一种调控鱼类红色素的基因pax7序列、方法及应用
文献发布时间:2024-04-18 19:58:21
技术领域
本发明属于生物技术领域,具体涉及一种调控鱼类红色素的基因pax7序列、方法及应用。
背景技术
鱼类具有六种主要的色素细胞,包括黑色素细胞、黄色素细胞、虹彩细胞、红色素细胞、白色素细胞和蓝色素细胞,这些色素细胞协同作用进而影响鱼类的体色。然而,除了模式鱼类斑马鱼和青鳉都具有的黑色素细胞、黄色素细胞和虹彩细胞,对其他色素细胞的研究较为少见。罗非鱼(Oreochromis niloticus)具有黑色素细胞、黄色素细胞、虹彩细胞以及模式鱼类所没有的红色素细胞,是研究红色素细胞的良好模型。但到目前为止,红色素细胞分化和形成的关键基因还未被鉴定。
红色是自然界常见的一种颜色,许多养殖鱼类特别是观赏鱼具有鲜艳的红色。罗非鱼是世界第三大淡水养殖鱼类,全球总产量约为610万吨。红罗非鱼是罗非鱼的一个变种,鲜红的体色使其具有更高的经济价值。红罗非鱼的红色主要由红色素细胞决定,因此鉴定影响红色素细胞分化和形成的关键基因对于创制出更加鲜红的罗非鱼具有指导意义。
基因编辑技术是目前进行基因功能鉴定的常用技术。由于真骨鱼类特有的基因组复制,pax7在多数鱼类均有两个拷贝,pax7a和pax7b。众所周知,pax7是影响斑马鱼和青鳉黄色素细胞分化的关键基因,但对红色素细胞的影响还不清楚。
发明内容
本发明利用CRISPR/Cas9基因编辑技术将罗非鱼pax7基因突变发现可以消除红色素细胞,突变红罗非鱼pax7基因可以获得红色消失的银白色罗非鱼。
本发明第一目的在于提供一种能调控鱼类红色素细胞分化的基因序列pax7,所述基因序列pax7包含两个序列,分别是pax7a(GeneID=100708659)和pax7b(GeneID=100696153)。
本发明另一目的在于提供一种上述基因序列pax7的两个用于gRNA体外合成的靶序列,核苷酸序列为SEQ ID NO.8和SEQ ID NO.9所示,分别来自pax7a基因和pax7b基因;或者SEQ ID NO.8和SEQ ID NO.9所示的核苷酸序列上几个碱基缺失或增加衍生产生的核苷酸序列。
本发明又一目的在于提供上述pax7基因序列在控制鱼类红色素细胞消除的方法。
一种pax7基因控制鱼类红色素细胞消除的方法,其特征在于:
(1)将pax7a和pax7b的gRNA混合并与Cas9 mRNA同时注射到野生型罗非鱼雌鱼与雄鱼交配获得的发育到I细胞期受精卵,采用PCR扩增-桑格尔测序和限制性内切酶消化的方法,筛选出雄性pax7a/b嵌合突变体;
(2)将雌鱼与雄性pax7a/b嵌合突变体交配,得到F1代鱼,采用罗非鱼PCR扩增-桑格测序和限制性内切酶消化的方法,筛选出基因型为pax7a/b
(3)将性成熟的pax7a/b
(4)对不同发育时期的pax7a/b
(5)使用相同方法对红鲫的pax7基因进行突变,获得F0代嵌合突变体。
进一步地,本发明提供一种pax7基因序列在控制鱼类红色素细胞消除中的应用。
本发明的有益效果:
本发明提供了一种pax7基因在控制鱼类红色素细胞分化中的应用,证实pax7基因是控制罗非鱼和红鲫红色素细胞分化和形成的关键基因,对创制更红或无红色素的罗非鱼提供了重要的指导意义。鱼类具有六种主要的色素细胞,包括黑色素细胞、黄色素细胞、虹彩细胞、红色素细胞、白色素细胞和蓝色素细胞,这些色素细胞协同作用进而影响鱼类的体色。然而,由于在斑马鱼和墨鱼等模式生物中没有红色素细胞,因此,对红色素细胞基因知之甚少,到目前为止,红色素细胞分化和形成的关键基因还未被鉴定。pax7基因前期已被证明与黄色素细胞分化相关,然而该基因是在没有红色素细胞的斑马鱼中被证实,对红色素细胞分化没有可预见性。
本发明利用基于CRISPR/Cas9基因编辑技术将pax7a和pax7b双突变,发现该基因为鱼类红色素细胞分化和形成的关键基因,pax7a/b突变导致罗非鱼和红鲫红色素细胞和黄色素细胞缺失,本发明鉴定的控制红色素细胞分化和形成的关键基因对创制出更具有经济价值的罗非鱼具有重要指导意义。
附图说明
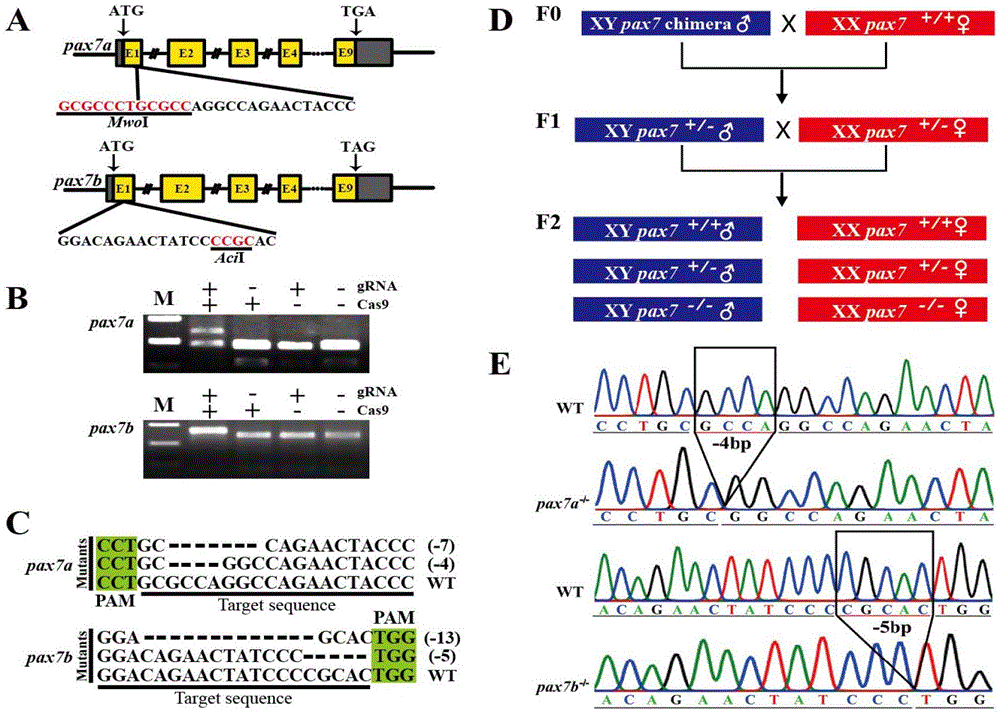
图1:CRISPR/Cas9技术突变罗非鱼的pax7基因(A:pax7a和pax7b的基因结构、突变靶位点和限制性酶切位点;B-C:通过酶切和Sanger测序验证突变;D:pax7a/b F0至F2代建系示意图示意图;E:野生型以及pax7a和pax7b纯合突变体基因测序结果比较)。
图2:pax7突变导致尼罗罗非鱼黄色素细胞缺失(A-D:受精后30天野生型、pax7a
图3:pax7突变导致尼罗罗非鱼红色素细胞缺失(A-D:受精后120天野生型、pax7a
图4:pax7突变导致尼罗罗非鱼和红罗非鱼红色消失(A-C:正常野生型的尼罗罗非鱼和两种红罗非鱼;D-F:pax7a/b
具体实施方式
1、基因敲除靶序列的选择和引物的合成
通过NCBI官网(https://www.ncbi.nlm.nih.gov/)分别对罗非鱼pax7a、pax7b的全基因序列进行下载。利用在线网站(http://zifit.partners.org/ZiFiT/)分别在两个基因的第一个外显子上设计基因编辑靶序列,靶序列核苷酸序列如SEQ ID No.8和SEQ IDNo.9所示。设计合成gRNA的正向引物和反向引物gRNA-pax7a-F、gRNA-pax7b-F和gRNA-R(具体序列见表1)。
表1:本发明中用于PCR扩增的引物
2、gRNA的体外合成
以gRNA质粒为模板(质粒信息见Chang et al.,Genome editing with RNA-guided Cas9nuclease in zebrafish embryos.Cell Res.2013Apr;23(4):465-72),分别用设计好的F引物(SEQ ID NO:1和SEQ ID NO:2)和通用R引物(SEQ ID NO:3)进行扩增并回收片段。扩增体系如下:50μL反应体系为2×Taq Master Mix 25μL;gRNA-F 1μL;gRNA-R 1μL;gRNA质粒(100ng/μL)0.3μL;补无酶水至总体积50μL。反应条件已试剂说明书为准,特别的,其中退火温度60℃;延伸时间10s。
以回收产物为模板进行体外转录,步骤如下:
(1)20μL反应体系为10×T7 Buffer2μL;T7 RNA聚合酶1μL;rNTP混合物1μL;上述扩增的gRNA模板800-1000ng;RNA酶抑制剂1μL;补无酶水至总体积20μL。37℃水浴反应2h;
(2)加入1μLDNase I(RNase free)除去体系中的DNA,37℃水浴反应15min;
(3)加入50μL 100%无水乙醇和2μL的pH为5.3的3mol/L NaAc;轻轻涡旋,-30℃保存2小时或者4℃过夜保存。
(4)4℃13000rpm离心30min;弃去上清;
(5)70%乙醇(DEPC水配制)清洗;
(6)4℃13000rpm离心15min,弃去上清,室温干燥5-10min;
(7)根据沉淀量加入适量无酶水溶解,测定浓度,电泳检测质量。gRNA应该具有一条100bp左右的单一亮带,合成的浓度应该在500ng/μL以上。-80℃保存备用,注意整个实验过程防止RNA酶的污染。
3、Cas9 mRNA的合成
质粒提取试剂盒提取Cas9质粒,利用XbaI对Cas9质粒进行线性化处理,37℃5h,加入5% SDS和80μg/μL蛋白酶K,50℃处理30min,Qiagen PCR产物直接回收试剂盒进行纯化,具体步骤参照TaKaRa Xba I酶切体系和Qiagen PCR产物直接回收试剂盒操作。用无酶水将线性化后的Cas9模板稀释至500-1000ng/μL。注射用mRNA按照T7 mMESSAGE
(1)20μL反应体系为10×Transcription buffer 2μL;T7 RNA聚合酶2μL;10×dNTP混合物2μL;Cas9模板500-1000ng;RNA酶抑制剂1μL;补无酶水至总体积20μL。
(2)37℃水浴反应3h;
(3)加入1μL TURBO DNase处理残余Cas9模板,37℃水浴反应10min;
(4)加入30μL LiCl和30μL无酶水,-80℃放置1-2h;
(5)4℃13000rpm离心30min;
(6)弃去上清,70%乙醇洗涤一次;
(7)4℃13000rpm离心15min,弃去上清,室温干燥5-10min;
(8)加入20μL无酶水溶解,Nanodrop 2000检测RNA浓度及OD值,电泳检测RNA质量。一般Cas9 mRNA的浓度要求要在1000ng/μL以上,电泳后应有一条在2kb以上的单一条带。-80℃保存备用。实验过程中注意防止RNA酶的污染。
4、显微注射
将pax7a和pax7b的gRNA混合并与Cas9 mRNA同时注射到野生型罗非鱼雌鱼与雄鱼交配获得的发育到I细胞期受精卵。将合成好的Cas9 mRNA浓度稀释至1000ng/μL,gRNA500ng/μL,按照gRNA/Cas9 mRNA终浓度为250/500ng/μL的比例混合,加入10%体积比的酚红作为指示剂。将罗非鱼受精卵置于无菌培养皿中,使用显微注射系统对单细胞期受精卵进行注射,注射剂量约为每个受精卵400~600pg mRNA,注射后的受精卵置于26℃恒温循环水孵化系统(CN 102177861A)中进行孵化。
5、基因组DNA的提取、目的片段扩增及突变检测
显微注射并孵化后72h,将20个受精卵混合并采用酚/氯仿法提取基因组DNA,采用酚/氯仿法提取基因组DNA,具体方法如下:从无水乙醇中取出预提基因组的组织并剪碎,加入0.65mL STNE(NaCl:100mmol/L,pH 8.0;EDTA:1mmol/L,pH 8.0;Tris-HCl,10mmol/L,pH8.0)、0.1mL 10% SDS和10-20μL 20mg/mL的蛋白酶K并温和混匀(颠倒数次,动作一定要轻,防止基因组DNA断裂),置于55℃摇床中(100rpm)消化过夜;加入650μL酚/氯仿/异戊醇(25:24:1)混合液,温和混匀,12000rpm离心10min,回收上清;重复酚/氯仿/异戊醇抽提过程2次,然后换成每管600μL氯仿抽提,回收的上清液加入等体积异丙醇,温和混匀后4℃静置2h,4℃13000rpm离心30min,弃上清;75%、100%乙醇各洗涤1次;自然晾干,DDW溶解,Nanodrop 2000检测OD及浓度。取30μg DNA于Eppendorf管中用200μL的吸头轻轻吹打随机剪切DNA,1.0%低熔点琼脂糖凝胶电泳,-20℃保存备用。通过设计的每个基因的扩增引物进行PCR扩增(引物见表1)。同时,提取野生型对照组个体的基因组DNA,扩增出目的条。通过1.5%琼脂糖凝胶电泳分离目的条带,然后进行胶回收。利用靶序列上的酶切位点对回收产物进行酶切,电泳检测是否存在未切开的条带并加以测序确定是否具有活性。
6、F0代阳性鱼个体的筛选
F0代阳性个体采用Mwo I和Aci I酶切方法,第一,提取实验鱼基因组DNA,第二,PCR扩增实验鱼pax7a和pax7b靶点基因组(引物见表1),同时扩增野生型基因组DNA作为对照。第三,酶切500ng基因组片段,1.5%琼脂糖电泳,溴化乙锭(EB)染色,拍照,突变个体DNA酶切会出现3条带,1条没有被切开的突变了的条带和2条切开的条带主要来自野生型,Quantity One软件(Bio-RAD,美国)计算酶切条带的光密度值,第四,为了确切得知F0代突变个体的突变序列,对没有切开的条带进行切胶回收、连接pMD19T载体后,进行亚克隆测序验证(如图1所示)。
7、pax7突变系构建流程
采用pax7a/b嵌合F0代的XY雌性个体建立突变系。建系流程如下:
(1)筛选精子中含移码突变的F0代XY雄鱼与野生型XX雌鱼交配,繁殖F1代;
(2)从F1代中筛选各种突变类型的XX和XY实验鱼,F1代杂合突变个体同样采用MwoI和Aci I酶切方法进行筛选,为确切得知F1代突变个体的突变序列,对没有切开的条带进行切胶回收、连接pMD19T载体后,进行亚克隆测序验证。
(3)选取突变类型为pax7a(-4bp)和pax7b(-5bp)的双杂合突变鱼交配,繁殖F2代。从中筛选出基因型为pax7a/b
8、红鲫pax7基因突变
使用相同方法对红鲫的pax7基因进行突变,获得F0代嵌合突变体。
9、图像采集与处理
用间氨基苯甲酸乙酯甲磺酸盐(MS-222)作为麻醉剂对0-90dpf尼罗罗非鱼进行麻醉,用莱卡体视显微镜对其进行图像采集。3月龄以上的罗非鱼采用尼康数码相机和奥林巴斯光学显微镜进行图象采集。为了对罗非鱼的黄色素细胞和黑色素细胞进行细胞计数,将其在放入含有肾上腺素的培养液中进行处理。使用Adobe photoshop CS6调整图像对比度和色彩平衡,对用作对照的图像进行相同的处理。使用Adobe Illustrator CS6进行图像拼接。
10、不同发育时期的pax7a/b
罗非鱼受精后30天野生型、pax7a
- NKIRAS2基因调控区序列、调控序列及其在鼻咽癌中的应用
- 两条特异性靶向猪Pax7基因的sgRNA导向序列及应用
- 两条特异性靶向猪Pax7基因的sgRNA导向序列及应用