基于第一性原理分子动力学和贝叶斯统计的熔点计算方法
文献发布时间:2023-06-19 19:30:30
技术领域
本发明属于计算材料领域,具体涉及一种基于第一性原理分子动力学和贝叶斯统计的熔点计算方法。
背景技术
对材料熔点的理论预测由来已久,在过去的二十年里,随着计算能力的提高,基于密度泛函理论(DFT)的第一性原理分子动力学(AIMD)模拟成为准确和普遍模拟方法,但基于DFT的熔点预测仍然相当具有挑战性的,因为受限于计算精度和时间的限制。目前常用于熔点计算的方法,例如,基于自由能的方法[ellers Michael S,Lísal Martin,BrennanJohn K.Free-energy calculations using classical molecular simulation:application to the determination of the melting point and chemical potentialof a flexible RDX model],熔点位于固体和液体自由能曲线的交点处,这种方法对吉布斯自由能计算精度要求很高,因为固液自由能曲线间的夹角很小,自由能计算的微小误差都会引起熔化温度的巨大误差;另一种主流方法,即固液共存法[AlfèDario.Temperatureof the inner-core boundary of the Earth:Melting of iron at high pressure fromfirst-principles coexistence simulations],熔点是通过使用AIMD的NVE系综模拟固液共存系统稳定时的温度确定的。由于固相和液相之间存在固液界面,系统保持稳定需要很大的晶胞(通常至少1000原子),并且经过长时间模拟(通常至少20ps以上)才能获取结果。
发明内容
针对上述现有模拟方法的不足,提出一种基于第一性原理分子动力学和贝叶斯统计的熔点计算方法,以解决计算熔点时间过长,成本太高问题。我们减小模拟系统尺寸,建立小尺寸的固液共存系统(200原子左右),在计算AIMD过程中使用机器学习力场,结合贝叶斯方法处理结果,以实现减小计算成本目的。
本发明的技术方案:
一种基于第一性原理分子动力学和贝叶斯统计的熔点计算方法,步骤如下:
S1、对初始单胞结构优化,并建立超胞;
单胞结构从开源材料数据库Materals Project下载或根据Perason's Handbook手动搭建;采用基于DFT的(Vienna Ab-initio Simulation Package)VASP软件对单胞结构进行第一性计算,获得优化后的单胞结构后再制备超胞结构,超胞边长大于
S2、计算估计熔点温度下的热膨胀;
使用NPT系综超胞放至实验温度或各组元平均温度弛豫10ps,将最终500~1000个构型的平均晶格常数作为热膨胀后的晶格常数;然后在相同温度下使用NVT系综弛豫5ps,使体系达到平衡;在第一性原理分子动力学(ab initio molecular dynamics,AIMD)计算过程中,若尺寸超过100个原子,使用Gamma参数来选择k网格点,布里渊区选取1×1×1的网格点;
S3、将热膨胀后的超胞沿Z方向扩胞,固定一半原子,在估计熔点温度以上1000K加热另一半超胞10ps,使其完全熔化,然后在相同温度继续弛豫4ps,每1ps取一个固-液共存构型,共取4个固-液共存构型为下一步弛豫做准备;
S4、将4个固-液共存构型都分别放到估计熔点温度范围(T
(T
其中,Y
这个键序参数是对粒子i周围的局部秩序的一种量化;其中q
就认为两个原子是“相连”的;式中:s
Bokeloh提出用二阶键序参数来区分固液原子,这个参数由s
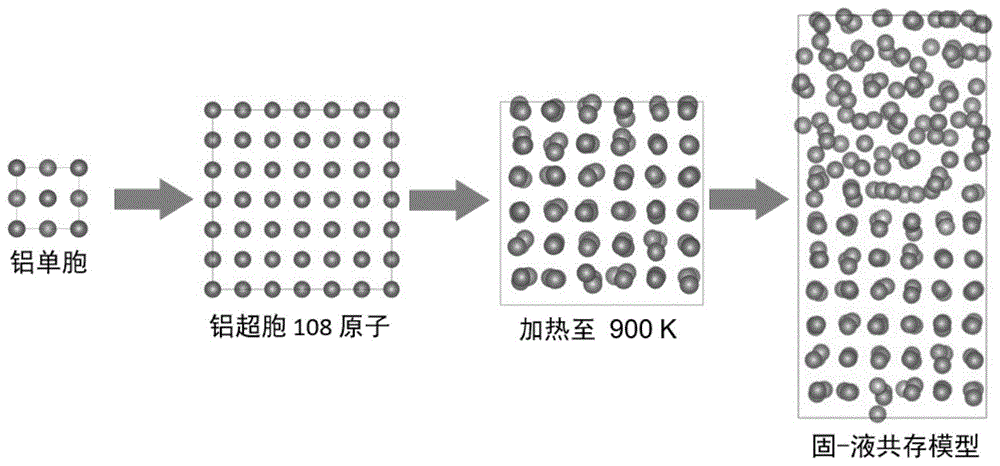
式中: 与现有的技术相比,本发明的有益效果: 本发明通过一种基于第一性原理分子动力学(AIMD)采用小尺寸固液共存构型系统(200个原子左右)计算熔点的方法,并在计算时结合机器学习力场,可以显著减少计算量。采用键序参数判断固液状态,再结合贝叶斯方法处理固液分布数据,可以得到熔化概率随温度变化的曲线。 附图说明 图1是共存相的模拟计算熔点的算法示意图; 以纯铝的熔点计算为例,首先对铝初始单胞结构优化,采用3×3×3方式扩胞;计算估计熔点温度下(900K);再延Z方向扩胞,并熔化半个超胞,制备固-液共存的构型。绿色为固体原子,棕色为液体原子。 图2是使用Steinhardt键序参数判断固液原子分布图; 首先使用Steinhardt键序参数计算固液共存构型的固体原子数,初始化avgthreshold参数,使得固体原子数为108,总原子数为216;然后就可以用此参数判断不同温度下构型的固液状态:例如,铝的实验熔点为933K,当共存构型在800K下弛豫10ps时固体原子数为216,超过了总原子数80%,可以判断为固体构型;在1100K下弛豫10ps时固体原子数为0,可以判断为液体构型。 图3是熔点拟合:(a)为不同温度下各构型±100K范围内的200个结构液体原子数的分布,判别液体原子数分别大于80%时,认为该构形继续计算后其状态是为液体。(b)为贝叶斯拟合温度曲线图,采用贝叶斯统计方法对处理后的数据进行计算。图中红色曲线对应50%概率时的温度为熔点。 具体实施方式 以下结合附图和技术方案,进一步说明本发明的具体实施方式。 以纯铝熔点举例计算,过程如下。 S1、对初始单胞结构优化,并建立超胞 从开源材料数据库Materals Project下载铝单胞结构,第一性计算采用基于DFT的Vienna Ab-initio Simulation Package(VASP)软件。电子交换关联泛函采用具有局域密度近似(LDA)进行计算。结构优化使用平面波截断能为400eV,能量变化收敛阈值10 S2、计算估计熔点温度下的热膨胀 将扩胞后的使用NPT系综将铝超胞放至估计温度弛豫10ps,统计最终500个构型的平均晶格常数作为热膨胀后的晶格常数,再使用NVT系综弛豫5ps,使体系达到平衡。使用Gamma方法来选择k网格点,布里渊区选取1×1×1的网格点。使用机器学习功能来加速计算。 S3、延Z方向扩胞,并熔化半个超胞,制备多个固-液共存的构型 将热膨胀后的超胞沿Z方向扩胞,固定一半铝原子,在2000K加热另一半超胞10ps,使其完全熔化,然后再继续弛豫4ps,每1ps取一个固液共存构型,共取4个构型为下一步做准备。 S4、收集固液分布结果,使用贝叶斯统计方法拟合熔点(T 将固-液共存的构型放到估计熔点温度范围(800~1100K)弛豫20ps,收集固液分布结果,使用基于python的pyscal软件包计算各构型的Steinhardt键序参数,得出各构型固体原子数。首先计算图2铝固液共存模型中的固体原子数,发现当average threshold为0.6时,图2固液共存模型的固体原子数为108,即为50%,average threshold校验完成。然后计算各个温度下弛豫完成的构型的固体原子数,如图2中铝的800K构型固体原子数为216;1100K构型的固体原子数为0。当固体原子数大于80%时,认为该构形继续计算后其状态是为固体,反之认为是液体,各构型的液体原子百分比如图3(a)所示。判断构型的固液状态后,将固体构型中的所有原子都视为固体原子,反之为液体原子,分别对应图3(b)中熔化概率为0和100的黑点。再使用贝叶斯统计方法对处理后的数据进行计算,拟合处理后的固体和液体原子数,得到熔化概率随温度变化的曲线。图3(b)中曲线对应50%概率时的温度941±3K为铝的熔点,与实验熔点933K基本一致。本方法在800~1100K范围内共计算了16个小尺寸的固液构型,消耗约600核时,与其他方法相比(通常5000核时以上)大幅缩短计算时间,不需要复杂的自由能计算,因而具有很好的应用潜力。