一种紫磷基光催化材料及其制备方法与应用
文献发布时间:2023-06-19 18:37:28
技术领域
本发明涉及纳米复合材料技术领域,尤其涉及一种紫磷基光催化材料及其制备方法与应用。
背景技术
氢气(H
目前,基于紫外光响应的无机半导体光催化剂得到了广泛的研究;然而,只有不到5%的太阳能存在于紫外光谱中。因此,在设计2D/2D范德华异质结光催化剂中,探索宽光谱响应的2D窄带半导体材料可进一步利用太阳能资源,在高效电荷分离的基础上,实现多策略协同作用,进而提高光催化转化效率。
近来,以黑磷(BP)为代表的二维烯材料因其层状结构以及带隙可调(0.3-2.0eV)等优异性能,成为一种具有宽光谱吸收的高效光催化材料,并且在构建2D/2D范德华异质结光催化领域有广泛的应用。然而,BP基材料在光催化体系中的稳定性制限制了其更广泛的应用,这主要源于O
发明内容
鉴于上述现有技术的不足,本发明的目的在于提供一种紫磷基光催化材料及其制备方法与应用,旨在解决现有黑磷基光催化材料稳定性差的问题。
一种紫磷基光催化材料的制备方法,其中,包括如下步骤:
将紫磷纳米材料与二维材料分散在有机溶剂中,超声处理,抽滤,洗涤干,燥后,得到紫磷基光催化材料。
可选地,所述的紫磷基光催化材料的制备方法,其中,所述紫磷纳米材料的制备,包括:
将紫磷块体放入研钵中研磨,得到紫磷粉料;
将所述紫磷粉料用所述有机溶剂转移至超声设备中,探头超声处理,离心筛分,得到紫磷纳米材料。
可选地,所述的紫磷基光催化材料的制备方法,其中,所述二维材料选自黑磷、g-C
可选地,所述的紫磷基光催化材料的制备方法,其中,所述有机溶剂选自N-甲基吡咯烷酮、N,N-二甲基甲酰胺、1,3-二甲基-2-咪唑啉酮、二甲基亚砜、异丙醇、仲丁醇、异丙胺、甲醇、乙醇和异丙醇中的一种或多种。
可选地,所述的紫磷基光催化材料的制备方法,其中,所述紫磷纳米材料与所述二维材料的摩尔比为0.01:1~100:1。
可选地,所述的紫磷基光催化材料的制备方法,其中,所述紫磷纳米材料的尺寸为1nm~5μm,厚度为0.5~500nm。
可选地,所述的紫磷基光催化材料的制备方法,其中,所述将紫磷块体放入研钵中研磨,得到紫磷粉料的步骤,具体包括:
将紫磷块体放入研钵中,加入一定量的所述有机溶剂,研磨得到紫磷粉料;其中,所述紫磷块体与所述有机溶剂的质量比为0.1:1~1:50。
可选地,所述的紫磷基光催化材料的制备方法,其中,所述探头超声处理,其中探头超声的功率为5W~100W,超声时间为0.1~10h。
一种紫磷基光催化材料,其中,所述紫磷基光催化材料采用上述所述的制备方法制备得到。
一种上述所述的紫磷基光催化材料,在光催化分解水制氢中的应用。
有益效果:与现有技术相比,本发明通过将紫磷构建成2D/2D复合材料,从材料组成上是首创;紫磷和其他二维材料在溶剂超声辅助条件下,可以通过相互吸引自组装,形成较紧密的复合结构,这也使得制备得到的光催化材料有较好的催化活性和较高的稳定性。
附图说明
图1为本发明实施例提供的紫磷基光催化材料合成示意图;
图2为本发明实施例提供的紫磷基光催化材料的扫描电镜图;
图3为本发明实施例提供的紫磷基光催化材料的Tauc图谱(用以计算材料的禁带宽度,确定材料的光响应能力);
图4为本发明实施例提供的紫磷基光催化材料的X射线光电子能谱全谱图(可以表明材料的表面同时具有C,N,P三种元素,意味着有较好的相互接触);
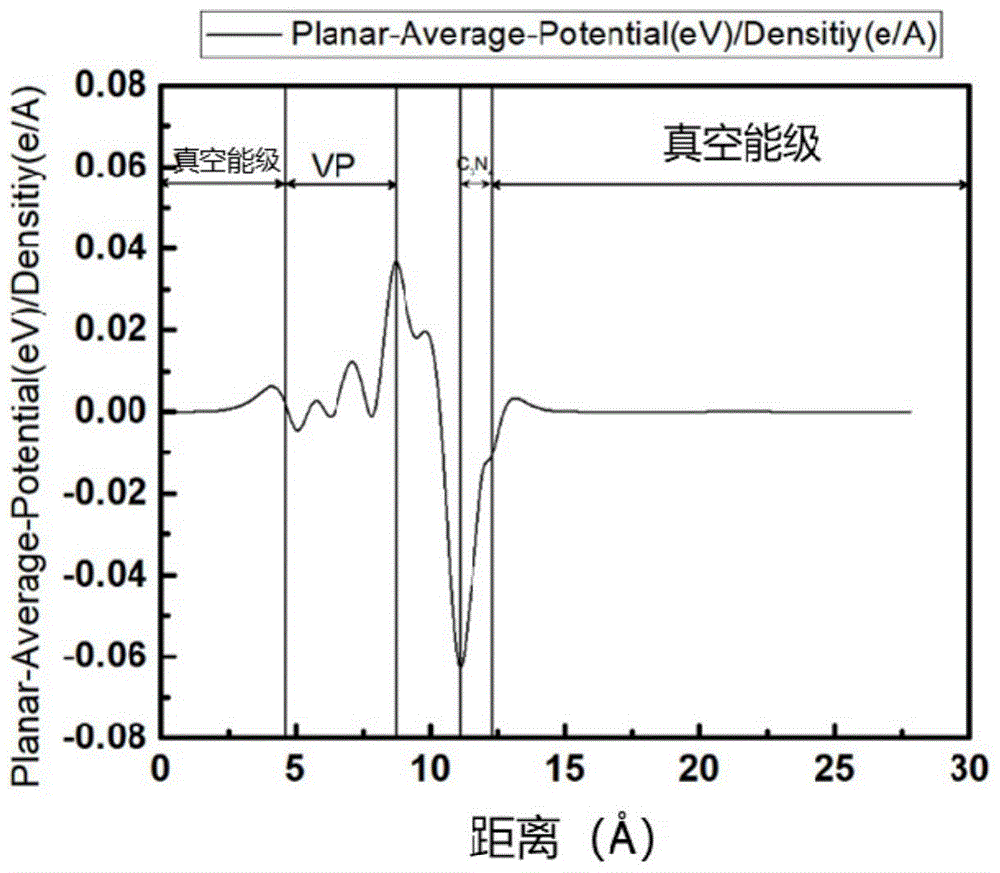
图5为本发明实施例提供的紫磷基光催化材料的面积分差分电荷密度(0以上代表正电荷聚集,下面部分代表负电荷聚集,界面处有大量的正负电荷聚集,有较强的相互作用)。
具体实施方式
本发明提供一种紫磷基光催化材料及其制备方法与应用,为使本发明的目的、技术方案及效果更加清楚、明确,以下对本发明进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
实施例1
(1)块状紫磷合成
将470mg红磷(阿拉丁,99.999%)与10mg的锡(Alfa Aesar,99.995%)以及18mg碘化锡(Alfa Aesar,99.998%)均匀混合,然后将混合物密封在一个14厘米长的石英管中(内径为10毫米,厚度2毫米),内部保持10
管式炉内未放置石英管的一端温度保持在450℃,保温6个小时,反应物部分保持室温,防止反应物转移回来。石英管内分为两个区域,一个区域是有反应物红磷,另外一个区域为产物回收区(这里称为空置区)。首先,将石英管升温,在8个小时的时间,同时将反应物一端温度升至650°C,而空置区域升温至530℃,并在此条件下保持5小时。然后,石英管两个区域分别在10小时时间降温到550℃和630℃,并保持30小时。最后,石英管内不同区域的温度都降到室温,需要100小时。将没有反应的原料洗涤清理,回收合成好的紫磷待进一步使用,紫磷的收益率为36%。
(2)紫磷纳米材料的尺寸调控
结合图1,称量适当质量的紫磷块体(200mg),放入玛瑙研钵中,滴入2-3滴N-甲基吡咯烷酮(NMP)溶剂,提高材料在研磨过程中的润滑性。研磨时间在20分钟。然后,将研磨好的紫磷材料用NMP转移到超声容器中,通过探头超声进一步剥离。超声时间为1小时,功率为20w,容器外为5℃冷井,防止温度过高对材料的影响。超声采用工作8秒,暂停2秒的程序工作。最后,将超声处理的紫磷材料通过离心的方法筛选分离,分别采用500转/分钟条件离心20分钟,得到尺寸为片径200-500nm,层数为200-800的紫磷纳米材料。紫磷纳米材料用乙醇和超纯水清洗后真空干燥过夜后回收备用。
实施例2
(1)块状紫磷的合成采用实施例1中的方法制备得到。
(2)紫磷纳米材料的尺寸调控
称量适当质量的紫磷块体(200mg),放入玛瑙研钵中,滴入2-3滴N-甲基吡咯烷酮(NMP)溶剂,提高材料在研磨过程中的润滑性。研磨时间在20分钟。然后,将研磨好的紫磷材料用NMP转移到超声容器中,通过探头超声进一步剥离。超声时间为1小时,功率为40w,容器外为5℃冷井,防止温度过高对材料的影响。超声采用工作8秒,暂停2秒的程序工作。最后,将超声处理的紫磷材料通过离心的方法筛选分离,分别采用1000转/分钟条件离心20分钟,得到尺寸为片径100-200nm,层数为50-200的紫磷纳米材料。紫磷纳米材料用乙醇和超纯水清洗后真空干燥过夜后回收备用。
实施例3
(1)块状紫磷的合成采用实施例1中的方法制备得到。
(2)紫磷纳米材料的尺寸调控
称量适当质量的紫磷块体(200mg),放入玛瑙研钵中,滴入2-3滴N-甲基吡咯烷酮(NMP)溶剂,提高材料在研磨过程中的润滑性。研磨时间在20分钟。然后,将研磨好的紫磷材料用NMP转移到超声容器中,通过探头超声进一步剥离。超声时间为1小时,功率为60w,容器外为5℃冷井,防止温度过高对材料的影响。超声采用工作8秒,暂停2秒的程序工作。最后,将超声处理的紫磷材料通过离心的方法筛选分离,分别采用2000转/分钟条件离心20分钟,得到尺寸为片径50-100nm,层数为10-50的紫磷纳米材料。紫磷纳米材料用乙醇和超纯水清洗后真空干燥过夜后回收备用。
实施例4
(1)块状紫磷的合成采用实施例1中的方法制备得到。
(2)紫磷纳米材料的尺寸调控
称量适当质量的紫磷块体(200mg),放入玛瑙研钵中,滴入2-3滴N-甲基吡咯烷酮(NMP)溶剂,提高材料在研磨过程中的润滑性。研磨时间在20分钟。然后,将研磨好的紫磷材料用NMP转移到超声容器中,通过探头超声进一步剥离。超声时间为1小时,功率为80w,容器外为5℃冷井,防止温度过高对材料的影响。超声采用工作8秒,暂停2秒的程序工作。最后,将超声处理的紫磷材料通过离心的方法筛选分离,分别采用5000转/分钟条件离心20分钟,得到尺寸为片径10-50nm,层数为5-10的紫磷纳米材料。紫磷纳米材料用乙醇和超纯水清洗后真空干燥过夜后回收备用。
实施例5
(1)块状紫磷的合成采用实施例1中的方法制备得到。
(2)紫磷纳米材料的尺寸调控
称量适当质量的紫磷块体(200mg),放入玛瑙研钵中,滴入2-3滴N-甲基吡咯烷酮(NMP)溶剂,提高材料在研磨过程中的润滑性。研磨时间在20分钟。然后,将研磨好的紫磷材料用NMP转移到超声容器中,通过探头超声进一步剥离。超声时间为1小时,功率为100w,容器外为5℃冷井,防止温度过高对材料的影响。超声采用工作8秒,暂停2秒的程序工作。最后,将超声处理的紫磷材料通过离心的方法筛选分离,分别采用10000转/分钟条件离心20分钟,得到尺寸为片径2-10nm,层数为1-5的紫磷纳米材料。紫磷纳米材料用乙醇和超纯水清洗后真空干燥过夜后回收备用。
实施例6
紫磷基复合光催化材料的制备,制备过程如图1所示。
称取10mg的上述实施例5制备得到的紫磷纳米材料,20mg的g-C
如图3所示,图3为VP,CN和VP/CN复合材料的Tauc图谱,通过切线的计算,可以得到材料的禁带宽度数值,表明材料对光的响应能力。特备是在VP与CN复合后,材料的光响应能力有一个明显的改变。
如图4所示,图4为VP,CN和VP/CN复合材料的X射线光电能谱图全图,可以看到不同材料表面元素的组成,特别是,复合后,VP/CN的表面同时具有N1s和P 2p的峰,说明两者较好的复合在一起。而且,在对C1s峰进行校准后,可以看到VP/CN的N1s和P 2p的峰对比VP都有位移,说明他们之间有较强的相互作用,而这也是材料性能优越的基础和原因之一。
如图5所示,图5为VP与CN复合材料的密度泛函数,可以分别看到VP和CN的真空能级,材料的电负性,VP在界面处带正电,CN带正电,两者结合处有较强的相互作用,同时通过模拟计算,以及复合材料接触后,费米能级匹配的原理,电荷会从CN向VP转移,实现光生电荷的高效分离和利用。
实施例7
紫磷基复合光催化材料的制备
称取10mg的上述实施例6制备得到的紫磷纳米材料,20mg的石墨烯,然后将他们同时放入约50毫升NMP中进行超声分散,反应条件设定为4小时,工作6秒,暂停4秒的工作程序,同时外面为5℃冷井。然后,将混合物抽滤、洗涤,在真空干燥过夜后回收,准备对其光催化性能进行评价。
实施例8
紫磷基复合光催化材料的制备
称取10mg的上述实施例6制备得到的紫磷纳米材料,20mg的硫化镉,然后将他们同时放入约50毫升NMP中进行超声分散,反应条件设定为4小时,工作6秒,暂停4秒的工作程序,同时外面为5℃冷井。然后,将混合物抽滤、洗涤,在真空干燥过夜后回收,准备对其光催化性能进行评价。
实施例9
紫磷基复合光催化材料的制备
称取10mg的上述实施例6制备得到的紫磷纳米材料,20mg的二硫化钼,然后将他们同时放入约50毫升NMP中进行超声分散,反应条件设定为4小时,工作6秒,暂停4秒的工作程序,同时外面为5℃冷井。然后,将混合物抽滤、洗涤,在真空干燥过夜后回收,准备对其光催化性能进行评价。
实施例10
紫磷基复合光催化材料的制备
称取10mg的上述实施例6制备得到的紫磷纳米材料,20mg的黑磷,然后将他们同时放入约50毫升NMP中进行超声分散,反应条件设定为4小时,工作6秒,暂停4秒的工作程序,同时外面为5℃冷井。然后,将混合物抽滤、洗涤,在真空干燥过夜后回收,准备对其光催化性能进行评价。
实施例11
紫磷基复合光催化材料的制备
称取10mg的上述实施例6制备得到的紫磷纳米材料,20mg的氮化硼,然后将他们同时放入约50毫升NMP中进行超声分散,反应条件设定为4小时,工作6秒,暂停4秒的工作程序,同时外面为5℃冷井。然后,将混合物抽滤、洗涤,在真空干燥过夜后回收,准备对其光催化性能进行评价。
对上述实施例中制备得到的VP材料、紫磷基复合光催化材料水分解性能进行分析。
光催化反应通过泊菲莱Labsolar-A6在线光催化反应设备进行评价,光源为300W泊菲莱PLS-SXE300D氙灯,通过顶部照射的方法进行反应。反应器为200毫升玻璃容器,上面通过石英玻璃密封。
具体来光催化测试步骤为:称取50mg样品分散于120ml体积分数为20%的甲醇水溶液中,然后通过涂抹真空脂将反应器密封好,然后连接到反应系统中。对系统抽真空,在5分钟内没有压力变化的情况下,准备开始光催化测试。反应期间,使用冷却水系统将反应物溶液保持在10℃左右。通过系统设置,每30分钟对反应器内产物组分进行分析,测试仪器为福立9790II气相色谱(配备有分子筛
光催化实验结果如下表所示:
表1不同层数VP构建复合光催化活性
表2不同比例VP/g-C
表3VP与不同二维材料复合(VP选用片径10-50nm,层数为5-10的紫磷纳米材料)
由上述实验结果可以看出,通过将紫磷纳米材料与二维材料进行复合,所得到的催化剂无论对析氢活性还是在CO
综上,本发明提供一种紫磷基光催化材料及其制备方法与应用,方法包括:将紫磷纳米材料与二维材料分散在有机溶剂中,超声处理,抽滤,洗涤干,燥后,得到紫磷基光催化材料。本发明通过将紫磷构建成2D/2D复合材料,从材料组成上是首创;紫磷和其他二维材料在溶剂超声辅助条件下,可以通过相互吸引自组装,形成较紧密的复合结构,这也使得制备得到的光催化材料有较好的催化活性和较高的稳定性。
应当理解的是,本发明的应用不限于上述的举例,对本领域普通技术人员来说,可以根据上述说明加以改进或变换,所有这些改进和变换都应属于本发明所附权利要求的保护范围。
- 一种二维碳氮基复合材料光催化剂及其制备方法、应用
- 一种污泥基除磷材料及其制备方法和含磷污水的处理方法
- 一种TiO2基/二维材料纳米复合光催化纤维膜及其制备方法
- 一种钙钛矿基复合纳米光催化材料及制备方法与用途
- 一种磷钼酸构筑多酸基镉金属杂化材料制备及光催化应用
- 一种磷钼酸构筑多酸基镉金属杂化材料制备及光催化应用