适于通过拉伸工序制备的微针材料的适合性测试方法以及包括其的微针的制备方法
文献发布时间:2023-06-19 12:07:15
技术领域
本发明涉及适于通过拉伸工序制备的微针材料的适合性测试方法以及包括其的微针的制备方法。
背景技术
已开发出许多用于治疗疾病的药物及生理活性物质等,但是在将药物和生理活性物质输送到体内的过程中,生物障碍(biological barrier,例如皮肤、口腔粘膜及脑血管屏障等)通过问题以及药物输送的效率问题仍有待改善。
药物和生理活性物质通常以片剂或胶囊剂形式口服给药,但是由于诸如在胃肠道中被消化或被吸收或者被肝机制损失等的原因,许多药物不能仅通过上述给药方法有效地输送。况且,一些药物不能通过肠粘膜而有效地扩散。而且患者的依从性也是一个问题(例如,对于应按规定间隔服药或无法服药的重症患者的情况等)。
用于输送药物和生物活性物质的另一种常用技术是使用以往的注射针(needle)。尽管与口服给药相比这种方法是有效的,但是存在在注射部位引起疼痛及皮肤局部损伤、出血以及在注射部位引起疾病感染等的问题。
为了解决上述口服给药和皮下注射的问题,使用了利用贴剂的透皮给药方法。使用贴剂的透皮给药副作用少,而且患者的依从性也高,并且易于保持药物的血药浓度。
为了解决如上所述的问题,已经开发了包括微针(microneedle)在内的各种微结构体。迄今为止开发的微针主要用于体内药物输送、体内血液以及体内分析物的检测等。
与以往的注射针不同,微针的特点是无痛的皮肤渗透和无创伤,在无痛的皮肤渗透的情况下,顶部(top)直径对于最小程度的侵袭性很重要。此外,由于微针必须穿透作为皮肤中最强大的障碍物的10-20μm的角质层(stratum corneum),因此需要具有足够的物理硬度。并且,还应考虑适当的长度以到达至毛细管来增加药物输送的效率。
以往提出了面内(In-plane)型微针(“Silicon-processed Microneedles”,Journal of microelectrochemical system 8,1999)之后,开发了各种类型的微针。在使用蚀刻方法的面外(out-of-plane)型固体微针(美国专利申请公开第2002138049号“Microneedle devices and methods of manufacture and use thereof”)的制备方法中生产出直径为50-100μm,长度为500μm的固体硅微针,但是无法实现无痛的皮肤渗透,而且很难将药物和美容成分输送到目标位置。
并且,美国佐治亚大学的普劳施尼茨(Prausnitz)提出了一种通过蚀刻玻璃或通过光刻法(photolithography)制备模具来制备可生物降解的高分子微针的方法。使用此方法的优点是可以自由加载能够制备成胶囊形式的药物,但是随药物加载量的增加,微针的硬度会变弱,因此在需要大量药物的情况下其应用受到限制。
2005年,由Nano Device and System公司提出吸收型微针(日本专利申请公开第2005154321号)。
这种吸收型微针旨用于药物输送或美容,而无需去除插入皮肤中的微针。在该方法中,将由麦芽糖(maltose)和药物混合而成的组合物添加到模具中并凝结来制备微针。上述日本专利提出通过将微针制成吸收型来进行药物的透皮吸收,但是当刺入皮肤时会伴随疼痛。并且,由于模具制备的技术限制,而无法制备出具有伴随无痛的适当的上端直径而且有效输送药物所需的长度即长度为1mm以上的微针。
2008年美国乔治亚大学的普劳施尼茨制备的可生物降解的微针是在聚二甲基硅氧烷(Polydimethylsiloxane:PDMS)模具中使用聚乙烯吡咯烷酮(Polyvinypyrrolidone:PVP)和甲基丙烯酸(Methacrylic acid:MAA)的混合物来制备的。并且,将羧甲基纤维素放入金字塔结构的模具中来制备微针。但是,使用模具的制备方法的局限性在于,为了调整微针的直径和长度,必须通过复杂的工序来制备新的模具和框架,而且其缺点在于,将材料放入模具中来制备微针的工序复杂且耗时长。
2008年,日本的Mukai等人通过美国申请的专利发布了一种使用针结构来制备皮肤针(skin needle)的装置及方法(美国专利申请公开US20080157421A1)。此方法使用通过使用销(pin)结构在基板上的基底上加热来拉动粘性物质的方法。由于此方法使用了利用销结构物来拉动被热熔化或具有粘性的物质的方法,因此需要根据所需的图案新制备销结构的工序,从而难以克服如下局限性:增加了生产成本,由于加热工序而难以加载对热敏感的各种生物药品(激素、疫苗、其他蛋白质药品等)的问题。
另一方面,皮肤由表皮的角质层(<20μm)、外皮(epidermis)(<100μm)及真皮(dermis)(300~2500μm)组成。因此,为了将药物和生理活性物质无痛地输送到特定的皮肤层,需要将微针的上端部的直径制成小于30μm,而且将有效长度制成用于穿透200~2000μm厚度的皮肤的足够硬度,由此有效地输送药物和皮肤美容成分。并且,为了通过可生物降解的固体微针输送药物和生理活性物质,有必要在微针制备过程中排除可能破坏药物和生理活性物质的活性的工序,例如高温热处理、有机溶剂处理等。
由于制备方法上的局限性,现有的固体微针仅限于硅、高分子、金属、玻璃等的材料,并且由于使用利用成型技术的制备方法,因而会产生如下缺点:因复杂且制备过程时间长而导致的药物变性、硬度不足以及药物损失等。因此,持续存在对穿透皮肤时能够实现无痛并且具有足够的长度以渗透到皮肤深处,且实现足够的硬度而对材料没有特别限制并且使药物损失最小化的微针的制备方法的需求。
为了解决如上所述的现有技术的问题,本发明人提出了称为鼓风拉伸方式(droplet air-born blowing)的新的微针的制备方法。本发明人提出的鼓风拉伸方式的各种微针的制备方法已通过韩国专利申请第10-2009-94018号、第10-2010-30127号、第10-2010-130169号、第10-2012-117936号、第10-2015-174066号及第10-2016-61903号等申请。上述本发明人的韩国专利申请的内容应被视为该整体并入本文。
在由本发明人提出的与现有的成型或微成型方法不同的鼓风拉伸方式的微针的制备方法中,采用粘性组合物作为微针材料。这是因为只有在具有粘性的材料的情况下,才可以在接触并拉伸后通过鼓风干燥制备成微针。
另一方面,在本发明的申请日之前,除了上述鼓风拉伸方式之外,已知几种拉伸方式的微针的制备方法。其中之一是称为离心光刻(centrifugal lithography)的工序。在离心光刻工序中,将粘性组合物提供给旋转体。粘性组合物被旋转体旋转时产生的离心力拉伸。当拉伸的粘性组合物的外侧端部接触位于旋转体外部的板时,形成沙漏形状并进行凝固。之后,沙漏形状的中间部分被切割而形成两个微针。与上述鼓风拉伸方式相比,其特征在于,在不经过单独的鼓风凝固工序而通过离心力拉伸的情况下,粘性组合物的水分蒸发的同时结构凝固。
如上所述,鼓风拉伸方式和离心光刻法之间在是否包括鼓风凝固工序方面存在差异,但是它们的相同之处在于通过拉伸粘性组合物来制备微针形状,这与上述成型方式不同。本发明以拉伸方式为前提而不是成型方式,并且本发明作为前提的这种拉伸方式应被理解为包括通过拉伸上述粘性组合物来成型微针的已知的所有方式,例如上述鼓风拉伸方式、离心光刻方式等。
如上所述,可以使用各种物质作为用于通过拉伸方式制备微针的粘性组合物。例如包含:透明质酸及其盐、聚乙烯吡咯烷酮、纤维素高分子(cellulose polymer)、葡聚糖、明胶、甘油、聚乙二醇、聚山梨酸酯、丙二醇、聚维酮、卡波姆(carbomer)、亚榄仁树胶(gumghatti)、瓜尔豆胶、葡糖酸、葡糖胺、达玛树脂(dammer resin)、凝乳酶酪蛋白(rennetcasein)、刺槐豆胶(locust bean gum)、微纤化纤维素(microfibrillated cellulose)、车前籽胶(psyllium seed gum)、黄原胶、阿拉伯半乳聚糖(arabino galactan)、阿拉伯胶、海藻酸、明胶、结冷胶(gellan gum)、卡拉胶、卡拉亚胶(karaya gum)、可得然胶(curdlan)、壳聚糖、甲壳质、他拉胶(tara gum)、罗望籽果胶(tamarind gum)、黄蓍树胶(tragacanthgum)、丹麦琼脂(furcelleran)、果胶(pectin)或者普鲁兰多糖(pullulan)。还可以使用除此之外的各种粘性物质,而不限于上述例示的物质。在与本发明人的上述鼓风拉伸方式有关的韩国专利申请的说明书中公开的粘性物质也可属于可用作微针材料的粘性组合物的候选组。
然而,不能断定具有粘性的材料就均适于拉伸方法。进而,拉伸方式中提供的材料不仅仅由已被确认适合用于形成微针的粘性组合物的材料组成。而是可以进一步包含各种生物活性药物,并且可以变更配比。如果以往已确认形成微针的适合性的材料进一步包含其他成分,或者变更配比,则应重新评估微针形成的适合性。
仅通过投入于实际工序中来形成各种长度的微针的实际过程投入之后通过评估过程,才能判断此材料是否适于通过拉伸方式的制备方法形成微针。
发明内容
技术问题
本发明的目的在于提供适于通过拉伸工序制备的微针材料、使用这种材料的微针的制备方法、微针材料的适合性测试方法以及包括其的微针的制备方法。
解决问题的手段
本发明一实施例的微针的制备方法包括:选择并提供微针材料的步骤,其中,使用粘弹性测定装置测定的上述微针材料的每个剪切速率下的粘性模量和弹性模量的值均属于预定的上限值与下限值之间;以及通过拉伸方式将在上述步骤中选择并提供的微针材料制备成微针的步骤。
在如上所述的微针的制备方法中,当选择微针材料时,可以进一步检查上述测定的粘性模量和弹性模量的值是否随着剪切速率的增加而持续减小。
另一方面,本发明一实施例的微针材料的适合性测试方法包括:使用粘弹性测定装置测定每个剪切数率下的微针材料的粘性模量和弹性模量的步骤;检查在上述每个剪切速率下测定的值是否均属于预定的上限值与下限值之间的步骤;以及当上述检查结果为肯定时,判断为上述微针材料适于通过拉伸方式成型微针的步骤。
在如上所述的微针材料的适合性测试方法中,可以进一步检查微针材料的测定的粘性模量和弹性模量的值是否随着剪切速率的增加而持续减小。
在上述微针的制备方法和其材料的适合性测试方法中,优选地,每个剪切速率下的粘性模量和弹性模量的预定上限值和下限值可以如下。
当剪切速率为0.03(1/s)时,上限值为47.17,下限值为6.67;
当剪切速率为0.04(1/s)时,上限值为35.07,下限值为4.86;
当剪切速率为0.06(1/s)时,上限值为25.37,下限值为3.77;
当剪切速率为0.10(1/s)时,上限值为18.40,下限值为3.06;
当剪切速率为0.16(1/s)时,上限值为13.47,下限值为2.56;
当剪切速率为0.25(1/s)时,上限值为10.06,下限值为2.18;
当剪切速率为0.40(1/s)时,上限值为7.71,下限值为1.88;
当剪切速率为0.63(1/s)时,上限值为5.99,下限值为1.65;
当剪切速率为1.01(1/s)时,上限值为4.71,下限值为1.46;
当剪切速率为1.62(1/s)时,上限值为4.08,下限值为1.32;以及
当剪切速率为2.59(1/s)时,上限值为2.89,下限值为1.09。
这些每个剪切速率下的上限值和下限值是本发明人通过重复实验得出的结果,并且是在实现本发明之前本发明所属技术领域的专家中未知的新边界值,可通过应用基于这种边界值的标准,可以轻松而准确地判断适于拉伸工序的微针材料的辨别。
并且,附加配置也可以包括在本发明中。
发明的效果
根据本发明,可以提供适于通过拉伸工序制备的微针材料、使用这种材料的微针的制备方法、微针材料的适合性测试方法及包括其的微针的制备方法。
更具体地,根据本发明,可以提供预先确认其制备适合性的微针材料,而无需通过投入于实际的拉伸工序来形成微针的工序,并且可以提供使用这种微针的微针制备方法。另一方面,根据本发明,当新接收用作微针材料的物质时,可以提供能够在投入于工序之前判断适合性的测试方法,并且可以提供包括这种测试方法的制备方法。由此,即使在添加新物质或变更配比等的情况下,也可以在投入于工序之前判断适合性。
如上所述,通过提供本发明的新颖且特征性的微针材料或者微针材料的测试方法或者使用上述微针材料的制备方法或者包括上述测试方法的制备方法,可以在投入于工序之前辨别适于拉伸工序的微针材料,从而提高整个工序的效率。由于可以客观有效地辨别合适的材料,因此可以提高利用拉伸法生产的微针的质量。
附图说明
图1为用于本发明的微针材料的适合性测试方法的粘弹性测定装置。
图2为对各种材料测定正切(tan delta)值的曲线图(x轴为粘弹性测定装置的剪切速率(1/s)值,y轴为正切值)。
图3为示出通过拉伸确认到制备适合性的正切值的范围的曲线图。
图4为示出对于透明质酸以外的各种高分子的正切值的测定结果的曲线图。
具体实施方式
以下对于本发明的详细说明可以参考通过示例的方式示出可以实施本发明的特定实施例的附图。对这些实施例的详细描述足以使本发明所属技术领域的普通技术人员能够实施本发明。应当理解为本发明的各种实施例相互不同,但不必相互排斥。例如,可以理解为本发明的说明书中描述的特定形状、结构和特性在不脱离本发明的思想和范围的情况下可以从一个实施例变更至另一个实施例。并且,应当理解为在不脱离本发明的思想和范围的情况下,也可以变更各个实施例中的个别结构要素的位置或布置。因此,下面将要说明的详细描述不是限制性的,并且本发明的范围应视为包括发明要求保护范围的权利要求请求的范围及与其等同的所有范围。
以下,参考附图对本发明的各种优选实施例进行详细说明,以使本发明所属技术领域的普通技术人员能够容易地实现本发明。
图1为用于本发明的微针材料的适合性测试方法的粘弹性测定装置。如图1所示的材料的粘弹性测定装置在本技术领域中被称为流变仪(rheometer)。流变仪是一种通过将试料加载到使用空气轴承来最大程度地减少摩擦的上板与固定的下板之间来测定试料的粘弹性的装置。
以如下方式进行测定,即,在测定试料的粘弹性之前,通过测定装置的摩擦力来在实际测定试料的粘弹性时排除设备本身的摩擦。上部的板呈平板形或圆锥形等形状,这些所使用的形状和大小可以根据试料的粘性或其他特性而变化。例如,当测定诸如粘性极低的水之类的物质时,存在只能通过使用较大面积的板来测定出的程度的电阻。在两个板或者圆锥形装置和板或者诸如杯和鲍勃(cup&bob)系统之类的其他类似几何形状之间加载试料。当扭矩施加到上部板时,旋转剪切应力会施加到物质上,并测定所得的应变或应变率(剪切率)。使用图1所示的粘弹性测定装置,即流变仪对本发明的说明书中公开的材料的粘弹性物性进行测定时的测定条件如下。
-温度:25℃
-几何间距:1mm
-剪切速率:0.03~2.59 1/s
-试料量:0.3mL
-使用的板:20mm的平板形
由本发明人使用图1所示的粘弹性测定设备测定的材料的固有值是介质损耗角正切(tangent of delta)值。简称正切(tan delta)值。本发明人发现,粘性组合物的正切值与是否可以使用上述粘性组合物来成型微针以及与可成型的微针的长度有关。即使为了制备微针而选择新的原料物质或选择现有物质,当所要选择新配比时,可以通过测定粘性组合物的正切值来预先确认微针成型的可能性。
高分子是同时具有粘性(viscosity)和弹性(elasticity)的粘弹性(viscoelasticity)物质。简而言之,具有充分粘性的物质在不受任何外力的情况下会改变形状。与此相反,具有充分弹性的物质对外力具有很强的抵抗力,并且当上述力消失时会立即恢复到原始状态。高分子的性质介于两种物质的中间一处。例如考虑橡胶球。当将橡胶球掉在地板上时,其会反弹到低于原始高度的位置。如此跳上来的是弹性。那么,不能恢复到原始高度的原因在于,其失去了对应的力量。确切地说,将力量以热的形式输出到外部,这就是粘性。
上述正切值定义为材料的粘性模量和弹性模量。较小的上述值表示弹性强而弹性模量高。上述值大表示粘性强而粘性模量高。
图2为对各种材料测定正切(tan delta)值的曲线图,其中x轴为用粘弹性测定装置测定时的剪切速率值(1/s),y轴为正切值。通过以各种配比混合高分子量透明质酸和低分子量透明质酸来制备图2的实验中使用的物质。
图2中的虚线或点划线所示的曲线图是不适合微针成型的材料的正切值的趋势。相反,实线所示的曲线图是适合微针成型的材料的正切值的趋势。
参照图2,可以确认根据适于微针成型的材料的剪切速率的增加的正切值的位移的特征。
第一个特征是正切值随着剪切速率的增加而减小。用虚线表示的不适合微针成型的材料之一表现出如下趋势,其中正切值增加到特定的剪切速率(约0.06 1/s),然后在该特定的剪切速率之后减少。表现出这种趋势的材料被判断为不适合通过拉伸方式成型为微针的材料。
第二个特征是正切值应在特定范围内。图2的曲线图中存在正切值随着剪切速率的增加而减小的两个点划线图,它们表现出正切值随着剪切率的增加而减小的趋势,但上述值不属于适合成型微针的范围。
本发明人对各种分子量的透明质酸进行反复实验以获得相当于上述第二特征的上限值和下限值,结果获得了图3所示的结果。
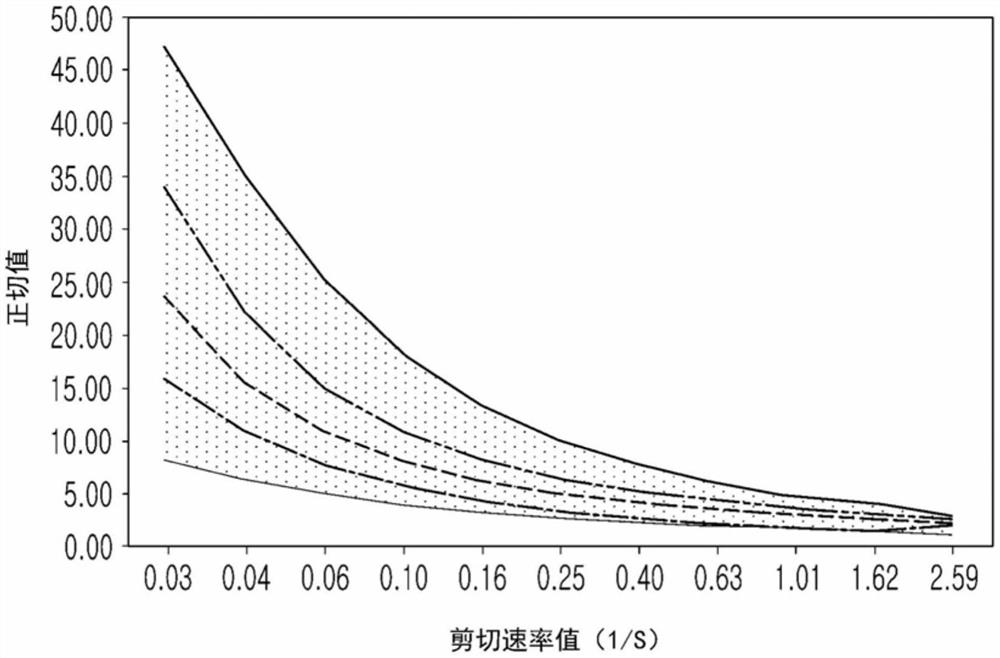
图3为示出通过拉伸确认到制备适合性的正切值的范围的曲线图。
在图3所示的曲线图中,顶部实线曲线图和底部实线曲线图分别是上限和下限,并且具有属于所表示的上限与下限之间区域的正切值的材料被判断为适于成型微针的材料。
在图3所示的曲线图中,每个剪切速率下的可成型为微针的上限值和下限值如下。
当剪切速率为0.03(1/s)时,上限值为47.17,下限值为6.67;
当剪切速率为0.04(1/s)时,上限值为35.07,下限值为4.86;
当剪切速率为0.06(1/s)时,上限值为25.37,下限值为3.77;
当剪切速率为0.10(1/s)时,上限值为18.40,下限值为3.06;
当剪切速率为0.16(1/s)时,上限值为13.47,下限值为2.56;
当剪切速率为0.25(1/s)时,上限值为10.06,下限值为2.18;
当剪切速率为0.40(1/s)时,上限值为7.71,下限值为1.88;
当剪切速率为0.63(1/s)时,上限值为5.99,下限值为1.65;
当剪切速率为1.01(1/s)时,上限值为4.71,下限值为1.46;
当剪切速率为1.62(1/s)时,上限值为4.08,下限值为1.32;以及
当剪切速率为2.59(1/s)时,上限值为2.89,下限值为1.09。
图3均示出对于透明质酸的正切值的测定结果,但是多个曲线图分别示出了具有不同分子量的透明质酸的测定结果。在图3中,顶部的实线曲线图表示最低分子量的透明质酸,底部的实线曲线图表示具有最高分子量的透明质酸。从图3的结果可以看出,分子量越大,随剪切速率而引起的正切值的变化越小,分子量越小,随剪切速率而引起的正切值的变化越大。更具体地,在图3的曲线图中,随剪切速率而引起的正切值的变化最大的透明质酸(分子量最小的透明质酸)至正切值的变化最小的透明质酸(分子量最大的透明质酸)的分子量依次为450kDa、510kDa、530kDa、540kDa、550kDa。
图3示出了在测定具有各种分子量的透明质酸的正切值的同时,对是否可以通过拉伸方式成型微针进行实验的结果。本发明人不限于透明质酸,而且还对其他物质进行了实验,以了解是否可以将相同正切值的范围用于判断通过拉伸方式成型微针的可能性。其结果在图4中示出。
图4为示出对于透明质酸以外的各种高分子物质的正切值的测定结果的曲线图。
在图4所示的曲线图中,每个剪切速率下的各种高分子物质的测定的正切值如下。
0.03 1/s:羟丙基甲基纤维素(HPMC)4.95,右旋糖酐(Dextran)7.52,软骨素(Chondroitin)6.7,聚乙烯聚吡咯烷酮(PVP)23.47
0.04 1/s:羟丙基甲基纤维素5.00,右旋糖酐5.39,软骨素4.89,聚乙烯聚吡咯烷酮15.61
0.06 1/s:羟丙基甲基纤维素5.13,右旋糖酐4.03,软骨素3.8,聚乙烯聚吡咯烷酮10.94
0.10 1/s:羟丙基甲基纤维素5.33,右旋糖酐3.09,软骨素3.09,聚乙烯聚吡咯烷酮8.11
0.16 1/s:羟丙基甲基纤维素5.59,右旋糖酐2.44,软骨素2.59,聚乙烯聚吡咯烷酮6.28
0.25 1/s:羟丙基甲基纤维素5.87,右旋糖酐1.98,软骨素2.21,聚乙烯聚吡咯烷酮5.05
0.40 1/s:羟丙基甲基纤维素6.14,右旋糖酐1.68,软骨素1.91,聚乙烯聚吡咯烷酮4.18
0.63 1/s:羟丙基甲基纤维素6.41,右旋糖酐1.32,软骨素1.68,聚乙烯聚吡咯烷酮3.54
1.01 1/s:羟丙基甲基纤维素6.72,右旋糖酐1.17,软骨素1.49,聚乙烯聚吡咯烷酮3.10
1.62 1/s:羟丙基甲基纤维素6.89,右旋糖酐1.14,软骨素1.35,聚乙烯聚吡咯烷酮2.58
2.59 1/s:羟丙基甲基纤维素6.97,右旋糖酐1.11,软骨素1.12,聚乙烯聚吡咯烷酮2.05
图4的实验目的在于,当基于正切值判断是否能够通过拉伸方式成型微针时,确认相同的标准是否可以应用于各种高分子物质,而不仅限于透明质酸。
为此,测定了羟丙基甲基纤维素(HydroxyPropyl MethylCellulose,HPMC)、右旋糖酐(Dextran)、软骨素(Chondroitin)、聚乙烯聚吡咯烷酮(Polyvinylpyrrolidone,PVP)等四种物质的粘弹性,在羟丙基甲基纤维素的情况下,表现出超过可成型范围的值,右旋糖酐、软骨素、聚乙烯聚吡咯烷酮满足了可成型标准。在这些四种材料中,羟丙基甲基纤维素、右旋糖酐、软骨素均为多糖(polysaccharide)系列,但聚乙烯聚吡咯烷酮属于高分子物质而不是多糖,因此包含在材料组中。
图4显示了用于判断适合性的正切值范围。示出用不同类型的线测定出的上述四种物质的正切值的趋势。在羟丙基甲基纤维素的情况下,不仅确认了不属于预定的正切值范围内的区域,而且与其他三种物质不同,正切值具有随着剪切速率的增加而持续减小的倾向,从而可以判断出不满足可成型标准。
实际上,使用每种物质作为材料通过拉伸方法进行了微针成型,结果,基于正切值获得了与预期相同的结论。即,实际上用羟丙基甲基纤维素无法进行成型,而右旋糖酐、软骨素和聚乙烯聚吡咯烷酮实际上能够进行成型。
通过本实验,确认到正切值可以用作拉伸过程中的是否可成型微针的标准,并且这不限于透明质酸,而且可以普遍应用于各种高分子物质中。
如上所述,确认了微针材料固有的粘弹性,更具体地,确认了正切值与是否可以成型为微针之间的相关关系。
尽管已经通过诸如结构要素之类的特定项和受限制的实施例以及附图来具体描述了本发明,但这仅仅是为了有助于更全面地理解本发明,而本发明不限于上述实施例,只要是本发明所属技术领域的普通技术人员,就可以根据这些描述进行各种修改和变形。
因此,本发明的思想不应限于上述所描述的实施例而定,并且除了后述的发明要求保护范围,与该发明要求保护范围均等或等同变化的所有范围均属于本发明的思想范围内。