一类具有近红外吸收的硼杂分子碳化合物及其制备方法与应用
文献发布时间:2023-06-19 19:28:50
技术领域
本发明属于近红外吸收材料技术领域,具体涉及一类具有近红外吸收的硼杂分子碳化合物及其制备方法与应用,尤其涉及该具有近红外吸收的硼杂分子碳化合物在制备具有近红外吸收的有机功能材料、双极性有机场效应晶体管材料、有机自旋电子器件材料中的应用。
背景技术
近红外光(Near Infrared,NIR)是介于可见光(ⅥS)和中红外光(MIR)之间,定义在波长780~2526nm范围内的电磁波。近红外区域是人们发现的最早的非可见光区域。近年来,近红外材料因其具有优异的光学、电学、磁学等性能,在激光屏蔽阻断、光电器件、医学治疗等领域展现出广泛的应用前景。
近红外材料通常是指能与近红外光存在吸收或反射,或在外界刺激下如光激发、电场作用等发出近红外光的一类物质。按作用机理可以分为近红外吸收材料和近红外发光材料,其中近红外吸收材料按组成成分可以分为无机材料和有机材料。在近红外有机材料中,如菁类染料、金属络合物染料、醌型染料和偶氮染料等是其主要的分子体系,它们具有良好的生物相容性和光吸收性能等特点,有望在生物医学、光电探测器等领域实现商业化应用。对于这些有机材料,主要通过改变酞菁类化合物侧链取代基、金属类络合物中心金属及在偶氮类化合物骨架上的给体或受体基团等,调控分子的吸收波长到近红外区域,实现优化近红外光吸收性能。目前,具有强近红外吸收能力的有机分子材料的种类仍然较少,限制了近红外有机材料的发展和应用。
硼原子具有空p轨道,在基于sp
发明内容
有鉴于此,为解决具有近红外吸收且稳定的含硼有机材料的设计困难、合成难度大等问题,本发明提供了一类具有近红外吸收的硼杂分子碳化合物及其制备方法与应用,该类具有近红外吸收的硼杂分子碳化合物为含有三配位硼结构单元或采用含三配位硼结构单元构筑的大尺寸有机硼稠环结构化合物。
本发明解决上述技术问题采取的技术方案如下。
本发明提供了一类具有近红外吸收的硼杂分子碳化合物,结构式为以下结构中的一种:
本发明还提供结构式为1的具有近红外吸收的硼杂分子碳化合物的制备方法,包括以下步骤:
步骤一、惰性气氛下,将反应物1c和三溴化硼反应,反应结束后,减压下除去剩余的三溴化硼,得到化合物1d;
所述化合物1c的结构式为:
步骤二、惰性气氛下,将10-溴-1,8-双(均三甲基苯氧基)蒽和正丁基锂于溶剂中反应,反应结束后,减压下除去溶剂,得到的黄色固体和化合物1d于溶剂中反应,反应结束后,减压除去溶剂,粗品依次用水、甲醇、正己烷洗涤,得棕黄色固体;将棕黄色固体和三氯化铁于溶剂中反应,反应结束后,除去溶剂,提纯,得到紫色固体粉末;
步骤三、惰性气氛下,将紫色固体粉末和N-溴代丁二酰亚胺于溶剂中反应,反应结束后,萃取,干燥,除去溶剂,提纯,得到紫黑色固体粉末,即结构式为1的具有近红外吸收的硼杂分子碳化合物。
本发明还提供结构式为2的具有近红外吸收的硼杂分子碳化合物的制备方法,包括以下步骤:
步骤一、惰性气氛下,将反应物1c和三溴化硼于溶剂中反应,反应结束后,减压下除去剩余的三溴化硼,得到化合物1d;
所述化合物1c的结构式为:
步骤二、惰性气氛下,将化合物1d和2,4,6-三异丙基苯溴化镁于溶剂中反应,反应结束后,减压除去溶剂,提纯,得黄绿色粉末;
步骤三、惰性气氛下,将黄绿色粉末和N-溴代丁二酰亚胺于溶剂中反应,反应结束后,萃取,干燥,除去溶剂,提纯,得到黄绿色粉末,即为结构式为2的具有近红外吸收的硼杂分子碳化合物。
本发明还提供结构式为3的具有近红外吸收的硼杂分子碳化合物的制备方法,包括以下步骤:
步骤一、惰性气氛下,将反应物3b和三溴化硼于溶剂中反应,反应结束后,减压下除去剩余的三溴化硼,得到化合物3c;
所述化合物3b的结构式如下:
步骤二、惰性气氛下,将10-溴-1,8-双(均三甲基苯氧基)蒽和正丁基锂于溶剂中反应,反应结束后,减压下除去溶剂,得到的黄色固体和化合物3c于溶剂中反应,反应结束后,减压除去溶剂,粗品依次用水、甲醇、正己烷洗涤,得棕黄色固体;将棕黄色固体和三氯化铁于溶剂中反应,反应结束后,除去溶剂,提纯,得到紫色固体粉末,即为结构式为3的具有近红外吸收的硼杂分子碳化合物。
本发明还提供结构式为4-10或12的具有近红外吸收的硼杂分子碳化合物的制备方法,包括以下步骤:
步骤一、惰性气氛下,将含有三配位硼结构单元、偶联单体、四三苯基膦钯、2-双环己基膦-2',6'-二甲氧基-1,1'-二联苯和磷酸钾于溶剂中进行偶联反应,反应结束后,萃取,干燥,除去溶剂,提纯,得到第一中间产物;
所述含有三配位硼结构单元为结构式为1-3的具有近红外吸收的硼杂分子碳化合物中的任意一个;
所述偶联单体为以下结构中的一种:
步骤二、惰性气氛下,将第一中间产物和均三甲基苯基溴化镁于溶剂中反应,反应结束后,萃取,干燥,得到的固体和三氟化硼乙醚于溶剂中反应,反应结束后,除去溶剂,得到第二中间产物;将第二中间产物和2,3-二氯-5,6-二氰基-1,4-苯醌于溶剂中反应,反应结束后,除去溶剂,提纯,得到具有近红外吸收的硼杂分子碳化合物;
或者,惰性气氛下,将第一中间产物、2,3-二氯-5,6-二氰基-1,4-苯醌和三氟甲磺酸于溶剂中反应,反应结束后,除去溶剂,提纯,得到具有近红外吸收的硼杂分子碳化合物;
或者,惰性气氛下,将第一中间产物和三氯化铁于溶剂中反应,反应结束后,除去溶剂,提纯,得到具有近红外吸收的硼杂分子碳化合物。
本发明还提供结构式为11或13的具有近红外吸收的硼杂分子碳化合物的制备方法,包括以下步骤:
步骤一、惰性气氛下,将反应单体、偶联单体、四三苯基膦钯、2-双环己基膦-2',6'-二甲氧基-1,1'-二联苯和磷酸钾于溶剂中进行偶联反应,反应结束后,萃取,干燥,除去溶剂,提纯,得到第一中间产物;
所述反应单体的结构式如下:
所述偶联单体为以下结构中的一种:
步骤二、惰性气氛下,将第一中间产物和三氯化铁于溶剂中反应,除去溶剂,提纯,得到具有近红外吸收的硼杂分子碳化合物。
本发明还提供上述具有近红外吸收的硼杂分子碳化合物在制备具有近红外吸收的有机功能材料中的应用。
本发明还提供上述具有近红外吸收的硼杂分子碳化合物在制备双极性有机场效应晶体管材料中的应用。
本发明还提供上述具有近红外吸收的硼杂分子碳化合物在制备有机自旋电子器件材料中的应用。
与现有技术相比,本发明的有益效果为:
1、本发明的具有近红外吸收的硼杂分子碳化合物(1-3),在空气中具有良好的水氧稳定性,对多种有机反应具有高容忍度,有助于进行结构拓展分子的设计与制备。
2、本发明的具有近红外吸收的硼杂分子碳化合物(4-13),经具有近红外吸收的硼杂分子碳化合物(1-3)扩展;分子的自旋密度主要分布在分子骨架上,而均三甲基苯基可以作为空间位阻进行保护较大自旋密度的碳原子,增加开壳化合物的稳定性;分子的大尺寸碳骨架能够增加分子的共轭程度,使分子带隙变小,实现调节分子的电子结构和能级带隙等;硼原子的引入既可以稳定自由基,也可以共轭骨架的电子分布、能级结构等;硼原子嵌入到分子骨架内,或在边缘时采用三异丙基苯基保护,能够提高硼原子的稳定性;均三甲基苯基或三异丙基苯基或烷基链的引入,显著提升了分子的溶解度。
3、本发明的具有近红外吸收的硼杂分子碳化合物(1-13),在多种有机溶剂中均具有良好的溶解度,如在二氯甲烷、四氢呋喃、甲苯溶剂中的溶解度均大于2mg mL
4、本发明的具有近红外吸收的硼杂分子碳化合物(1-13),合成方法简单,产率较高,提纯工艺便捷,有利于大量合成。
5、本发明的具有近红外吸收的硼杂分子碳化合物(1-13),具有近红外吸收性质,光吸收范围、光吸收能力和能级带隙可通过共轭结构进行调控,有望面向不同的近红外材料的应用需求进行订制设计,有发展成新型近红外有机材料的潜力。
附图说明
为了更清楚地说明本发明实施例中的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其它的附图。
图1为本发明实施例4的具有近红外吸收的硼杂分子碳化合物的紫外可见近红外吸收光谱;
图2为本发明实施例10的具有近红外吸收的硼杂分子碳化合物的紫外可见近红外吸收光谱;
图3为本发明实施例11的具有近红外吸收的硼杂分子碳化合物的紫外可见近红外吸收光谱;
图4为本发明实施例11的具有近红外吸收的硼杂分子碳化合物的电化学测试循环伏安曲线;
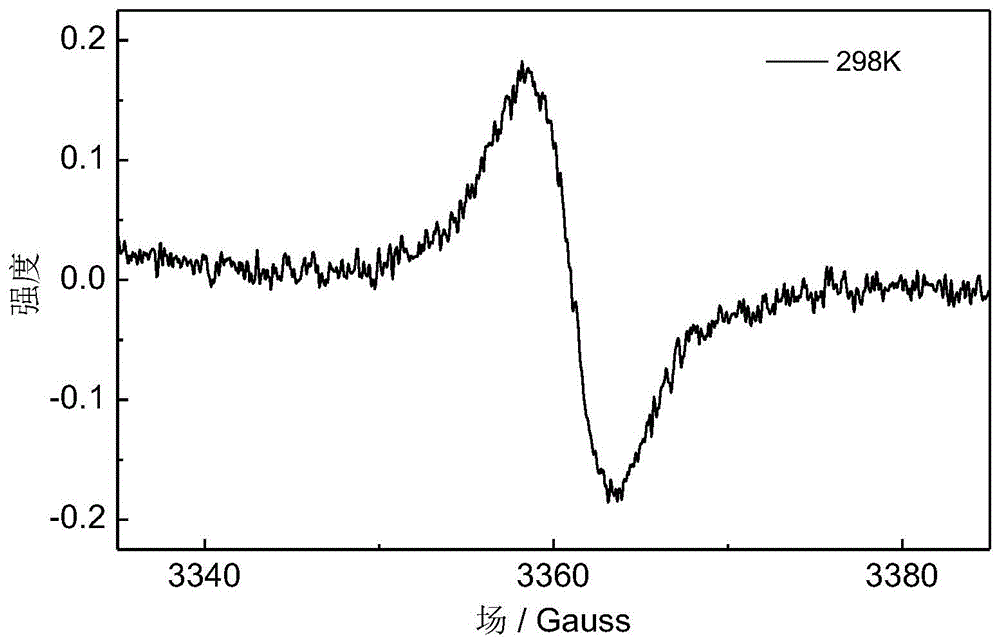
图5为本发明实施例4的具有近红外吸收的硼杂分子碳化合物的电子自旋共振测试图谱。
具体实施方式
为了进一步理解本发明,下面对本发明优选实施方案进行描述,但是应当理解,这些描述只是为进一步说明本发明的特征和优点,而不是对本发明权利要求的限制。
本发明的具有近红外吸收的硼杂分子碳化合物,结构式为以下结构中的一种:
上述技术方案中,1-3为含有三配位硼结构单元,4-13为采用含三配位硼结构单元(1-3)构筑的大尺寸有机硼稠环结构化合物。其中,分子的自旋密度主要分布在分子骨架上,而均三甲基苯基可以作为空间位阻进行保护较大自旋密度的碳原子,增加开壳化合物的稳定性;分子的大尺寸碳骨架能够增加分子的共轭程度,使分子带隙变小,实现调节分子的电子结构和能级带隙等;硼原子的引入既可以稳定自由基,也可以共轭骨架的电子分布、能级结构等;硼原子嵌入到分子骨架内,或在边缘时采用三异丙基苯基保护,能够提高硼原子的稳定性;均三甲基苯基或三异丙基苯基或烷基链的引入,显著提升了分子的溶解度。故而,本发明的具有近红外吸收的硼杂分子碳化合物可以通过调节硼杂分子碳化合物的开壳程度,实现材料的近红外光吸收能力;还可以通过调节硼杂分子碳化合物的共轭尺寸和共轭程度,实现材料的近红外光吸收能力。
本发明的具有近红外吸收的硼杂分子碳化合物,结构式为1-3,即含有三配位硼结构单元时,合成路线如下:
本发明的结构式为4-10或12的具有近红外吸收的硼杂分子碳化合物的制备方法,包括以下步骤:
步骤一、惰性气氛下,将含有三配位硼结构单元、偶联单体、四三苯基膦钯、2-双环己基膦-2',6'-二甲氧基-1,1'-二联苯和磷酸钾于溶剂中进行偶联反应,反应结束后,萃取,干燥,除去溶剂,提纯,得到第一中间产物;
其中,偶联单体为以下结构中的一种:
步骤二、惰性气氛下,将第一中间产物和均三甲基苯基溴化镁于溶剂中反应,反应结束后,萃取,干燥,得到的固体和三氟化硼乙醚于溶剂中反应,反应结束后,除去溶剂,得到第二中间产物;将第二中间产物和2,3-二氯-5,6-二氰基-1,4-苯醌于溶剂中反应,反应结束后,除去溶剂,提纯,得到具有近红外吸收的硼杂分子碳化合物;
或者,惰性气氛下,将第一中间产物、2,3-二氯-5,6-二氰基-1,4-苯醌和三氟甲磺酸于溶剂中反应,反应结束后,除去溶剂,提纯,得到具有近红外吸收的硼杂分子碳化合物;
或者,惰性气氛下,将第一中间产物和三氯化铁于溶剂中反应,反应结束后,除去溶剂,提纯,得到具有近红外吸收的硼杂分子碳化合物。
本发明的结构式为11或13的具有近红外吸收的硼杂分子碳化合物的制备方法,包括以下步骤:
步骤一、惰性气氛下,将反应单体、偶联单体、四三苯基膦钯、2-双环己基膦-2',6'-二甲氧基-1,1'-二联苯和磷酸钾于溶剂中进行偶联反应,反应结束后,萃取,干燥,除去溶剂,提纯,得到第一中间产物;
其中,反应单体的结构式如下:
其中,偶联单体为以下结构中的一种:
步骤二、惰性气氛下,将第一中间产物和三氯化铁于溶剂中反应,除去溶剂,提纯,得到具有近红外吸收的硼杂分子碳化合物。
上述技术方案中,结构式为1-13具有近红外吸收的硼杂分子碳化合物的制备过程中,溶剂没有特殊限制,为能够溶解相应反应原料的溶剂即可。反应原料配比本领域技术人员根据待制备具有近红外吸收的硼杂分子碳化合物的结构式能够确定。惰性气氛通常为氮气,需要说明的是,本领域技术人员公知的其他惰性气氛也可。
本发明还提供上述具有近红外吸收的硼杂分子碳化合物在制备具有近红外吸收的有机功能材料中的应用。
本发明还提供上述具有近红外吸收的硼杂分子碳化合物在制备双极性有机场效应晶体管材料中的应用。
本发明还提供上述具有近红外吸收的硼杂分子碳化合物在制备有机自旋电子器件材料中的应用。
在本发明中所使用的术语,一般具有本领域普通技术人员通常理解的含义,除非另有说明。为了使本领域的技术人员更好地理解本发明的技术方案,下面将结合实施例对本发明作进一步的详细介绍。
在以下实施例中,未详细描述的各种过程和方法是本领域中公知的常规方法。下述实施例中所用的材料、试剂、装置、仪器、设备等,如无特殊说明,均可从商业途径获得。
以下结合实施例进一步说明本发明。
实施例1化合物1的合成
步骤一、将化合物1a(1.0g,2.99mmol),1,8-二溴萘(941mg,3.29mmol),四(三苯基膦)钯(346mg,0.30mmol),碳酸钾(4.14g,29.91mmol)投入干燥的带有冷凝管的反应瓶中,进行三次抽真空、通氩气的抽换气操作后,依次注入通气除氧的四氢呋喃(20mL),通气除氧的水(4mL),密封体系随后升温至90℃反应2h。反应结束后,将反应混合物投入水中,用二氯甲烷萃取水相三次,所得的二氯甲烷相水洗一次后合并有机相用无水硫酸钠干燥。减压除去二氯甲烷后,粗品采用200-300目硅胶作为固定相,石油醚作为洗脱剂进行柱层析分离,得到白色固体,记作化合物1b。产量831mg,产率56%。
质谱分析确定的分子离子(C
步骤二、将化合物1b(445mg,0.90mmol)投入严格烘烤的反应瓶中,进行三次抽真空、通氩气的抽换气操作后,溶于20mL干燥的乙醚中。反应体系降温至0℃稳定片刻后,逐滴滴加正丁基锂溶液(1.6M,1.1ml,1.80mmol)。在0℃条件下反应2h后,逐滴滴加二氯二甲基硅烷(0.10mL,1.15mmol),反应逐渐升至25℃下反应6h。反应结束后,加入3ml饱和氯化铵溶液搅拌30min,随后将反应混合物投入水中,用乙酸乙酯萃取水相三次,所得的乙酸乙酯相水洗一次后合并有机相后用无水硫酸钠干燥。减压除去乙酸乙酯后,粗品采用200-300目硅胶作为固定相,石油醚作为洗脱剂进行柱层析分离,得到淡黄色固体,记作化合物1c。产量198mg,产率56%。
质谱分析确定的分子离子(C
步骤三、将化合物1c(106mg,0.27mmol)投入严格烘烤的带有冷凝管的反应瓶中,进行三次抽真空、通氩气的抽换气操作后,加入三溴化硼(0.86ml,8.24mmol),随后25℃反应8h后减压下除去剩余的三溴化硼,得到化合物1d,将其溶解分散于5ml干燥的甲苯溶液中并将体系温度降至0℃。
将10-溴-1,8-双(均三甲基苯氧基)蒽(159mg,0.30mmol)投入严格烘烤的反应瓶中,进行三次抽真空、通氩气的抽换气操作后,溶于8mL干燥的乙醚中。反应体系降温至0℃稳定片刻后,逐滴滴加正丁基锂溶液(1.6M,0.18ml,0.30mmol),25℃反应30min。反应结束后减压下除去乙醚溶剂,得黄色固体,溶解分散于5ml干燥的甲苯溶液并将其逐滴滴加至1d的0℃甲苯溶液中,反应30min后升至25℃下反应12h。反应结束后,减压下除去甲苯溶液,粗品依次用水、甲醇、正己烷洗涤,得棕黄色固体(108mg)。
将所得棕黄色固体投入严格烘烤的反应瓶中,进行三次抽真空、通氩气的抽换气操作后,溶于35mL干燥的二氯甲烷中。反应体系降温至0℃稳定片刻后,将三氯化铁(176mg,1.10mmol)溶于3ml硝基甲烷后逐滴加入反应体系,0℃条件下反应30min。随后加入3ml甲醇淬灭,终止反应。减压下除去溶剂,粗品采用200-300目硅胶作为固定相,二氯甲烷比石油醚=1:5(体积比)作为洗脱剂进行柱层析分离,得到紫色固体粉末,记作化合物1e。产量41mg,产率38%。
质谱分析确定的分子离子(C
步骤四、将化合物1e(156mg,0.20mmol),N-溴代丁二酰亚胺(42mg,0.24mmol)投入带有冷凝管的反应瓶中,进行三次抽真空、通氩气的抽换气操作后,依次注入二氯甲烷(25mL),冰醋酸(5mL),密封体系随后升温50℃反应2h。反应结束后,将反应混合物投入水中,用二氯甲烷萃取水相三次,所得的二氯甲烷相水洗一次后合并有机相用无水硫酸钠干燥。减压除去二氯甲烷后,粗品采用200-300目硅胶作为固定相,二氯甲烷比石油醚=1:5(体积比)作为洗脱剂进行柱层析分离,得到紫黑色固体粉末即为化合物1。产量122mg,产率70%。
质谱分析确定的分子离子(C
实施例2化合物2的合成
步骤一、将实施例1中的化合物1c(106mg,0.27mmol)投入严格烘烤的带有冷凝管的反应瓶中,进行三次抽真空、通氩气的抽换气操作后,加入三溴化硼(0.86ml,8.24mmol),随后25℃反应8h后减压下除去剩余的三溴化硼,得到化合物1d,将其溶解分散于5ml干燥的甲苯溶液中。反应体系降至0℃稳定片刻后,逐滴滴加2,4,6-三异丙基苯溴化镁(0.4M,1.37ml,0.55mmol),反应30min后升至25℃反应2h。反应结束后减压下除去甲苯溶剂,粗品采用200-300目硅胶作为固定相,石油醚作为洗脱剂进行柱层析分离,得黄绿色粉末,记作化合物2a。产量68mg,产率45%。
质谱分析确定的分子离子(C
步骤二、将化合物2a(300mg,0.55mmol),N-溴代丁二酰亚胺(116mg,0.65mmol)投入带有冷凝管的反应瓶中,进行三次抽真空、通氩气的抽换气操作后,依次注入二氯甲烷(30mL),冰醋酸(5mL),密封体系随后升温50℃反应12h。反应结束后,将反应混合物投入水中,用二氯甲烷萃取水相三次,所得的二氯甲烷相水洗一次后合并有机相用无水硫酸钠干燥。减压除去二氯甲烷后,粗品采用200-300目硅胶作为固定相,石油醚作为洗脱剂进行柱层析分离,得到黄绿色粉末即为化合物2。产量192mg,产率56%。
质谱分析确定的分子离子(C
实施例3化合物3的合成
步骤一、将化合物3a(1.0g,3.47mmol),2,6-二叔丁基-4-甲基吡啶(1.1g,5.21mmol),1,3,5-三氯苯(5ml)投入干燥的带有冷凝管的反应瓶中,进行三次抽真空、通氩气的抽换气操作后,注入三溴化硼(0.67ml,6.94mmol),密封体系后升温至200℃反应12h。反应结束后,向体系里边滴加3ml甲醇,随后采用200-300目硅胶作为固定相,将乙酸乙酯作为洗脱剂进行柱层析分离,减压下除去有机溶剂。粗品用正己烷洗涤一次,得浅棕褐色固体,记作化合物3b。产量490mg,产率45%。
质谱分析确定的分子离子(C
步骤二、将化合物3b(100mg,0.32mmol)投入严格烘烤的带有冷凝管的反应瓶中,进行三次抽真空、通氩气的抽换气操作后,加入三溴化硼(0.92ml,9.55mmol),随后25℃反应8h后减压下除去剩余的三溴化硼,得到化合物3c,将其溶解分散于5ml干燥的甲苯溶液中并将体系温度降至0℃。
将10-溴-1,8-双(均三甲基苯氧基)蒽(183mg,0.35mmol)投入严格烘烤的反应瓶中,进行三次抽真空、通氩气的抽换气操作后,溶于8mL干燥的乙醚中。反应体系降温至0℃稳定片刻后,逐滴滴加正丁基锂溶液(1.6M,0.22ml,0.35mmol),25℃反应30min。随后减压下除去乙醚溶剂,得黄色固体,溶解分散于5ml干燥的甲苯溶液并将其逐滴滴加至3c的0℃甲苯溶液中,反应30mins后升至25℃下反应12h。反应结束后,减压下除去甲苯溶液,粗产品依次用水、甲醇、正己烷洗涤,得棕黄色固体(118mg)。
步骤三、将所得棕黄色固体投入严格烘烤的反应瓶中,进行三次抽真空、通氩气的抽换气操作后,溶于35mL干燥的二氯甲烷中。反应体系降温至0℃稳定片刻后,将三氯化铁(203mg,1.27mmol)溶于3ml硝基甲烷后逐滴加入反应体系,0℃条件下反应30min。随后加入3ml甲醇淬灭,终止反应。减压下除去溶剂,粗品采用200-300目硅胶作为固定相,二氯甲烷比石油醚=1:5(体积比)作为洗脱剂进行柱层析分离,得到紫色固体粉末即为化合物3。产量43mg,产率37%。
质谱分析确定的分子离子(C
实施例4化合物4的合成
步骤一、将化合物1(66mg,0.08mmol),化合物4a(59mg,0.24mmol),四三苯基膦钯(4.4mg,0.004mmol),2-双环己基膦-2',6'-二甲氧基-1,1'-二联苯(3mg,0.008mmol),磷酸钾(37mg,0.38mmol)投入带有冷凝管的反应瓶中,进行三次抽真空、通氩气的抽换气操作后,依次注入通气除氧的甲苯(12mL),通气除氧的乙醇(3mL),通气除氧的水(3mL)。随后升温至90℃反应2h。反应结束后,将反应混合物投入水中,用乙酸乙酯萃取水相三次,所得的乙酸乙酯相水洗一次后合并有机相用无水硫酸钠干燥。减压除去乙酸乙酯后,粗品采用200-300目硅胶作为固定相,乙酸乙酯:石油醚=1:12(体积比)作为洗脱剂进行柱层析分离,得到紫红色固体,记作化合物4b。产量45mg,产率65%。
质谱分析确定的分子离子(C
步骤二、严格无水无氧条件下,将化合物4b(56mg,0.06mmol)分散在25mL干燥的四氢呋喃中。在25℃下逐滴加入均三甲基苯基溴化镁(1.0M,0.6mL,0.60mmol),反应30min。反应结束后,反应混合物投入水中,用乙酸乙酯萃取水相。乙酸乙酯层干燥、旋干,得到的固体置于干燥的反应瓶中,溶于25mL干燥的二氯甲烷,25℃条件下逐滴加入三氟化硼乙醚(0.07mL,0.6mmol),反应30min后用2ml甲醇淬灭,终止反应。减压下除去反应溶剂,得到紫黑色固体,记作化合物4c。产量41mg,产率60%。
质谱分析确定的分子离子(C
步骤三、严格无水条件下,将化合物4c(20mg,0.02mmol)溶于8mL干燥的甲苯中。将2,3-二氯-5,6-二氰基-1,4-苯醌(8mg,0.04mmol)溶于2mL干燥的甲苯中。将2,3-二氯-5,6-二氰基-1,4-苯醌的甲苯溶液逐滴转移至反应体系,随后25℃反应30min。反应结束后,体系通过中性氧化铝柱层析(洗脱剂为甲苯)。减压下除去甲苯后,粗品采用200-300目硅胶作为固定相,用甲苯:石油醚=1:3(体积比)作为洗脱剂进行柱层析分离,得到黑紫色固体,记作化合物4。产量为10mg,产率50%。
质谱分析确定的分子离子(C
实施例5化合物5的合成
步骤一、将化合物1(66mg,0.08mmol),化合物5a(53mg,0.23mmol),四三苯基膦钯(5mg,0.004mmol),2-双环己基膦-2',6'-二甲氧基-1,1'-二联苯(3mg,0.008mmol),磷酸钾(37mg,0.38mmol)投入带有冷凝管的反应瓶中,进行三次抽真空、通氩气的抽换气操作后,依次注入通气除氧的甲苯(12mL),通气除氧的乙醇(3mL),通气除氧的水(3mL)。随后升温至90℃反应2h。反应结束后,将反应混合物投入水中,用乙酸乙酯萃取水相三次,所得的乙酸乙酯相水洗一次后合并有机相用无水硫酸钠干燥。减压除去乙酸乙酯后,粗品采用200-300目硅胶作为固定相,乙酸乙酯:石油醚=1:15(体积比)作为洗脱剂进行柱层析分离,得到紫红色固体,记作化合物5b。产量39mg,产率58%。
质谱分析确定的分子离子(C
步骤二、无水无氧条件下,将化合物5b(35mg,0.04mmol)分散在25mL干燥的四氢呋喃中。在25℃下逐滴加入均三甲基苯基溴化镁(1.0M,0.4mL,0.40mmol),反应30min。反应结束后,反应混合物投入水中,用乙酸乙酯萃取水相。乙酸乙酯层干燥、旋干,得到的固体置于干燥的反应瓶中,溶于25mL干燥的二氯甲烷,25℃条件下逐滴加入三氟化硼乙醚(0.05mL,0.40mmol),反应30min后用2ml甲醇淬灭,终止反应。反应体系旋干,得到紫黑色固体,记作化合物5c。产量23mg,产率60%。
质谱分析确定的分子离子(C
步骤三、无水条件下,将化合物5c(30mg,0.03mmol)溶于9mL干燥的甲苯中。将2,3-二氯-5,6-二氰基-1,4-苯醌(13mg,0.06mmol)溶于2mL干燥的甲苯中。将2,3-二氯-5,6-二氰基-1,4-苯醌的甲苯溶液逐滴转移至反应体系,随后升温80℃反应5h。反应结束体系恢复至室温后,通过中性氧化铝柱层析(洗脱剂为甲苯)。减压下除去甲苯后,粗品采用200-300目硅胶作为固定相,用二硫化碳作为洗脱剂进行柱层析分离,得到黑紫色固体,记作化合物5。产量为9mg,产率30%。
质谱分析确定的分子离子(C
实施例6化合物6的合成
步骤一、将化合物2(90mg,0.14mmol),化合物4a(111mg,0.42mmol),四三苯基膦钯(8mg,0.007mmol),2-双环己基膦-2',6'-二甲氧基-1,1'-二联苯(10mg,0.014mmol),磷酸钾(99mg,0.72mmol)投入带有冷凝管的反应瓶中,进行三次抽真空、通氩气的抽换气操作后,依次注入通气除氧的甲苯(15mL),通气除氧的乙醇(5mL),通气除氧的水(5mL)。随后升温至90℃反应3h。反应结束后,将反应混合物投入水中,用乙酸乙酯萃取水相三次,所得的乙酸乙酯相水洗一次后合并有机相用无水硫酸钠干燥。减压除去乙酸乙酯后,粗品采用200-300目硅胶作为固定相,乙酸乙酯:石油醚=1:12(体积比)作为洗脱剂进行柱层析分离,得到杏黄色固体,记作化合物6a。产量52mg,产率53%。
质谱分析确定的分子离子(C
步骤二、无水无氧条件下,将化合物6a(42mg,0.06mmol)分散在25mL干燥的四氢呋喃中。在25℃下逐滴加入均三甲基苯基溴化镁(1.0M,0.6mL,0.60mmol),反应30min。反应结束后,反应混合物投入水中,用乙酸乙酯萃取水相。乙酸乙酯层干燥、旋干,得到的固体置于干燥的反应瓶中,溶于25mL干燥的二氯甲烷,25℃条件下逐滴加入三氟化硼乙醚(0.07mL,0.6mmol),反应30min后用2ml甲醇淬灭,终止反应。反应体系旋干,得到棕黄色固体,记作化合物6b。产量36mg,产率66%。
质谱分析确定的分子离子(C
步骤三、无水条件下,将化合物6b(25mg,0.03mmol)溶于8mL干燥的甲苯中。将2,3-二氯-5,6-二氰基-1,4-苯醌(13mg,0.06mmol)溶于2mL干燥的甲苯中。将2,3-二氯-5,6-二氰基-1,4-苯醌的甲苯溶液逐滴转移至反应体系,随后升温80℃反应3h。反应结束体系恢复至室温后,,通过中性氧化铝柱层析(洗脱剂为甲苯)。减压下除去甲苯后,粗品采用200-300目硅胶作为固定相,用二硫化碳作为洗脱剂进行柱层析分离,得到蓝黑色固体,记作化合物6。产量为12mg,产率46%。
质谱分析确定的分子离子(C
实施例7化合物7的合成
步骤一、将化合物2(50mg,0.08mmol),化合物5a(53mg,0.23mmol),四三苯基膦钯(5mg,0.004mmol),2-双环己基膦-2',6'-二甲氧基-1,1'-二联苯(3mg,0.008mmol),磷酸钾(37mg,0.38mmol)投入干燥的带有冷凝管的反应瓶中,进行三次抽真空、通氩气的抽换气操作后,依次注入干燥的甲苯(12mL),通气除氧的乙醇(3mL),通气除氧的水(3mL)。随后升温至90℃反应2h。反应结束后,将反应混合物投入水中,用乙酸乙酯萃取水相三次,所得的乙酸乙酯相水洗一次后合并有机相用无水硫酸钠干燥。减压除去乙酸乙酯后,粗品采用200-300目硅胶作为固定相,乙酸乙酯:石油醚=1:15(体积比)作为洗脱剂进行柱层析分离,得到杏黄色固体,记作化合物7a。产量33mg,产率63%。
质谱分析确定的分子离子(C
步骤二、无水无氧条件下,将化合物7a(26mg,0.04mmol)分散在25mL干燥的四氢呋喃中。在25℃下逐滴加入均三甲基苯基溴化镁(1.0M,0.4mL,0.40mmol),反应30min。反应结束后,反应混合物投入水中,用乙酸乙酯萃取水相。乙酸乙酯层干燥、旋干,得到的固体置于干燥的反应瓶中,溶于25mL干燥的二氯甲烷,25℃条件下逐滴加入三氟化硼乙醚(0.05mL,0.40mmol),反应30min后用2ml甲醇淬灭,终止反应。反应体系旋干,得到浅棕黄色固体,记作化合物7b。产量18mg,产率58%。
质谱分析确定的分子离子(C
步骤三、无水条件下,将化合物7b(22mg,0.03mmol)溶于9mL干燥的甲苯中。将2,3-二氯-5,6-二氰基-1,4-苯醌(14mg,0.06mmol)溶于2mL干燥的甲苯中。将2,3-二氯-5,6-二氰基-1,4-苯醌的甲苯溶液逐滴转移至反应体系,随后升温80℃反应2h。反应结束体系恢复至室温后,通过中性氧化铝柱层析(洗脱剂为甲苯)。减压下除去甲苯后,粗品采用200-300目硅胶作为固定相,用二硫化碳作为洗脱剂进行柱层析分离,得到棕褐色固体,记作化合物7。产量为3mg,产率13%。
质谱分析确定的分子离子(C
实施例8化合物8的合成
将化合物1(95mg,0.11mmol),4,9-二频哪醇硼酯-2,7-二叔丁基芘(30mg,0.05mmol),四三苯基膦钯(3mg,0.003mmol),2-双环己基膦-2',6'-二甲氧基-1,1'-二联苯(2mg,0.006mmol),磷酸钾(56mg,0.26mmol)投入带有冷凝管的反应瓶中,进行三次抽真空、通氩气的抽换气操作后,依次注入通气除氧的甲苯(12mL),通气除氧的乙醇(3mL),通气除氧的水(3mL)。随后升温至90℃反应5h。反应结束后,将反应混合物投入水中,用乙酸乙酯萃取水相三次,所得的乙酸乙酯相水洗一次后合并有机相用无水硫酸钠干燥。减压除去乙酸乙酯后,粗品采用200-300目硅胶作为固定相,二氯甲烷:石油醚=1:5(体积比)作为洗脱剂进行柱层析分离,得到紫色固体(56mg)。
将所得紫色固体与2,3-二氯-5,6-二氰基-1,4-苯醌(20mg,0.09mmol)投入严格烘烤的反应瓶中,进行三次抽真空、通氩气的抽换气操作后,溶于30mL干燥的二氯甲烷中。反应体系降温至0℃稳定片刻后,将三氟甲磺酸(0.08ml)逐滴加入,随后升至25℃反应1h。随后加入3ml甲醇溶液,减压下除去溶剂后,粗品采用200-300目硅胶作为固定相,二氯甲烷比石油醚=1:1(体积比)作为洗脱剂进行柱层析分离,得到紫黑色固体粉末即化合物8。产量15mg,产率31%。
质谱分析确定的分子离子(C
实施例9化合物9的合成
按照实施例8的反应条件及工艺,使用化合物1和(2,6-双(己氧基)萘-1,5-二基)二硼酸为原料,得到紫黑色粉末即为化合物9。
质谱分析确定的分子离子(C
实施例10化合物10的合成
将化合物3(78mg,0.11mmol),4,9-二频哪醇硼酯-2,7-二叔丁基芘(30mg,0.05mmol),四三苯基膦钯(3mg,0.003mmol),2-双环己基膦-2',6'-二甲氧基-1,1'-二联苯(3mg,0.006mmol),磷酸钾(56mg,0.26mmol)投入干燥的带有冷凝管的反应瓶中,进行三次抽真空、通氩气的抽换气操作后,依次注入干燥的甲苯(8mL),通气除氧的乙醇(2mL),通气除氧的水(2mL)。随后升温至100℃反应8h。反应结束后,将反应混合物投入水中,用乙酸乙酯萃取水相三次,所得的乙酸乙酯相水洗一次后合并有机相用无水硫酸钠干燥。减压除去乙酸乙酯后,粗品采用200-300目硅胶作为固定相,二氯甲烷:石油醚=1:6(体积比)作为洗脱剂进行柱层析分离,得到紫色固体(51mg)。
将所得紫黑色固体投入严格烘烤的带有冷凝管的反应瓶中,进行三次抽真空、通氩气的抽换气操作后,溶于30mL干燥的二氯甲烷中。反应体系降温至0℃稳定片刻后,将三氯化铁(78mg,0.48mmol)溶于3ml硝基甲烷后逐滴加入反应体系,0℃反应30min。随后加入3ml甲醇淬灭,终止反应,减压下除去溶剂后,粗品采用200-300目硅胶作为固定相,二氯甲烷比石油醚=1:5(体积比)作为洗脱剂进行柱层析分离,得到紫黑色固体粉末,即为化合物10。产量2mg,产率5%。
质谱分析确定的分子离子(C
实施例11化合物11的合成
按照实施例10的反应条件及工艺,使用反应单体和4,9-二频哪醇硼酯-2,7-二叔丁基芘为原料,得到紫色粉末即为化合物11。其中,反应单体的结构式如下:
/>
质谱分析确定的分子离子(C
实施例12化合物12的合成
按照实施例10的反应条件及工艺,使用化合物3和2',5'-二溴-4,4'-二叔丁基-1,1':4',1”-三联苯为原料,得到紫黑色粉末即为化合物12。
质谱分析确定的分子离子(C
实施例13化合物13的合成
按照实施例10的反应条件及工艺,使用反应单体和6,13-二频哪醇硼酯-2-(叔丁基)-9b硼萘并[1,8-ab]苝为原料,得到紫色粉末即为化合物13。其中,反应单体的结构式如下:
质谱分析确定的分子离子(C
对实施例4中合成的硼杂分子碳化合物4、实施例10中合成的硼杂分子碳化合物10以及实施例11中合成的硼杂分子碳化合物11进行紫外可见吸收光谱分析,结果如图1、图2、图3所示,从图中可以看出,硼杂分子碳化合物4的主吸收峰位在953nm、858nm、783nm、606nm,表现出可见、近红外吸收性质;硼杂分子碳化合物10的主吸收峰位在685nm、575nm、513nm,表现出近红外吸收性质;硼杂分子碳化合物11的主吸收峰位在585nm、546nm、490nm,相比于硼杂分子碳化合物10,其最大吸收蓝移100nm,说明共轭π骨架扩展增大明显可以使化合物的吸收光谱红移至近红外。本发明的硼杂分子碳化合物具有明显的近红外吸收性质,且这些化合物能通过硅胶柱,进行柱层析分离提纯,说明其具备良好的稳定性,有制备近红外材料的应用潜力。
对实施例11中合成的硼杂分子碳化合物11进行电化学测试分析,结果如图4所示,计算可得硼杂分子碳化合物11的HOMO能级和LUMO能级分别为-5.20eV和-3.03eV,带隙为2.17eV。说明本发明的硼杂分子碳化合物的能级结构和窄带隙性质,表明其具有作为双极性有机场效应晶体管材料的潜力。
对实施例4中合成的硼杂分子碳化合物4进行电子顺磁共振(ESR)分析,结果如图5所示,从图中可以看出,硼杂分子碳化合物4在25℃就有ESR信号响应,g
显然,上述实施方式仅仅是为清楚地说明所作的举例,而并非对实施例的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有实施例予以穷举。而由此所引申出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
- 具有聚集诱导发光性质的近红外二区荧光化合物及制备方法、纳米粒胶束及其应用
- 一种碳60-双齿氮杂环卡宾钯(0)化合物及其制备方法和应用
- 一种可靶向线粒体的近红外二区荧光化合物及其制备方法与应用
- 一种具有IDO1/TDO双靶点的小分子化合物及其制备方法与应用
- 一种近红外比率型荧光分子转子、制备方法及其在检测细胞内黏度和蛋白变性中的应用
- 一种具有近红外吸收和近红外发光特性的改性碳纳米点、其制备方法及其应用
- 一种具有近红外吸收和近红外发光特性的改性碳纳米点、其制备方法及其应用