手性苯并芳杂环并二氢吡喃酮类化合物及其制备方法
文献发布时间:2023-06-19 13:45:04
技术领域
本发明涉及不对称合成技术领域,尤其涉及手性苯并芳杂环并二氢吡喃酮类化合物及其制备方法。
背景技术
苯并芳杂环类衍生物(例如苯并噻吩、吲哚等),广泛存在于天然产物和药物分子中,具有多样性的生物活性,如:抗疟疾、抗结核、抗癌等。通过对苯并芳杂环结构的修饰,可不同程度地改善该类化合物的药理性质,从而有利于新药先导化合物的发现。手性苯并芳杂环并二氢吡喃酮类化合物,作为一种很重要的苯并芳杂环衍生物,在许多具有生物活性的天然产物和药物中被发现,是新药研发的理想结构骨架,因此,构建光学纯的苯并芳杂环并二氢吡喃酮骨架一直是有机化学家和药物化学家研究的热点领域。
据调研,绝大多数的药物由手性分子构成,不同构型的手性分子可能具有明显不同的生物活性,其在人体内的药理、代谢、毒性及疗效存在着显著的差异。不对称合成手性药物作为一种高效、便捷的方法,近年来得到了快速的发展。通过研究和发展新的合成方法,借此获得光学纯的手性药物,在有机化学和药物化学领域具有十分重要的研究价值。
然而,经过发明人的文献调研发现,高对映选择性地合成光学纯的苯并芳杂环并二氢吡喃酮类化合物的方法很少。因此,鉴于苯并芳杂环并二氢吡喃酮类骨架的重要性,开发一类具有反应条件温和、易操作、底物稳定易得、应用范围广、对映选择性高等特点的简单高效的合成方法,用于构建一系列光学纯的苯并芳杂环并二氢吡喃酮类化合物,以期作为新药筛选的候选分子,丰富先导化合物库具有非常重要的意义。
发明内容
本发明的目的之一,就在于提供一类新型的手性苯并芳杂环并二氢吡喃酮类化合物,以解决上述问题。
为了实现上述目的,本发明采用的技术方案如下:一类新型的手性苯并芳杂环并二氢吡喃酮类化合物,具有如下结构式(Ⅳ)所示的结构:
上述结构式Ⅳ中,R
本发明提供了一类新型的合成光学纯的苯并芳杂环并二氢吡喃酮类化合物,该类化合物具有一个六元内酯骨架单元,其3,4位具有两个连续的立体中心。
本发明的化合物的应用价值在于:现有的苯并芳杂环并二氢吡喃酮类衍生物普遍具有很好的生物活性,因此,本发明所提供的一大类新的化合物具有极大的潜在应用价值,为药物候选分子的发现及筛选尤其是高通量筛选提供充足的化合物源,丰富了该类先导化合物库。另外,本发明方法能够以高对映选择性的构建一系列苯并芳杂环并二氢吡喃酮类骨架,丰富了该类手性骨架单元的合成路线。
本发明的目的之二,在于提供一种上述的手性苯并芳杂环并二氢吡喃酮类化合物的制备方法,采用的技术方案为,
包括如下步骤:
将苯并芳杂环2-烯-3-酮(Ⅰ),丁烯酸内酯(Ⅱ)或吖内酯(Ⅲ)溶解于有机溶剂中,然后加入手性催化剂,在0℃-60℃下搅拌反应12-72h,待反应完全后,直接分离纯化得到产物(Ⅳ);
其中,所述苯并芳杂环2-烯-3-酮(Ⅰ)具有如下结构:
所述丁烯酸内酯(Ⅱ)具有如下结构:
所述吖内酯(Ⅲ)具有如下结构:
合成路线为:
步骤(1)中的手性催化剂,优选如下方框内所示的双功能催化剂,后文实施例中的有机催化剂也是指双功能催化剂;
需要说明的是,实施例中所述的“手性催化剂A”、“手性催化剂B”和“手性催化剂C”,是指与上式方框内的A、B、C对应的取代化合物。另外,本领域技术人员可以理解的,本申请所采用的手性催化剂,并不限于上面的三种,凡是以金鸡纳碱和手性二胺为母体的催化剂,也可以实现本发明。
本发明采用上述的合成方法,合成了一系列新的苯并芳杂环并二氢吡喃酮类衍生物。
作为优选的技术方案:所述反应溶剂选自甲苯、均三甲苯、二氯甲烷、氯仿、四氢呋喃、乙醚、乙腈、甲基叔丁基醚、1,4-二氧六环、氯苯中的至少一种。
作为优选的技术方案:所述手性催化剂为金鸡纳碱-方酰胺催化剂。
作为优选的技术方案:所述催化剂用量为20mol%。
作为优选的技术方案:所述反应温度为0℃至60℃。
作为优选的技术方案:所述分离纯化方法为柱层析。
与现有技术相比,本发明的优点在于:本发明采用苯并芳杂环2-烯-3-酮和丁烯酸内酯或吖内酯发生串联[4+2]Michael/酯交换反应,高立体选择性地实现了具有两个连续手性中心的苯并芳杂环并二氢吡喃酮类型化合物的构建;本方法制得的化合物丰富了苯并芳杂环并二氢吡喃酮化合物的种类,从而为先导化合物及药物候选分子的筛选提供充足的化合物源;本发明的制备方法具有新颖、简洁、操作简单、反应条件温和、底物普适性好、立体选择性高等优点(可达>20:1dr,>99%ee)。
附图说明
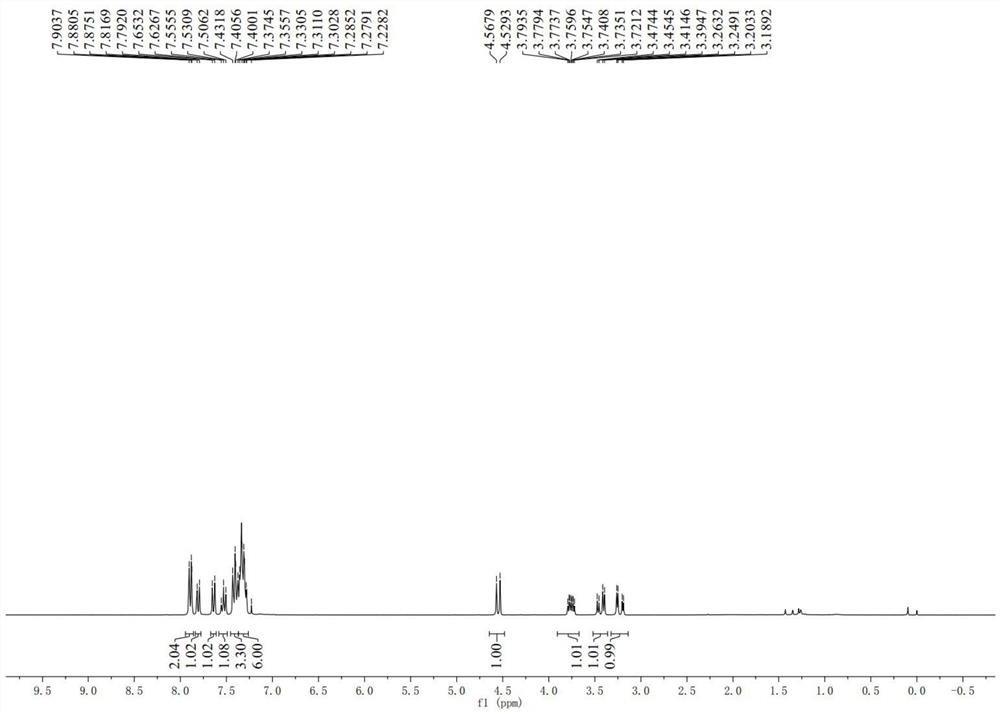
图1为实施例1制得的Ⅳ-a的氢谱图;
图2为实施例1制得的Ⅳ-a的碳谱图;
图3为实施例1制得的Ⅳ-a的单晶图。
具体实施方式
下面将结合附图对本发明作进一步说明。
实施例1:合成化合物(Ⅳ-a)
化合物Ⅳ-a的合成:
在一根干燥的反应试管中加入丁烯酸内酯Ⅱ-a(0.1mmol),苯并噻吩环2-烯-3-酮Ⅰ-a(0.12mmol),手性催化剂A-C,然后加入2.0mL溶剂,反应完全后,粗产品经柱色谱分离纯化得化合物Ⅳ-a,不同的反应条件如表1所示,具体反应过程如下:
表1不同的反应条件
表1中,“x”是指所用溶剂的量。
从表1中可见,以丁烯酸内酯Ⅱ-a作为模板底物进行条件筛选,不同类型的催化剂对反应的活性和立体选择性都略有影响,通过比较,奎宁衍生的方酰胺催化剂反应效果最好;同时,溶剂对反应的非对映选择性影响较大,通过比较,甲苯的非对映选择性最好;此外,分子筛的加入对反应的活性和立体选择性均没有起到正向的作用;另外,需要说明的是,对于吖内酯底物Ⅲ而言,溶剂选择二氯甲烷时反应的立体选择性最好;最终,丁烯酸内酯Ⅱ的反应采用催化剂A20mol%、2mL甲苯作溶剂、反应温度为0℃,是更为优选的方案;而吖内酯Ⅲ的反应采用催化剂A20mol%、2mL二氯甲烷作溶剂、反应温度为0℃,是更为优选的方案。
在最优方案下,得到的Ⅳ-a情况如下:
白色固体,35.0mg,收率88%;11.4:1dr,99%ee;[α]
Ⅳ-a的氢谱、碳谱分别如图1-2所示。
单晶图谱测定结果如图3所示,表明化合物Ⅳ-a的绝对构型为(C9S,C10R)。
实施例2:合成化合物Ⅳ-b
在一根干燥的反应试管中加入丁烯酸内酯Ⅱ-b0.1mmol,苯并噻吩环2-烯-3-酮Ⅰ-b0.12mmol和手性催化剂A 0.02mmol,然后加入2.0mL甲苯,反应混合物在0℃下搅拌反应;反应完全后,粗产品经柱色谱分离纯化得化合物Ⅳ-b。黄色固体;29.9mg,收率72%;8.0:1dr,99%ee;[α]
实施例3:合成化合物Ⅳ-c
在一根干燥的反应试管中加入丁烯酸内酯Ⅱ-c0.1mmol,苯并噻吩环2-烯-3-酮Ⅰ-c0.12mmol和手性催化剂A 0.02mmol,然后加入2.0mL甲苯,反应混合物在0℃下搅拌反应;反应完全后,粗产品经柱色谱分离纯化得化合物Ⅳ-c。黄色固体;30.7mg,收率为71%;9.5:1dr,99%ee;[α]
实施例4:合成化合物Ⅳ-d
在一根干燥的反应试管中加入丁烯酸内酯Ⅱ-d 0.1mmol,苯并噻吩环2-烯-3-酮Ⅰ-d 0.12mmol和手性催化剂A 0.02mmol,然后加入2.0mL甲苯,反应混合物在0℃下搅拌反应;反应完全后,粗产品经柱色谱分离纯化得化合物Ⅳ-d。黄色固体;36.2mg,收率为88%;8.1:1dr,99%ee;[α]
实施例5:合成化合物Ⅳ-e
在一根干燥的反应试管中加入丁烯酸内酯Ⅱ-e0.1mmol,苯并噻吩环2-烯-3-酮Ⅰ-e0.12mmol和手性催化剂A 0.02mmol,然后加入2.0mL甲苯,反应混合物在0℃下搅拌反应;反应完全后,粗产品经柱色谱分离纯化得化合物Ⅳ-e。粉色固体;35.4mg,收率为82%;9.4:1dr,99%ee;[α]
实施例6:合成化合物Ⅳ-f
在一根干燥的反应试管中加入丁烯酸内酯Ⅱ-f 0.1mmol,苯并噻吩环2-烯-3-酮Ⅰ-f 0.12mmol和手性催化剂A 0.02mmol,然后加入2.0mL甲苯,反应混合物在0℃下搅拌反应;反应完全后,粗产品经柱色谱分离纯化得化合物Ⅳ-f。黄色固体;34.3mg,收率为83%;9.1:1dr,99%ee;[α]
实施例7:合成化合物Ⅳ-g
在一根干燥的反应试管中加入丁烯酸内酯Ⅱ-g0.1mmol,苯并噻吩环2-烯-3-酮Ⅰ-g0.12mmol和手性催化剂A 0.02mmol,然后加入2.0mL甲苯,反应混合物在0℃下搅拌反应;反应完全后,粗产品经柱色谱分离纯化得化合物Ⅳ-g。黄色固体;32.3mg,收率为78%;8.0:1dr,99%ee;[α]
实施例8:合成化合物Ⅳ-h
在一根干燥的反应试管中加入丁烯酸内酯Ⅱ-h 0.1mmol,苯并噻吩环2-烯-3-酮Ⅰ-h 0.12mmol和手性催化剂C 0.02mmol,然后加入2.0mL甲苯,反应混合物在0℃下搅拌反应;反应完全后,粗产品经柱色谱分离纯化得化合物Ⅳ-h。黄色固体;36.7mg,收率为82%;12.1:1dr,99%ee;[α]
实施例9:合成化合物Ⅳ-i
在一根干燥的反应试管中加入吖内酯Ⅲ-i0.1mmol,苯并噻吩环2-烯-3-酮Ⅰ-i0.12mmol和手性催化剂A 0.02mmol,然后加入2.0mL二氯甲烷,反应混合物在0℃下搅拌反应;反应完全后,粗产品经柱色谱分离纯化得化合物Ⅳ-i。黄色固体;40.7mg,收率为93%;8.3:1dr,93%ee;[α]
HPLC(Chiralpak IA色谱柱,乙醇/正己烷=50/50,流速=1.0mL/min,λ=254nm)t
实施例10:合成化合物Ⅳ-j
在一根干燥的反应试管中加入吖内酯Ⅲ-j0.1mmol,苯并噻吩环2-烯-3-酮Ⅰ-j0.12mmol和手性催化剂A 0.02mmol,然后加入2.0mL二氯甲烷,反应混合物在0℃下搅拌反应;反应完全后,粗产品经柱色谱分离纯化得化合物Ⅳ-j。淡黄色固体;45.6mg,收率为88%;14.6:1dr,97%ee;[α]
实施例11:合成化合物Ⅳ-k
在一根干燥的反应试管中加入吖内酯Ⅲ-k0.1mmol,苯并噻吩环2-烯-3-酮Ⅰ-k0.12mmol和手性催化剂A 0.02mmol,然后加入2.0mL二氯甲烷,反应混合物在0℃下搅拌反应;反应完全后,粗产品经柱色谱分离纯化得化合物Ⅳ-k。红色固体;49.3mg,收率为94%;>20:1dr,98%ee;[α]
实施例12:合成化合物Ⅳ-l
在一根干燥的反应试管中加入吖内酯Ⅲ-l0.1mmol,苯并噻吩环2-烯-3-酮Ⅰ-l0.12mmol和手性催化剂A 0.02mmol,然后加入2.0mL二氯甲烷,反应混合物在0℃下搅拌反应;反应完全后,粗产品经柱色谱分离纯化得化合物Ⅳ-l。淡黄色固体;45mg,收率为89%;8.8:1dr,96%ee;[α]
实施例13:合成化合物Ⅳ-m
在一根干燥的反应试管中加入丁烯酸内酯Ⅱ-m0.1mmol,吲哚环2-烯-3-酮Ⅰ-m0.12mmol和手性催化剂A 0.02mmol,然后加入2.0mL甲苯,反应混合物在0℃下搅拌反应;反应完全后,粗产品经柱色谱分离纯化得化合物Ⅳ-m。黄色固体;40mg,收率为75%;9.2:1dr,99%ee;[α]
实施例14:合成化合物Ⅳ-n
实施例15:合成化合物Ⅳ-o
在一根干燥的反应试管中加入丁烯酸内酯Ⅱ-o 0.1mmol,吲哚环2-烯-3-酮Ⅰ-o0.12mmol和手性催化剂A 0.02mmol,然后加入2.0mL甲苯,反应混合物在0℃下搅拌反应;反应完全后,粗产品经柱色谱分离纯化得化合物Ⅳ-o。黄色固体;45mg,收率为89%;8.8:1dr,96%ee;[α]
实施例16:合成化合物Ⅳ-p
在一根干燥的反应试管中加入吖内酯Ⅱ-p 0.1mmol,吲哚环2-烯-3-酮Ⅰ-p0.12mmol和手性催化剂A 0.02mmol,然后加入2.0mL二氯甲烷,反应混合物在0℃下搅拌反应;反应完全后,粗产品经柱色谱分离纯化得化合物Ⅳ-p。黄色固体;45.2mg,收率为72%;7.4:1dr,99%ee;[α]
实施例17:合成化合物Ⅳ-q
在一根干燥的反应试管中加入吖内酯Ⅱ-q0.1mmol,吲哚环2-烯-3-酮Ⅰ-q0.12mmol和手性催化剂A 0.02mmol,然后加入2.0mL二氯甲烷,反应混合物在0℃下搅拌反应;反应完全后,粗产品经柱色谱分离纯化得化合物Ⅳ-q。黄色固体;55.2mg,收率为86%;7.1:1dr,99%ee;[α]
实施例18:合成化合物Ⅳ-r
在一根干燥的反应试管中加入吖内酯Ⅱ-r0.1mmol,吲哚环2-烯-3-酮Ⅰ-r0.12mmol和手性催化剂A 0.02mmol,然后加入2.0mL二氯甲烷,反应混合物在0℃下搅拌反应;反应完全后,粗产品经柱色谱分离纯化得化合物Ⅳ-r。黄色固体;52.9mg,收率为80%;>20:1dr,99%ee;[α]
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
- 手性苯并芳杂环并二氢吡喃酮类化合物及其制备方法
- 具有视黄酸衍生物类生物活性的四氢萘、苯并二氢吡喃、二氢苯并噻喃和1,2,3,4-四氢喹啉羧酸的芳基或杂芳基酰胺