使用GFP和TXR滤镜(filter cube),使用LAS X软件通过时间推移显微镜检查每30分钟拍摄图像,持续20小时。在成像期间,将细胞在37℃和5%CO2下保持在加湿室(Okolab Cage孵育系统)中。
为了使单个细胞中染色质-Cajal小体关联的动态可视化(图38-41、44和45),在第0天,用编码PGK-ABI-BFP-dCas9和sgLacO的慢病毒转染表达较低水平的PYL1-sfGFP-Coilin的U2OS 2-6-3细胞,在第1天用嘌呤霉素处理,并接种在ibidi 96孔u板中。将每个孔在共聚焦显微镜下成像以聚焦于所选细胞中的ABI-BFP-dCas9标记的LacO基因座。在ABA处理之前捕获图像以进行比较。在共聚焦显微镜下不移动样品的情况下,将10倍含ABA的培养基加入到成像孔中以达到1mM ABA的最终浓度,然后在加入ABA后立即对含有先前聚焦的LacO基因座的相同细胞成像。在ABA加入后拍摄的第一个图像被给出t=0,并且所有其他图像相应地通过捕获时间进行比对。
在Fiji(图像J)(Schindelin等人,2012)或MetaMorph(Molecular Devices,CA)中进行图像处理。在本文附图中显示了显示标记的基因组基因座的最大荧光的单个显微镜平面,或显示最大基因座荧光的两个/三个相邻Z平面的平均值。使用Fiji中的“平滑”函数处理一些图像以仅减少用于可视化的噪声。
使用Fiji中的“Analyze/Plot Profile”功能进行线扫描,在Excel中分析并在GraphPad Prism(7.00版,Mac OS,GraphPad Software,La Jolla California USA,www.graphpad.com)中作图。相对于沿着线的最大(=1)和最小(=0)荧光强度,将沿着线的每个点处的荧光强度归一化。
为了测定活U2OS细胞中的周边募集功效,通过CRISPR成像标记Chr3,Chr13和Chr1/LacO基因座,并通过TRF1-mCherry标记端粒,而通过PYL1-sfGFP-Emerin标记核膜。在扫描共聚焦平面的Z堆叠之后,在切片观察器(NIS元件观察器)中观察每个标记基因座的位置,以确定其在XY,XZ和YZ平面中的位置。在没有双重计数任何基因座的情况下,将基因座分为三类:与PYL1-GFP-Emerin在XY,YZ和YZ平面中共定位的直接位于核周边的基因座,不与PYL1-GFP-Emerin共定位的基因座,和不在核周边的与内部PYL1-GFP-Emerin共定位的基因座(在罕见的情况下)。记录每个单独细胞的每个类别中的基因座的数目。只有与PYL1-GFP-Emerin共定位于核包膜的第一类基因座被计数为核周边定位的基因座。对含有至少一个位于核周边的基因座的细胞进行定量。
为了测定固定的U2OS细胞(例如,Chr7、ChrX、PTEN、CXCR4、XIST)中的周边募集功效,用FISH标记靶向的基因组基因座,并用DAPI染色细胞核。在扫描共聚焦平面的Z堆叠之后,在3D空间中观察每个标记基因座的位置,以确定其在XY,XZ和YZ平面中的位置。在3D空间中位于细胞核(DAPI)边缘的基因组基因座被归类为周边定位的基因座。否则,它被认为是内部定位的轨迹。记录每个单独细胞的每个类别中的基因座的数目。还对含有至少一个位于核周边的基因座的细胞进行定量。
为了测定Cajal小体在固定的U2OS 2-6-3细胞中的共定位功效,用FISH标记靶向的LacO基因座,用Hoechst 33342染色细胞核,用PYL1-GFP-Coilin标记Cajal小体。在扫描共聚焦平面的Z叠堆之后,我们在3D空间中鉴定每个LacO基因座的位置。在没有双重计数的情况下,基因座分为两类:与PYL1-GFP-Coilin共定位的基因座,和不与PYL1-GFP-Coilin共定位的基因座。记录每个单独细胞的每个类别中的基因座的数目。还对含有至少一个Cajal小体共定位基因座的细胞进行定量。
为了定量CFP-SKL表达,用靶向lacO基因座的sgRNA或非靶向sgRNA转导含有ABI-dCas9-P2A-mCherry和PYL1-sfGFP-Emerin或PYL1-sfGFP-Coilin的U2OS 2-6-3细胞,用3mM的ABA处理2天,然后用50ng/ml的强力霉素诱导40小时(核周边细胞束缚)或24小时(Cajal小体束缚)。处理后,使用0.25%胰蛋白酶EDTA(Life Technologies)解离U2OS 2-6-3细胞,并使用405nm、488nm和561nm激光器在CytoFlex S(Beckman Coulter Life Sciences)上通过流式细胞术进行分析。对每个样品分析至少8,000个细胞。门控阳性dCas9(mCherry)和Emerin(GFP)表达的细胞。使用405nm激光器和450/45过滤器检测CFP-SKL荧光。为了定量相对荧光,将未处理的(没用Dox和ABA)细胞的平均总荧光设为0,而将强力霉素诱导的细胞(仅用Dox)的平均总荧光设为1。报道了在3个独立实验中的技术重复。
进行实时RT-PCR以确定基因组重组后邻近靶向Chr3基因座的PPP1R2和ACAP2基因的表达变化。对于每个样品,使用RNeasy Plus Mini试剂盒(Qiagen Cat 74134)分离总RNA,并根据制造商的方案使用iScript cDNA合成试剂盒(BioRad,Cat 1708890)合成cDNA。使用SYBR Green Master Mix(BioRad),利用PrimePCR测定进行定量PCR,并根据制造商的说明书在Biorad CFX384实时系统(C1000 Touch Thermal Cycler)上运行。Cq值用于定量基因表达。将PPP1R2和ACAP2基因的相对表达相对于GAPDH对照归一化。为了计算相对mRNA表达水平,通过将非ABA处理的样品中的平均值设定为1来归一化各处理的相对表达。报道了在3个实验中的重复。
使用Alamar蓝细胞活力试剂(ThermoFisher Scientific)进行细胞活力测定,其测量细胞的代谢活性。对于每种条件,将用和不用ABA处理的100μl细胞以相等的浓度(500-1000个细胞/孔)接种在相同的96孔板中。在检测时,将10μl的Alamar蓝试剂加入到每个孔中,并将板在37℃孵育1小时。然后,在Synergy H1微板读数器(Biotek Inc.)中使用540nm处的激发波长和585nm处的发射波长测量荧光强度。使用仅含有100μl培养基(含有和不含ABA)的孔的平均荧光强度作为空白。对于每个孔,通过从其原始荧光强度减去背景(空白孔的平均强度)来计算相对荧光强度。为了计算相对细胞活力,通过将非ABA处理的孔中的平均值设定为1来归一化每个孔中的相对荧光强度。报道了在3个实验中的重复。
为了定量端粒核周边束缚如何影响细胞周期进程,用编码sgTelomere和TRF1-mCherry的慢病毒混合物或编码非靶向sgRNA的慢病毒处理含有核周边束缚系统的U2OS细胞。在ABA处理2天后通过显微镜检查证实端粒束缚。在ABA处理3天后,对照和处理的细胞用0.25%胰蛋白酶EDTA解离,用Hoechst 33342以1:1000稀释液染色1小时,并通过流式细胞术在CytoFlex S(Beckman Coulter Life Sciences)上使用405nm激光器进行分析。对每个样品分析至少20,000个细胞。使用FlowJO进行细胞周期分析。
使用软件Tandem Repeats Finder(Benson,1999)鉴定来自人类基因组(hg38)的14个核苷酸或更长序列的所有串联重复序列。含有10个或更多个相同串联重复序列的区域被定义为“重复序列簇”。这些重复序列簇针对每条人类染色体。使用BEDTools套件计算重复序列簇和基因之间的距离。
使用TrackMate插件(Tineve等人,2017)在Fiji中进行基因组基因座追踪。为了追踪基因组基因座,将估计的Blob直径设定为0.5-1μm。将连接最大距离设定为2μm,将间隙闭合距离设定为3μm,并且将间隙闭合最大框架设定为2。测量每个基因座(x
为了比较步距,对19个内部定位的Chr3基因座的1696个步距和14个周边定位的Chr3基因座的1669个步距进行了分析。进行具有不等方差的双侧t检验。使用Excel中的直方图函数分析直方图,并在GraphPad Prism 7中作图。
为了定量重新定位的功效(图8、9、16、18-21、30、33、36、38和39),使用GraphPad中的费希尔精确检验计算p值,并且误差条显示根据Bernoulli分布计算的平均值的标准误差(SEM)。计数的基因座/细胞的数目列在每个图的底部。对于图26、42、43、46-51、53、56和57,p值使用Excel中的具有不等方差的双侧t检验来计算,并且误差条显示标准偏差。分析3个实验中的重复。对于图27,拟合γ分布使用R包fitdistrplus以最大似然拟合,并使用Kolmogorov-Smirnov检验(对于周边基因座p=0.06&对于内部基因座p=0.77)检验拟合优度。
V.参考文献
Ballister,E.R.,Aonbangkhen,C.,Mayo,A.M.,Lampson,M.A.,and Chenoweth,D.M.(2014).Localized light-induced protein dimerization in living cells usinga photocaged dimerizer.Nat Commun 5,5475.
Barrangou,R.,Fremaux,C.,Deveau,H.,Richards,M.,Boyaval,P.,Moineau,S.,Romero,D.A.,and Horvath,P.(2007).CRISPR provides acquired resistance againstviruses in prokaryotes.Science 315,1709-1712.
Benson,G.(1999).Tandem repeats finder:a program to analyze DNAsequences.Nucleic Acids Res 27,573-580.
Berk,J.M.,Tifft,K.E.,and Wilson,K.L.(2013).The nuclear envelope LEM-domain protein emerin.Nucleus 4,298-314.
Bernardi,R.,and Pandolfi,P.P.(2003).Role of PML and the PML-nuclearbody in the control of programmed cell death.Oncogene 22,9048-9057.
Bickmore,W.A.(2013).The spatial organization of the human genome.AnnuRev Genomics Hum Genet 14,67-84.
Bonev,B.,and Cavalli,G.(2016).Organization and function of the 3Dgenome.Nat Rev Genet 17,772.
Chen,B.,Gilbert,L.A.,Cimini,B.A.,Schnitzbauer,J.,Zhang,W.,Li,G.W.,Park,J.,Blackburn,E.H.,Weissman,J.S.,Qi,L.S.,et al.(2013).Dynamic imaging ofgenomic loci in living human cells by an optimized CRISPR/Cas system.Cell155,1479-1491.
Cheutin,T.,McNairn,A.J.,Jenuwein,T.,Gilbert,D.M.,Singh,P.B.,andMisteli,T.(2003).Maintenance of stable heterochromatin domains by dynamicHP1binding.Science 299,721-725.
Clowney,E.J.,LeGros,M.A.,Mosley,C.P.,Clowney,F.G.,Markenskoff-Papadimitriou,E.C.,Myllys,M.,Barnea,G.,Larabell,C.A.,and Lomvardas,S.(2012).Nuclear aggregation of olfactory receptor genes governs their monogenicexpression.Cell 151,724-737.
Cong,L.,Ran,F.A.,Cox,D.,Lin,S.,Barretto,R.,Habib,N.,Hsu,P.D.,Wu,X.,Jiang,W.,Marraffini,L.A.,et al.(2013).Multiplex genome engineering usingCRISPR/Cas systems.Science 339,819-823.
Crabbe,L.,Cesare,A.J.,Kasuboski,J.M.,Fitzpatrick,J.A.,and Karlseder,J.(2012).Human telomeres are tethered to the nuclear envelope duringpostmitotic nuclear assembly.Cell Rep 2,1521-1529.
Cristofari,G.,Adolf,E.,Reichenbach,P.,Sikora,K.,Terns,R.M.,Terns,M.P.,and Lingner,J.(2007).Human telomerase RNA accumulation in Cajal bodiesfacilitates telomerase recruitment to telomeres and telomere elongation.MolCell 27,882-889.
de Koning,A.P.,Gu,W.,Castoe,T.A.,Batzer,M.A.,and Pollock,D.D.(2011).Repetitive elements may comprise over two-thirds of the human genome.PLoSGenet 7,e1002384.
Dekker,J.,Marti-Renom,M.A.,and Mirny,L.A.(2013).Exploring the three-dimensional organization of genomes:interpreting chromatin interactiondata.Nat Rev Genet 14,390-403.
Dekker,J.,Rippe,K.,Dekker,M.,and Kleckner,N.(2002).Capturingchromosome conformation.Science 295,1306-1311.
Denker,A.,and de Laat,W.(2016).The second decade of 3C technologies:detailed insights into nuclear organization.Genes Dev 30,1357-1382.
Finlan,L.E.,Sproul,D.,Thomson,I.,Boyle,S.,Kerr,E.,Perry,P.,Ylstra,B.,Chubb,J.R.,and Bickmore,W.A.(2008).Recruitment to the nuclear periphery canalter expression of genes in human cells.PLoS Genet 4,e1000039.
Gall,J.G.(2000).Cajal bodies:the first 100 years.Annu Rev Cell DevBiol 16,273-300.
Gao,Y.,Xiong,X.,Wong,S.,Charles,E.J.,Lim,W.A.,and Qi,L.S.(2016).Complex transcriptional modulation with orthogonal and inducible dCas9regulators.NatMethods 13,1043-1049.
Gilbert,L.A.,Horlbeck,M.A.,Adamson,B.,Villalta,J.E.,Chen,Y.,Whitehead,E.H.,Guimaraes,C.,Panning,B.,Ploegh,H.L.,Bassik,M.C.,et al.(2014).Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation.Cell159,647-661.
Grimm,J.B.,English,B.P.,Chen,J.,Slaughter,J.P.,Zhang,Z.,Revyakin,A.,Patel,R.,Macklin,J.J.,Normanno,D.,Singer,R.H.,et al.(2015).A general methodto improve fluorophores for live-cell and single-molecule microscopy.NatMethods 12,244-250,243 p following 250.
Gross,S.P.,Vershinin,M.,and Shubeita,G.T.(2007).Cargo transport:twomotors are sometimes better than one.Curr Biol 17,R478-486.
Guan,J.,Liu,H.,Shi,X.,Feng,S.,and Huang,B.(2017).Tracking MultipleGenomic Elements Using Correlative CRISPR Imaging and Sequential DNAFISH.Biophys J 112,1077-1084.
Hilton,I.B.,D'Ippolito,A.M.,Vockley,C.M.,Thakore,P.I.,Crawford,G.E.,Reddy,T.E.,and Gersbach,C.A.(2015).Epigenome editing by a CRISPR-Cas9-basedacetyltransferase activates genes from promoters and enhancers.Nat Biotechnol33,510-517.
Hoogenraad,C.C.,Wulf,P.,Schiefermeier,N.,Stepanova,T.,Galjart,N.,Small,J.V.,Grosveld,F.,de Zeeuw,C.I.,and Akhmanova,A.(2003).Bicaudal Dinduces selective dynein-mediated microtubule minus end-directedtransport.The EMBO Journal22,6004-6015.
Jady,B.E.,Richard,P.,Bertrand,E.,and Kiss,T.(2006).Cell cycle-dependent recruitment of telomerase RNA and Cajal bodies to humantelomeres.Mol Biol Cell 17,944-954.
Jinek,M.,Chylinski,K.,Fonfara,I.,Hauer,M.,Doudna,J.A.,andCharpentier,E.(2012).A programmable dual-RNA-guided DNA endonuclease inadaptive bacterial immunity.Science 337,816-821.
Jinek,M.,East,A.,Cheng,A.,Lin,S.,Ma,E.,and Doudna,J.(2013).RNA-programmed genome editing in human cells.Elife 2,e00471.
Kaiser,T.E.,Intine,R.V.,and Dundr,M.(2008).De novo formation of asubnuclear body.Science 322,1713-1717.
Kapitein,L.C.,Schlager,M.A.,van der Zwan,W.A.,Wulf,P.S.,Keijzer,N.,and Hoogenraad,C.C.(2010).Probing intracellular motor protein activity usingan inducible cargo trafficking assay.Biophysical Journal 99,2143-2152.
Kearns,N.A.,Pham,H.,Tabak,B.,Genga,R.M.,Silverstein,N.J.,Garber,M.,and Maehr,R.(2015).Functional annotation of native enhancers with a Cas9-histone demethylase fusion.Nat Methods 12,401-403.
Knight,S.C.,Xie,L.,Deng,W.,Guglielmi,B.,Witkowsky,L.B.,Bosanac,L.,Zhang,E.T.,El Beheiry,M.,Masson,J.B.,Dahan,M.,et al.(2015).Dynamics ofCRISPR-Cas9 genome interrogation in living cells.Science 350,823-826.
Komor,A.C.,Kim,Y.B.,Packer,M.S.,Zuris,J.A.,and Liu,D.R.(2016).Programmable editing of a target base in genomic DNA without double-strandedDNA cleavage.Nature 533,420-424.
Kosak,S.T.,Skok,J.A.,Medina,K.L.,Riblet,R.,Le Beau,M.M.,Fisher,A.G.,and Singh,H.(2002).Subnuclear compartmentalization of immunoglobulin lociduring lymphocyte development.Science 296,158-162.
Kumaran,R.I.,and Spector,D.L.(2008).A genetic locus targeted to thenuclear periphery in living cells maintains its transcriptional competence.JCell Biol 180,51-65.
Langer-Safer,P.R.,Levine,M.,and Ward,D.C.(1982).Immunological methodfor mapping genes on Drosophila polytene chromosomes.Proc Natl Acad Sci U S A79,4381-4385.
Levine,M.,Cattoglio,C.,and Tjian,R.(2014).Looping back to leapforward:transcription enters a new era.Cell 157,13-25.
Liang,F.S.,Ho,W.Q.,and Crabtree,G.R.(2011).Engineering the ABA plantstress pathway for regulation of induced proximity.Sci Signal 4,rs2.
Ma,H.,Tu,L.C.,Naseri,A.,Huisman,M.,Zhang,S.,Grunwald,D.,and Pederson,T.(2016).Multiplexed labeling of genomic loci with dCas9 and engineeredsgRNAs using CRISPRainbow.Nat Biotechnol 34,528-530.
Machyna,M.,Neugebauer,K.M.,and Stanek,D.(2015).Coilin:The first25years.RNA Biol 12,590-596.
Mali,P.,Yang,L.,Esvelt,K.M.,Aach,J.,Guell,M.,DiCarlo,J.E.,Norville,J.E.,and Church,G.M.(2013).RNA-guided human genome engineering viaCas9.Science339,823-826.
Mao,Y.S.,Zhang,B.,and Spector,D.L.(2011).Biogenesis and function ofnuclear bodies.Trends Genet 27,295-306.
McCaffrey,M.W.,and Lindsay,A.J.(2012).Roles for myosin Va in RNAtransport and turnover.Biochem Soc Trans 40,1416-1420.
Morgan,S.L.,Mariano,N.C.,Bermudez,A.,Arruda,N.L.,Wu,F.,Luo,Y.,Shankar,G.,Jia,L.,Chen,H.,Hu,J.F.,et al.(2017).Manipulation of nucleararchitecture through CRISPR-mediated chromosomal looping.Nat Commun 8,15993.
Neugebauer,K.M.(2017).Special focus on the Cajal Body.RNA Biol 14,669-670.
Qi,L.S.,Larson,M.H.,Gilbert,L.A.,Doudna,J.A.,Weissman,J.S.,Arkin,A.P.,and Lim,W.A.(2013).Repurposing CRISPR as an RNA-guided platform forsequence-specific control of gene expression.Cell 152,1173-1183.
Reddy,K.L.,Zullo,J.M.,Bertolino,E.,and Singh,H.(2008).Transcriptionalrepression mediated by repositioning of genes to the nuclear lamina.Nature452,243-247.
Schmitt,A.D.,Hu,M.,and Ren,B.(2016).Genome-wide mapping and analysisof chromosome architecture.Nat Rev Mol Cell Biol 17,743-755.
Schreer,A.,Tinson,C.,Sherry,J.P.,and Schirmer,K.(2005).Application ofAlamar blue/5-carboxyfluorescein diacetate acetoxymethyl ester as anoninvasive cell viability assay in primary hepatocytes from rainbowtrout.Anal Biochem 344,76-85.
Shachar,S.,Voss,T.C.,Pegoraro,G.,Sciascia,N.,and Misteli,T.(2015).Identification of Gene Positioning Factors Using High-Throughput ImagingMapping.Cell162,911-923.
Shevtsov,S.P.,and Dundr,M.(2011).Nucleation of nuclear bodies byRNA.Nat Cell Biol 13,167-173.
Schindelin,J.,Arganda-Carreras,I.,Frise,E.,Kaynig,V.,Longair,M.,Pietzsch,T.,Preibisch,S.,Rueden,C.,Saalfeld,S.,Schmid,B.,et al.(2012).Fiji:anopen-source platform for biological-image analysis.Nat Methods 9,676-682.
Smogorzewska,A.,and de Lange,T.(2002).Different telomere damagesignaling pathways in human and mouse cells.EMBO J 21,4338-4348.
Smoyer,C.J.,and Jaspersen,S.L.(2014).Breaking down the wall:thenuclear envelope during mitosis.Curr Opin Cell Biol 26,1-9.
Takei,Y.,Shah,S.,Harvey,S.,Qi,L.S.,and Cai,L.(2017).MultiplexedDynamic Imaging of Genomic Loci by Combined CRISPR Imaging and DNA SequentialFISH.Biophys J 112,1773-1776.
Tanenbaum,M.E.,Gilbert,L.A.,Qi,L.S.,Weissman,J.S.,and Vale,R.D.(2014).A protein-tagging system for signal amplification in gene expressionand fluorescence imaging.Cell 159,635-646.
Tinevez,J.Y.,Perry,N.,Schindelin,J.,Hoopes,G.M.,Reynolds,G.D.,Laplantine,E.,Bednarek,S.Y.,Shorte,S.L.,and Eliceiri,K.W.(2017).TrackMate:Anopen and extensible platform for single-particle tracking.Methods 115,80-90.
Tsuchiya,Y.,Hase,A.,Ogawa,M.,Yorifuji,H.,and Arahata,K.(1999).Distinct regions specify the nuclear membrane targeting of emerin,theresponsible protein for Emery-Dreifuss muscular dystrophy.Eur J Biochem 259,859-865.
van Steensel,B.,and Belmont,A.S.(2017).Lamina-Associated Domains:Links with Chromosome Architecture,Heterochromatin,and Gene Repression.Cell169,780-791.
Vernier,M.,Bourdeau,V.,Gaumont-Leclerc,M.F.,Moiseeva,O.,Begin,V.,Saad,F.,Mes-Masson,A.M.,and Ferbeyre,G.(2011).Regulation of E2Fs andsenescence by PML nuclear bodies.Genes Dev 25,41-50.
Wang,Q.,Sawyer,I.A.,Sung,M.H.,Sturgill,D.,Shevtsov,S.P.,Pegoraro,G.,Hakim,O.,Baek,S.,Hager,G.L.,and Dundr,M.(2016).Cajal bodies are linked togenome conformation.Nat Commun 7,10966.
Williams,R.R.,Azuara,V.,Perry,P.,Sauer,S.,Dvorkina,M.,Jorgensen,H.,Roix,J.,McQueen,P.,Misteli,T.,Merkenschlager,M.,et al.(2006).Neural inductionpromotes large-scale chromatin reorganisation of the Mash1 locus.J Cell Sci119,132-140.
Yu,M.,and Ren,B.(2017).The Three-Dimensional Organization ofMammalian Genomes.Annu Rev Cell Dev Biol 33,265-289.
Zhu,L.,and Brangwynne,C.P.(2015).Nuclear bodies:the emergingbiophysics of nucleoplasmic phases.Curr Opin Cell Biol 34,23-30.
Zuleger,N.,Kelly,D.A.,Richardson,A.C.,Kerr,A.R.,Goldberg,M.W.,Goryachev,A.B.,and Schirmer,E.C.(2011).System analysis shows distinctmechanisms and common principles of nuclear envelope protein dynamics.J CellBiol 193,109-123.
Zullo,J.M.,Demarco,I.A.,Pique-Regi,R.,Gaffney,D.J.,Epstein,C.B.,Spooner,C.J.,Luperchio,T.R.,Bernstein,B.E.,Pritchard,J.K.,Reddy,K.L.,et al.(2012).DNA sequence-dependent compartmentalization and silencing of chromatinat the nuclear lamina.Cell 149,1474-1487.
VI.示例性实施方案
根据当前公开的主题提供的示例性实施方案包括但不限于权利要求书和以下实施方案:
1.控制靶多核苷酸在细胞的隔室中的空间和时间定位的系统,所述系统包括:
(a)与第一二聚化结构域连接的隔室特异性蛋白;和
(b)靶向所述靶多核苷酸的致动器部分,其中所述致动器部分与第二二聚化结构域连接,所述第二二聚化结构域能够与所述第一二聚化结构域组装成二聚体。
2.如实施方案1所述的系统,其中所述靶多核苷酸包括基因组DNA。
3.如实施方案1所述的系统,其中所述靶多核苷酸包括RNA。
4.如实施方案1-3中任一项所述的系统,其中所述致动器部分包括Cas蛋白,并且其中所述系统还包括:
(c)与所述致动器部分复合并与所述靶多核苷酸杂交的导向RNA。
5.如实施方案1-3中任一项所述的系统,其中所述致动器部分包括RNA结合蛋白,其中所述系统还包括:
(c)与所述致动器部分复合并与所述靶多核苷酸杂交的导向RNA,并且其中所述系统任选地还包括:
(d)与所述导向RNA复合的Cas蛋白。
6.如实施方案4或5所述的系统,其中所述Cas蛋白基本上缺乏DNA切割活性。
7.如实施方案4-6中任一项所述的系统,其中所述Cas蛋白是Cas9蛋白、Cas12蛋白、Cas13蛋白、CasX蛋白或CasY蛋白。
8.如实施方案7所述的系统,其中所述Cas12蛋白选自Cas12a、Cas12b、Cas12c、Cas12d和Cas12e。
9.如实施方案7所述的系统,其中所述Cas13蛋白选自Cas13a、Cas13b、Cas13c和Cas13d。
10.如实施方案5所述的系统,其中所述RNA结合蛋白是ADAR1或ADAR2,所述导向RNA包括ADAR募集RNA(arRNA)。
11.如实施方案1-3中任一项所述的系统,其中所述致动器部分包含与所述靶多核苷酸杂交的结合蛋白,其中所述结合蛋白是锌指核酸酶或TALE核酸酶。
12.如实施方案1-3中任一项所述的系统,其中所述致动器部分包含与导向多核苷酸复合的Argonaute蛋白,其中所述导向多核苷酸是导向RNA或导向DNA,并且其中所述导向多核苷酸与所述靶多核苷酸杂交。
13.如实施方案1-12中任一项所述的系统,其中所述隔室特异性蛋白选自隔室内源性的蛋白、调节蛋白、马达蛋白、DNA修复蛋白和以上的组合。
14.如实施方案1-13中任一项所述的系统,其中所述隔室是核隔室。
15.如实施方案14所述的系统,其中所述核隔室包括内核膜。
16.如实施方案15所述的系统,其中所述隔室特异性蛋白包括Emerin、Lap2β、核纤层蛋白B或以上的组合。
17.如实施方案14所述的系统,其中所述核隔室包括Cajal小体。
18.如实施方案17所述的系统,其中所述隔室特异性蛋白包括coilin、SMN、Gemin3、SmD1、SmE或以上的组合。
19.如实施方案14所述的系统,其中所述核隔室包括核小点。
20.如实施方案19所述的系统,其中所述隔室特异性蛋白包括SC35。
21.如实施方案14所述的系统,其中所述核隔室包括PML小体。
22.如实施方案21所述的系统,其中所述隔室特异性蛋白包括PML、SP100或以上的组合。
23.如实施方案14所述的系统,其中所述核隔室包括核芯复合物。
24.如实施方案23所述的系统,其中所述隔室特异性蛋白包括Nup50、Nup98、Nup53、Nup153、Nup62或以上的组合。
25.如实施方案14所述的系统,其中所述核隔室包括核仁。
26.如实施方案25所述的系统,其中所述隔室特异性蛋白包括核仁蛋白B23。
27.如实施方案14所述的系统,其中所述核隔室包括异染色质。
28.如实施方案27所述的系统,其中所述隔室特异性蛋白包括HP1、KRAB-ZFP、以上的截短形式或以上的组合。
29.如实施方案14所述的系统,其中所述核隔室包括核小体。
30.如实施方案29所述的系统,其中所述隔室特异性蛋白包括53BP1、Rad51或以上的组合。
31.如实施方案1-13中任一项所述的系统,其中所述隔室是细胞质隔室。
32.如实施方案31所述的系统,其中所述细胞质隔室包括细胞骨架成分。
33.如实施方案32所述的系统,其中所述隔室特异性蛋白包括驱动蛋白、动力蛋白、肌球蛋白或以上的组合。
34.如实施方案1-33中任一项所述的系统,其中所述隔室特异性蛋白还与荧光蛋白连接。
35.如实施方案1-34中任一项所述的系统,其中所述致动器部分还与荧光蛋白连接。
36.如实施方案1-35中任一项所述的系统,其中所述第一二聚化结构域和所述第二二聚化结构域仅在配体、光或酶的存在下组装形成二聚体。
37.如实施方案36所述的系统,其中所述第一二聚化结构域和所述第二二聚化结构域各自在配体存在下结合配体。
38.如实施方案36或37所述的系统,其中所述配体是化学诱导物或光生诱导物。
39.控制靶多核苷酸在细胞的隔室中的空间和时间定位的方法,所述方法包括:
(a)提供与第一二聚化结构域连接的隔室特异性蛋白;
(b)提供与第二二聚化结构域连接的致动器部分;
(c)形成包含所述致动器部分和所述靶多核苷酸的复合物;和
(d)组装包含所述第一二聚化结构域和所述第二二聚化结构域的二聚体,从而将所述靶多核苷酸定位在所述隔室中。
40.如实施方案39所述的方法,其中所述靶多核苷酸不是所述隔室内源性的。
41.如实施方案39或40所述的方法,其中所述靶多核苷酸的定位包括调节所述靶多核苷酸的表达。
42.如实施方案41所述的方法,其中所述调节包括降低所述靶多核苷酸的表达。
43.如实施方案41所述的方法,其中所述调节包括增加所述靶多核苷酸的表达。
44.如实施方案39-43中任一项所述的方法,其中所述靶多核苷酸的定位还包括调节所述隔室内源性的一种或多种另外的多核苷酸的表达。
45.如实施方案39-44中任一项所述的方法,其中所述靶多核苷酸的定位包括改变细胞功能、细胞命运、细胞生长、细胞凋亡和/或细胞分化。
46.如实施方案45所述的方法,其中所述靶多核苷酸包含端粒。
47.如实施方案39-46中任一项所述的方法,其中所述靶多核苷酸的定位还包括在细胞内产生一个或多个另外的隔室。
48.如实施方案39-47中任一项所述的方法,其中所述靶多核苷酸的定位还包括修复DNA断裂。
49.如实施方案48所述的方法,其中所述修复包括引入外源DNA。
50.如实施方案49所述的方法,其中所述引入包括重组,非同源末端连接或同源引导的修复。
51.如实施方案39-50中任一项所述的方法,其中所述靶多核苷酸的定位诱导相分离以形成隔室。
52.如实施方案51所述的方法,其中所述隔室是包含蛋白质、RNA、DNA或以上的组合的人工聚集体。
53.如实施方案51或52所述的方法,其中所述隔室是核小体或细胞体。
54.如实施方案39-53中任一项所述的方法,其中所述靶多核苷酸的定位诱导促进DNA修复并提高基因编辑效率的核小体的形成。
55.如实施方案39-54中任一项所述的方法,其中所述靶多核苷酸包括基因组DNA。
56.如实施方案39-54中任一项所述的方法,其中所述靶多核苷酸包括RNA。
57.如实施方案39-56中任一项所述的方法,其中所述致动器部分包括Cas蛋白,并且其中所述方法还包括:
(c)提供与所述致动器部分复合并与所述靶多核苷酸杂交的导向RNA。
58.如实施方案39-56中任一项所述的方法,其中所述致动器部分包括RNA结合蛋白,其中所述方法还包括:
(c)提供与所述致动器部分复合并与所述靶多核苷酸杂交的导向RNA,并且其中所述方法任选地还包括:
(d)提供与所述导向RNA复合的Cas蛋白。
59.如实施方案57或58所述的方法,其中所述Cas蛋白基本上缺乏DNA切割活性。
60.如实施方案57-59中任一项所述的方法,其中所述Cas蛋白是Cas9蛋白、Cas12蛋白、Cas13蛋白、CasX蛋白或CasY蛋白。
61.如实施方案60所述的方法,其中所述Cas12蛋白选自Cas12a、Cas12b、Cas12c、Cas12d和Cas12e。
62.如实施方案60所述的方法,其中所述Cas13蛋白选自Cas13a、Cas13b、Cas13c和Cas13d。
63.如实施方案58所述的方法,其中所述RNA结合蛋白是ADAR1或ADAR2,并且所述导向RNA包括ADAR募集RNA(arRNA)。
64.如实施方案39-56中任一项所述的方法,其中所述致动器部分包含与所述靶多核苷酸杂交的结合蛋白,其中所述结合蛋白是锌指核酸酶或TALE核酸酶。
65.如实施方案39-56中任一项所述的方法,其中所述致动器部分包含与导向多核苷酸复合的Argonaute蛋白,其中所述导向多核苷酸是导向RNA或导向DNA,并且其中所述导向多核苷酸与所述靶多核苷酸杂交。
66.如实施方案39-65中任一项所述所述的方法,其中所述隔室特异性蛋白选自隔室内源性的蛋白、调节蛋白、马达蛋白、DNA修复蛋白和以上的组合。
67.如实施方案39-66中任一项所述的方法,其中所述隔室是核隔室。
68.如实施方案67所述的方法,其中所述核隔室包括内核膜。
69.如实施方案68所述的方法,其中所述隔室特异性蛋白包括Emerin、Lap2β、核纤层蛋白B或以上的组合。
70.如实施方案67所述的方法,其中所述核隔室包括Cajal小体。
71.如实施方案70所述的方法,其中所述隔室特异性蛋白包括coilin、SMN、Gemin3、SmD1、SmE或以上的组合。
72.如实施方案67所述的方法,其中所述核隔室包括核小点。
73.如实施方案72所述的方法,其中所述隔室特异性蛋白包含SC35。
74.如实施方案67所述的方法,其中所述核隔室包括PML小体。
75.如实施方案74所述的方法,其中所述隔室特异性蛋白包括PML、SP100或以上的组合。
76.如实施方案67所述的方法,其中所述核隔室包括核芯复合物。
77.如实施方案76所述的方法,其中所述隔室特异性蛋白包括Nup50、Nup98、Nup53、Nup153、Nup62或以上的组合。
78.如实施方案67所述的方法,其中所述核隔室包括核仁。
79.如实施方案78所述的方法,其中所述隔室特异性蛋白包括核仁蛋白B23。
80.如实施方案67所述的方法,其中所述核隔室包括异染色质。
81.如实施方案80所述的方法,其中所述隔室特异性蛋白包括HP1、KRAB-ZFP、以上的截短形式或以上的组合。
82.如实施方案67所述的方法,其中所述核隔室包括核小体。
83.如实施方案82所述的方法,其中所述隔室特异性蛋白包含53BP1、Rad51或以上的组合。
84.如实施方案39-66中任一项所述的方法,其中所述隔室是细胞质隔室。
85.如实施方案84所述的方法,其中所述细胞质隔室包括细胞骨架成分。
86.如实施方案85所述的方法,其中所述隔室特异性蛋白包括驱动蛋白、动力蛋白、肌球蛋白或以上的组合。
87.如实施方案39-86中任一项所述的方法,其中所述隔室特异性蛋白还与荧光蛋白连接。
88.如实施方案39-87中任一项所述的方法,其中所述致动器部分还与荧光蛋白连接。
89.如实施方案39-88中任一项所述的方法,其中所述组装仅在配体,光或酶存在下发生。
90.如实施方案89所述的方法,其中所述第一二聚化结构域和所述第二二聚化结构域在所述配体存在下各自结合所述配体。
91.如实施方案89或90所述的方法,其中所述配体是化学诱导物或光生诱导物。
尽管为了清楚理解的目的,已经通过说明和实施例的方式详细描述了上述发明,但是本领域技术人员将理解,在所附权利要求的范围内可以实施某些改变和修改。此外,本文提供的每篇参考文献通过引用以其整体并入本文,其程度如同每篇参考文献单独通过引用并入本文一样。
权利要求书(按照条约第19条的修改)
1.控制靶多核苷酸在细胞的隔室中的空间和时间定位的系统,所述系统包括:
(a)与第一二聚化结构域连接的隔室特异性蛋白;和
(b)靶向所述靶多核苷酸的致动器部分,其中所述致动器部分与第二二聚化结构域连接,所述第二二聚化结构域能够与所述第一二聚化结构域组装成二聚体。
2.如权利要求1所述的系统,其中所述靶多核苷酸包括基因组DNA。
3.如权利要求1所述的系统,其中所述靶多核苷酸包括RNA。
4.如权利要求1-3中任一项所述的系统,其中所述致动器部分包括Cas蛋白,并且其中所述系统还包括:
(c)与所述致动器部分复合并与所述靶多核苷酸杂交的导向RNA。
5.如权利要求1-3中任一项所述的系统,其中所述致动器部分包括RNA结合蛋白,其中所述系统还包括:
(c)与所述致动器部分复合并与所述靶多核苷酸杂交的导向RNA,并且其中所述系统任选地还包括:
(d)与所述导向RNA复合的Cas蛋白。
6.如权利要求4所述的系统,其中所述Cas蛋白基本上缺乏DNA切割活性。
7.如权利要求4所述的系统,其中所述Cas蛋白是Cas9蛋白、Cas12蛋白、Cas13蛋白、CasX蛋白或CasY蛋白。
8.如权利要求7所述的系统,其中所述Cas12蛋白选自Cas12a、Cas12b、Cas12c、Cas12d和Cas12e。
9.如权利要求7所述的系统,其中所述Cas13蛋白选自Cas13a、Cas13b、Cas13c和Cas13d。
10.如权利要求5所述的系统,其中所述RNA结合蛋白是ADAR1或ADAR2,以及所述导向RNA包括ADAR募集RNA(arRNA)。
11.如权利要求1-3中任一项所述的系统,其中所述致动器部分包含与所述靶多核苷酸杂交的结合蛋白,其中所述结合蛋白是锌指核酸酶或TALE核酸酶。
12.如权利要求1-3中任一项所述的系统,其中所述致动器部分包含与导向多核苷酸复合的Argonaute蛋白,其中所述导向多核苷酸是导向RNA或导向DNA,并且其中所述导向多核苷酸与所述靶多核苷酸杂交。
13.如权利要求1所述的系统,其中所述隔室特异性蛋白选自隔室内源性的蛋白、调节蛋白、马达蛋白、DNA修复蛋白和以上的组合。
14.如权利要求1所述的系统,其中所述隔室是核隔室。
15.如权利要求14所述的系统,其中所述核隔室包括内核膜。
16.如权利要求15所述的系统,其中所述隔室特异性蛋白包括Emerin、Lap2β、核纤层蛋白B或以上的组合。
17.如权利要求14所述的系统,其中所述核隔室包括Cajal小体。
18.如权利要求17所述的系统,其中所述隔室特异性蛋白包括coilin、SMN、Gemin 3、SmD1、SmE或以上的组合。
19.如权利要求14所述的系统,其中所述核隔室包括核小点。
20.如权利要求19所述的系统,其中所述隔室特异性蛋白包括SC35。
21.如权利要求14所述的系统,其中所述核隔室包括PML小体。
22.如权利要求21所述的系统,其中所述隔室特异性蛋白包括PML、SP100或以上的组合。
23.如权利要求14所述的系统,其中所述核隔室包括核芯复合物。
24.如权利要求23所述的系统,其中所述隔室特异性蛋白包括Nup50、Nup98、Nup53、Nup153、Nup62或以上的组合。
25.如权利要求14所述的系统,其中所述核隔室包括核仁。
26.如权利要求25所述的系统,其中所述隔室特异性蛋白包括核仁蛋白B23。
27.如权利要求14所述的系统,其中所述核隔室包括异染色质。
28.如权利要求27所述的系统,其中所述隔室特异性蛋白包括HP1、KRAB-ZFP、以上的截短形式或以上的组合。
29.如权利要求14所述的系统,其中所述核隔室包括核小体。
30.如权利要求29所述的系统,其中所述隔室特异性蛋白包括53BP1、Rad51或以上的组合。
31.如权利要求1所述的系统,其中所述隔室是细胞质隔室。
32.如权利要求31所述的系统,其中所述细胞质隔室包括细胞骨架成分。
33.如权利要求32所述的系统,其中所述隔室特异性蛋白包括驱动蛋白、动力蛋白、肌球蛋白或以上的组合。
34.如权利要求1所述的系统,其中所述隔室特异性蛋白还与荧光蛋白连接。
35.如权利要求1所述的系统,其中所述致动器部分还与荧光蛋白连接。
36.如权利要求1所述的系统,其中所述第一二聚化结构域和所述第二二聚化结构域仅在配体、光或酶的存在下组装形成二聚体。
37.如权利要求36所述的系统,其中所述第一二聚化结构域和所述第二二聚化结构域各自在配体存在下结合配体。
38.如权利要求36或37所述的系统,其中所述配体是化学诱导物或光生诱导物。
39.控制靶多核苷酸在细胞的隔室中的空间和时间定位的方法,所述方法包括:
(a)提供与第一二聚化结构域连接的隔室特异性蛋白;
(b)提供与第二二聚化结构域连接的致动器部分;
(c)形成包含所述致动器部分和所述靶多核苷酸的复合物;和
(d)组装包含所述第一二聚化结构域和所述第二二聚化结构域的二聚体,从而将所述靶多核苷酸定位在所述隔室中。
40.如权利要求39所述的方法,其中所述靶多核苷酸不是所述隔室内源性的。
41.如权利要求39或40所述的方法,其中所述靶多核苷酸的定位包括调节所述靶多核苷酸的表达。
42.如权利要求41所述的方法,其中所述调节包括降低所述靶多核苷酸的表达。
43.如权利要求41所述的方法,其中所述调节包括增加所述靶多核苷酸的表达。
44.如权利要求39所述的方法,其中所述靶多核苷酸的定位还包括调节所述隔室内源性的一种或多种另外的多核苷酸的表达。
45.如权利要求39所述的方法,其中所述靶多核苷酸的定位包括改变细胞功能、细胞命运、细胞生长、细胞凋亡和/或细胞分化。
46.如权利要求45所述的方法,其中所述靶多核苷酸包含端粒。
47.如权利要求39所述的方法,其中所述靶多核苷酸的定位还包括在细胞内产生一个或多个另外的隔室。
48.如权利要求39所述的方法,其中所述靶多核苷酸的定位还包括修复DNA断裂。
49.如权利要求48所述的方法,其中所述修复包括引入外源DNA。
50.如权利要求49所述的方法,其中所述引入包括重组,非同源末端连接或同源引导的修复。
51.如权利要求39所述的方法,其中所述靶多核苷酸的定位诱导相分离以形成隔室。
52.如权利要求51所述的方法,其中所述隔室是包含蛋白质、RNA、DNA或以上的组合的人工聚集体。
53.如权利要求51或52所述的方法,其中所述隔室是核小体或细胞体。
54.如权利要求39所述的方法,其中所述靶多核苷酸的定位诱导促进DNA修复并提高基因编辑效率的核小体的形成。
55.如权利要求39所述的方法,其中所述靶多核苷酸包括基因组DNA。
56.如权利要求39所述的方法,其中所述靶多核苷酸包括RNA。
57.如权利要求39所述的方法,其中所述致动器部分包括Cas蛋白,并且其中所述方法还包括:
(c)提供与所述致动器部分复合并与所述靶多核苷酸杂交的导向RNA。
58.如权利要求39所述的方法,其中所述致动器部分包括RNA结合蛋白,其中所述方法还包括:
(c)提供与所述致动器部分复合并与所述靶多核苷酸杂交的导向RNA,并且其中所述方法任选地还包括:
(d)提供与所述导向RNA复合的Cas蛋白。
59.如权利要求57或58所述的方法,其中所述Cas蛋白基本上缺乏DNA切割活性。
60.如权利要求57所述的方法,其中所述Cas蛋白是Cas9蛋白、Cas12蛋白、Cas13蛋白、CasX蛋白或CasY蛋白。
61.如权利要求60所述的方法,其中所述Cas12蛋白选自Cas12a、Cas12b、Cas12c、Cas12d和Cas12e。
62.如权利要求60所述的方法,其中所述Cas13蛋白选自Cas13a、Cas13b、Cas13c和Cas13d。
63.如权利要求58所述的方法,其中所述RNA结合蛋白是ADAR1或ADAR2,并且所述导向RNA包括ADAR募集RNA(arRNA)。
64.如权利要求39所述的方法,其中所述致动器部分包含与所述靶多核苷酸杂交的结合蛋白,其中所述结合蛋白是锌指核酸酶或TALE核酸酶。
65.如权利要求39所述的方法,其中所述致动器部分包含与导向多核苷酸复合的Argonaute蛋白,其中所述导向多核苷酸是导向RNA或导向DNA,并且其中所述导向多核苷酸与所述靶多核苷酸杂交。
66.如权利要求39所述所述的方法,其中所述隔室特异性蛋白选自隔室内源性的蛋白、调节蛋白、马达蛋白、DNA修复蛋白和以上的组合。
67.如权利要求39所述的方法,其中所述隔室是核隔室。
68.如权利要求67所述的方法,其中所述核隔室包括内核膜。
69.如权利要求68所述的方法,其中所述隔室特异性蛋白包括Emerin、Lap2β、核纤层蛋白B或以上的组合。
70.如权利要求67所述的方法,其中所述核隔室包括Cajal小体。
71.如权利要求70所述的方法,其中所述隔室特异性蛋白包括coilin、SMN、Gemin 3、SmD1、SmE或以上的组合。
72.如权利要求67所述的方法,其中所述核隔室包括核小点。
73.如权利要求72所述的方法,其中所述隔室特异性蛋白包含SC35。
74.如权利要求67所述的方法,其中所述核隔室包括PML小体。
75.如权利要求74所述的方法,其中所述隔室特异性蛋白包括PML、SP100或以上的组合。
76.如权利要求67所述的方法,其中所述核隔室包括核芯复合物。
77.如权利要求76所述的方法,其中所述隔室特异性蛋白包括Nup50、Nup98、Nup53、Nup153、Nup62或以上的组合。
78.如权利要求67所述的方法,其中所述核隔室包括核仁。
79.如权利要求78所述的方法,其中所述隔室特异性蛋白包括核仁蛋白B23。
80.如权利要求67所述的方法,其中所述核隔室包括异染色质。
81.如权利要求80所述的方法,其中所述隔室特异性蛋白包括HP1、KRAB-ZFP、以上的截短形式或以上的组合。
82.如权利要求67所述的方法,其中所述核隔室包括核小体。
83.如权利要求82所述的方法,其中所述隔室特异性蛋白包含53BP1、Rad51或以上的组合。
84.如权利要求39所述的方法,其中所述隔室是细胞质隔室。
85.如权利要求84所述的方法,其中所述细胞质隔室包括细胞骨架成分。
86.如权利要求85所述的方法,其中所述隔室特异性蛋白包括驱动蛋白、动力蛋白、肌球蛋白或以上的组合。
87.如权利要求39所述的方法,其中所述隔室特异性蛋白还与荧光蛋白连接。
88.如权利要求39所述的方法,其中所述致动器部分还与荧光蛋白连接。
89.如权利要求39所述的方法,其中所述组装仅在配体、光或酶存在下发生。
90.如权利要求89所述的方法,其中所述第一二聚化结构域和所述第二二聚化结构域在所述配体存在下各自结合所述配体。
91.如权利要求89或90所述的方法,其中所述配体是化学诱导物或光生诱导物。
92.如权利要求1所述的系统,其中所述隔室是包含能够促进同源性定向修复(HDR)的蛋白质的核隔室。
93.如权利要求1所述的系统,其中所述隔室特异性蛋白包括能够促进同源性定向修复(HDR)的蛋白质。
94.如权利要求92或93所述的系统,其中所述能够促进HDR的蛋白质包括:RPA、Rad51、Rad52、MRN复合物、MRX复合物、Ct1P、Sae2、Sgsl、BRCA2、Exol、OsExol、ATM、BRCA2、RAD54或MDC1。
95.如权利要求92或93所述的系统,还包括用于HDR的模板多核苷酸。
96.如权利要求95所述的系统,其中所述模板多核苷酸是外源性多核苷酸。
97.如权利要求95所述的系统,其中所述模板多核苷酸是内源性多核苷酸。
98.如权利要求97所述的系统,其中所述内源性多核苷酸是染色单体。
99.如权利要求95所述的系统,其中所述模板多核苷酸与所述第一二聚化结构域融合。
100.如权利要求1所述的系统,其中所述隔室特异性蛋白能够促进非同源末端连接(NHEJ)。
101.如权利要求100所述的系统,其中所述隔室特异性蛋白包括:KU70、KU80、Artemis、PKcs、LigIV或XRCC4。
102.如权利要求1所述的系统,其中所述二聚体的组装影响所述靶多核苷酸定位到所述隔室。
103.如权利要求1所述的系统,其中所述二聚体的组装影响所述隔室定位到所述靶多核苷酸。
104.如权利要求39所述的方法,其中所述隔室是包含能够促进同源性定向修复(HDR)的蛋白质的核隔室。
105.如权利要求39所述的方法,其中所述隔室特异性蛋白包括能够促进同源性定向修复(HDR)的蛋白质。
106.如权利要求104或105所述的方法,其中所述能够促进HDR的蛋白质包括:RPA、Rad51、Rad52、MRN复合物、MRX复合物、Ct1P、Sae2、Sgsl、BRCA2、Exol、OsExol、ATM、BRCA2、RAD54或MDC1。
107.如权利要求104或105所述的方法,其还包括提供用于HDR的模板多核苷酸。
108.如权利要求107所述的方法,其中所述模板多核苷酸是外源行多核苷酸。
109.如权利要求107所述的方法,其中所述模板多核苷酸是内源性多核苷酸。
110.如权利要求109所述的方法,其中所述内源行多核苷酸是染色单体。
111.如权利要求107所述的方法,其中所述模板多核苷酸与所述第一二聚化结构域融合。
112.如权利要求39所述的方法,其中所述隔室特异性蛋白能够促进非同源末端连接(NHEJ)。
113.如权利要求112所述的方法,其中所述隔室特异性蛋白包括:KU70、KU80、Artemis、PKcs、LigIV或XRCC4。
114.如权利要求39所述的方法,其中所述二聚体的组装影响所述靶多核苷酸定位到所述隔室。
115.如权利要求39所述的方法,其中所述二聚体的组装影响所述隔室定位到所述靶多核苷酸。
116.用于共定位细胞隔室和靶多核苷酸的方法,所述方法包括:
(a)提供与第一二聚化结构域连接的隔室特异性蛋白,其中所述隔室特异性蛋白是(i)所述隔室的一部分或(ii)足以实现所述隔室的形成;
(b)提供与第二二聚化结构域连接的致动器部分,其中所述致动器部分能够与所述靶多核苷酸形成复合物;和
(c)组装包含所述第一二聚化结构域和所述第二二聚化结构域的二聚体以实现所述隔室和所述靶多核苷酸的共定位。
117.用于在细胞中共定位靶多核苷酸和模板多核苷酸的方法,所述方法包括:
(a)提供与第一二聚化结构域连接的第一致动器部分,其中所述第一致动器部分能够与所述模板多核苷酸形成第一复合物;
(b)提供与第二二聚化结构域连接的第二致动器部分,其中所述第二致动器部分能够与所述靶多核苷酸形成第二复合物;和
(c)组装包含所述第一二聚结构域和所述第二二聚结构域的二聚体,以实现所述靶多核苷酸和所述模板多核苷酸的共定位。
118.如权利要求117所述的方法,其还包括:形成(i)包含所述第一致动器部分和所述模板多核苷酸的所述第一复合物,以及(ii)包含所述第二致动器部分和所述靶多核苷酸的所述第二复合物。
- 用于多核苷酸空间组织的系统和方法
- 用于关于参考多核苷酸序列标注样本多核苷酸序列中的变异的方法和系统